
Abstract
Phagocytic engulfment and digestion of extracellular material are essential for debris removal, metabolic recycling, tissue remodeling, and inflammation control after brain injury. However, the coordination of phagocytosis across cell types and disease contexts remains incompletely understood. Ju et al. demonstrate that remote ischemic conditioning enhances CD36-dependent phagocytic activity in recruited monocytes/macrophages, which contributes to improved stroke recovery. Building on these important findings, we suggest that effective brain repair requires coordinated phagocytosis across multiple cell types, extending beyond apoptotic neurons to diverse injury-associated substrates. We further argue that intracellular signaling, aging, sex, and circadian biology shape phagocytic efficiency and therapeutic potential.
Phagocytosis, the cellular process of cargo engulfment and digestion, is typically initiated by cytokines and chemokines that recruit immune cells towards injured tissues. A subset of the infiltrating phagocytes then acquires elevated phagocytic capacities, characterized by upregulation of substrate recognition receptors, cytoskeleton remodeling, and activation of lysosomal enzymes. In this process, surface receptors determine which substrates are engulfed, whereas lysosomal function dictates how effectively internalized debris is processed. Efferocytosis is a specialized form of phagocytosis that refers to the recognition and clearance of apoptotic cells. Successful digestion by phagocytes helps prevent secondary necrosis and release of damage-associated molecular patterns (DAMPs), thereby suppressing additional pro-inflammatory mediators, such as IL-1β, IL-6, and TNF-α. Lysosomal degradation generates lipid-derived metabolites that activate pro-resolving transcriptional programs and promote anti-inflammatory cytokines and trophic factors, such as IL-10, TGF-β, and osteopontin, to restore immune homeostasis for functional recovery.1,2 Although phagocytosis is generally considered helpful for brain repair in the stroke research field, 2 its overall impact, including efferocytosis of dying neurons, is likely to depend on the understudied biological factors, such as age, sex, metabolic resources, the intracellular context of signal transduction, and the surrounding chemical milieu.
Recent work by Ju et al. provides a major mechanistic breakthrough by identifying efferocytosis as a central pathway through which remote ischemic conditioning (RIC) promotes stroke recovery. Their study demonstrates that RIC induced a shift in circulating monocytes toward a CD36high/Ly6Chigh phenotype and enhanced their chemotaxis into the injured brain, where infiltrating cells markedly amplified CD36-dependent clearance of apoptotic neurons. Genetic deletion of CD36 abolished RIC-enhanced efferocytosis and eliminated its protective effects against delayed transneuronal degeneration, firmly establishing CD36-dependent clearance as essential for functional recovery. 3 These findings reveal a targetable immune mechanism through which a simple, non-invasive intervention can drive neurorepair for stroke.
Although Ju et al. primarily examines efferocytosis of apoptotic neurons, their work highlights the broader importance of considering other phagocytic substrates critical for network integrity. The early study by Chang et al. showed that erythrocyte clearance drives reparative macrophage polarization and improves outcomes after intracerebral hemorrhage. 4 Emerging evidence indicates that efficient phagocytosis of myelin debris is essential for white matter repair, as inadequate clearance by microglia and macrophages leads to debris accumulation and impaired remyelination and axonal regeneration, particularly in the aged brain.5,6 In a mouse model of Alzheimer’s disease, perivascular cell-derived osteopontin induces a distinct phagocytic microglial state that promotes synaptic engulfment. 1 Beyond neural debris, removal of infiltrating immune cells represents an additional mechanism whereby phagocytosis may restrain inflammatory amplification in the injured brain.
While Ju et al. focus on CD36-dependent clearance mediated by recruited monocytes/macrophages, their findings raise broader questions on the coordination of debris removal from the injured brain. The diversity of phagocytic substrates also implies that post-injury clearance is unlikely to be restricted to microglia and macrophages. Besides professional phagocytes, perivascular astrocytes and other related vascular-associated cells drive microglia toward specialized phagocytic states that enhance synaptic and debris clearanc. 1 Notably, astrocytes exhibit distinct metabolic programs that shape their responses to injury and energy demands in the brain. 7 In addition, microvascular endothelial cells engulf myelin debris via IgG-dependent mechanisms and process the debris through the autophagy-lysosome pathway, thereby modulating inflammation, angiogenesis, and tissue remodeling after CNS injury. 8
Although Ju et al. established CD36 as a mediator of RIC-enhanced monocyte/macrophage efferocytosis, their findings open exciting opportunities to further explore intracellular signaling pathways that connect CD36 engagement to cytoskeletal remodeling and inflammation resolution. Prior studies indicates that CD36 interfaces with multiple receptors and signaling pathways, including lipid-sensing and inflammatory networks. TAM receptors, including MerTK and Axl, cooperate with CD36 to facilitate lipid uptake from apoptotic membranes through bridging molecules such as Gas6 and Protein S. Activation of TAM signaling enhances engulfment efficiency and actively promotes a pro-resolving microglial phenotype by suppressing inflammatory pathways.2,4 In parallel, other lipid-sensing receptors may also contribute to processing lipid-rich debris generated after brain injury.
Beyond ligand recognition, CD36 functions as a signaling integrator that links cargo uptake to intracellular reprogramming. Engagement of CD36 by apoptotic membranes promotes cytoskeletal remodeling required for engulfment and generates lipid-derived signals that activate nuclear receptor-dependent programs favoring inflammation resolution. CD36-mediated uptake also intersects with STAT6/Arg1-associated anti-inflammatory pathways that support efferocytosis and limit secondary tissue injury. 2 We argue that CD36-dependent clearance reflects coordinated signaling and metabolic adaptation that ultimately determine whether phagocytosis resolves post-injury inflammation.
Aging and sex are important biological factors determining phagocytic capacities. Although aged phagocytes retain myelin uptake, they exhibit defective lipid metabolism and lysosomal processing, leading to debris accumulation and failure to resolve inflammation, acting as a repair bottleneck. Aging also alters microglial phagocytic efficiency and regulation.6,9 Female microglia display higher intrinsic phagocytic activities than male microglia under baseline conditions, yet exhibit reduced flexibility in adapting to inflammatory conditions. 9 Another important but underappreciated factor is circadian biology, as intrinsic clocks regulate macrophages and microglia metabolism and inflammation, with circadian control of redox reactions influencing immune responses. 10 These observations suggest that the efficacy of phagocytosis-based therapies, including RIC, may vary according to different circadian phases, an important consideration for both experimental design and clinical translation.
Collectively, these findings frame efferocytosis as a gateway to broader phagocytosis-driven brain repair, extending beyond apoptotic neurons to white matter debris, into a multicellular phagocytic network involving not only microglia and other professional phagocytes, but also astrocytes and endothelial cells. Targeting specific stages of phagocytosis, particularly post-engulfment metabolic and degradative pathways, may refine and improve the precision of therapeutic strategies against brain injury (Figure 1).
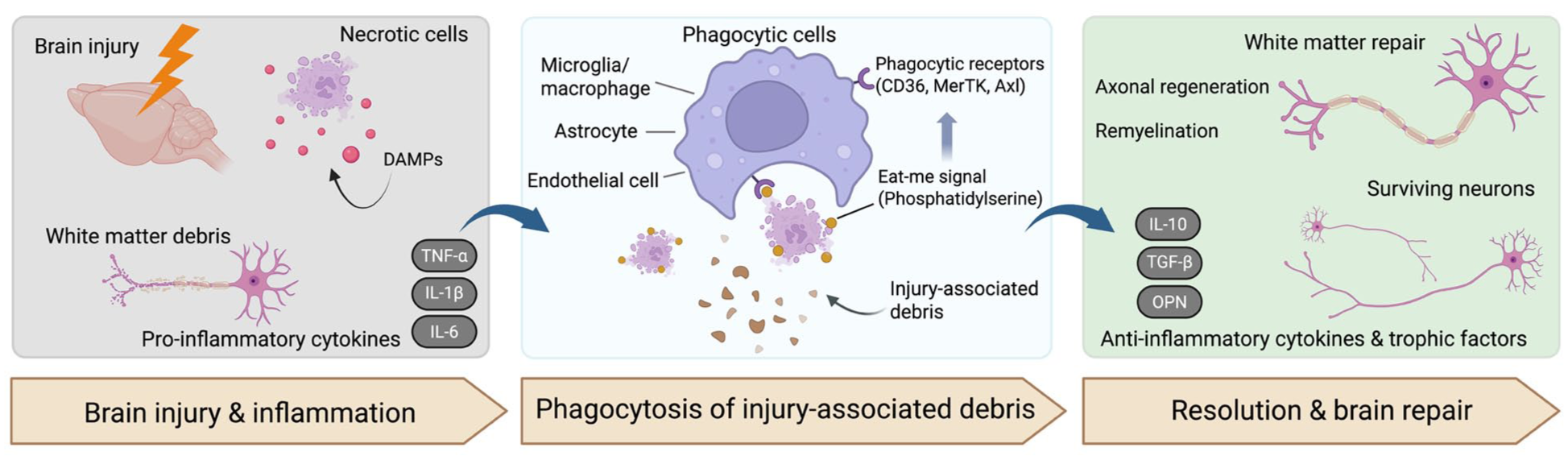
Schematic illustration of phagocytosis in coordinating inflammatory resolution and brain repair following acute brain injury. Brain injury from stroke or trauma results in necrotic cell death and tissue damage, leading to the release of DAMPs, the accumulation of myelin and axonal debris, and the production of pro-inflammatory cytokines (e.g. TNF-α, IL-1β, and IL-6), which together propagate neuroinflammation. In the subacute phase, multiple phagocytic cell types, including microglia/macrophages, astrocytes, and endothelial cells, recognize injury-associated debris via phagocytic receptors such as CD36, MerTK, and Axl in response to “eat-me” signals like phosphatidylserine. Efficient clearance of cellular and myelin debris promotes the resolution of inflammation. It is accompanied by the production of anti-inflammatory cytokines and trophic factors (e.g. IL-10, TGF-β, and OPN). These coordinated processes support white matter repair (axonal repair, regeneration, and remyelination) and the preservation of stressed neurons, thereby supporting brain repair and functional recovery.
Footnotes
Author contributions
JL drafted the manuscript. RL, WZ, and W-C W provided critical revisions and intellectual input. JC conceptualized and supervised the work. All authors reviewed and approved the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the VA SRCS Award (821-RC-NB-30556) and VA merit review grants (I01 BX005290 and I01BX003377), the AHA Transformational Award (969858), and NIH grants (NS138326 and NS124673).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
This Commentary does not report original data, samples, or models. Therefore, data availability is not applicable.