
Abstract
Futile reperfusion is a phenomenon of inadequate perfusion despite successful recanalization after acute ischemic stroke (AIS). It is associated with poor patient outcomes and has received increasing interest due to its clinical diagnosis becoming more common. However, the underlying mechanisms remain elusive, and experimental studies are focused on the pathological background of futile reperfusion. Our recent study has confirmed that poor primary collateralization plays a crucial role in the insufficiency of reperfusion after AIS in mice. Specifically, the absence of primary collaterals in the circle of Willis (CoW) promoted the development of spreading depolarizations (SDs) during AIS. In our experimental stroke model, the occurrence of SDs during ischemia always predicted futile reperfusion. Conversely, in mice with a complete CoW, no SDs were observed, and reperfusion was complete. Importantly, the human CoW displays variation in the primary collaterals in approximately 50% of the population. Therefore, futile reperfusion may result from SD evolution in AIS patients. Our purpose here is to emphasize the crucial role of SD in the development of futile reperfusion. We propose that adequate collateral recruitment can prevent SD occurrence, leading to improved reperfusion and AIS outcomes.
Keywords
Incomplete circle of Willis (CoW) predisposes to futile reperfusion after acute ischemic stroke (AIS)
Futile reperfusion refers to inadequate perfusion that occurs after acute ischemic stroke (AIS), despite the removal of the obstructing thrombus from the cerebral vessel through recanalization therapy.1,2 Despite the technically successful recanalization, patients with AIS may still have poor outcomes and increased mortality rates when diagnosed with futile reperfusion.1,3 Although an unfavorable clinical diagnosis is often associated with futile reperfusion, our understanding of the underlying mechanisms is limited and elusive. Experimentally identified pathologies behind futile reperfusion include capillary pericyte constriction,4,5 neutrophil obstruction of capillaries, 6 and the localization of the region affected by the ischemic injury. 7 Recent studies have highlighted the decisive role of cerebrovascular anatomical variations and collateralization in the restoration of cerebral blood flow after a stroke.8,9 A recent article of ours confirms that incomplete primary posterior collateralization (incomplete circle of Willis) can lead to spreading depolarization (SD) and futile reperfusion after AIS. 10 The purpose of this opinion paper is to explore the role of poor perfusion in the development of SD and the mechanistic background of SD-related futile reperfusion.
Spreading depolarization is a slowly propagating (2–6 mm/min) wave of almost complete neuronal and glial depolarization accompanied by a progressive reduction in cerebral blood flow, leaving arterioles and capillaries permanently constricted. 11 The pathological potential of SD lies in the associated vasoconstriction and cytotoxic edema of glial and neuronal cells,12,13 which trigger the maturation of primary brain infarcts and cause the transformation of the ischemic penumbra into an ischemic core after AIS. 14 In our experimental setting, futile reperfusion (referred to as “reperfusion failure” in the original article) was causally related to the prior occurrence of SD (Figure 1). 10 Importantly, both the spontaneous occurrence of SD during ischemia and the subsequent futile reperfusion after AIS were dependent on the richness of primary collateralization at the level of the circle of Willis (Figure 1(a)). Indeed, the ipsilateral absence of the P1 segment of the posterior cerebral artery (incomplete circle of Willis) invariably led to a profound drop in CBF (<25% of baseline perfusion), SD occurrence and futile reperfusion of the ipsilateral hemisphere (Figure 1(b) and (c)). In contrast, neither SD nor futile reperfusion was observed on the contralateral side when the circle of Willis was complete (P1 segment was present). 10 Based on our data, we propose a detrimental role of primary collateralization in the development of SD and futile reperfusion after AIS.
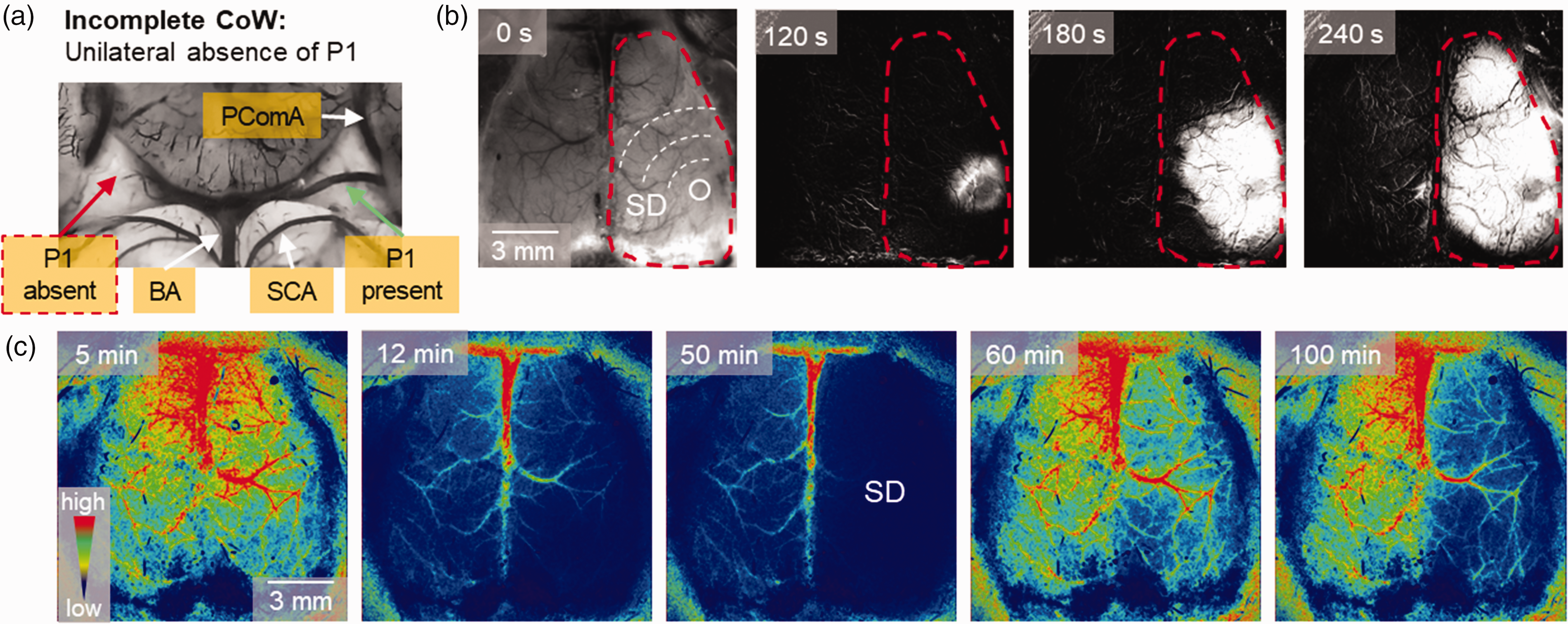
Incomplete circle of Willis (CoW) promotes SD evolution and futile reperfusion in C57BL/6 laboratory mice. (a) A representative image of the mouse CoW after carbon black ink perfusion demonstrates the unilateral presence (left, green arrow) or absence (right, red arrow) of the P1 segment of the posterior cerebral artery. Abbreviations: BA: basilar artery, PComA: posterior communicating artery, SCA: superior cerebellar artery. (b) Representative background subtracted IOS image sequences show the unilateral propagation of SD in the mouse cortex. Note, that SD developed in the hemisphere ipsilateral to the absence of P1. (c) Representative color-coded CBF maps of the same preparation demonstrate futile reperfusion in the hemisphere ipsilateral to SD.
Incomplete CoW is detrimental to SD incidence
In species with a closed circulatory system, the collateral circulation provides an essential secondary reservoir to maintain blood supply in the event of occlusion of the main vessel. 15 Obstruction of arterial blood flow due to thromboembolism or various hemodynamic disturbances leads to the recruitment of the collateral circulation.9,15 Collateral recruitment refers to the increased blood flow through anastomotic connections to compensate for the reduced perfusion resulting from blood vessel occlusion. 15 In the brain, collateral recruitment depends on the diameter and patency of the primary and secondary collateral vessels.9,15 The circle of Willis forms the basis for primary collaterals, which redirect perfusion to poorly perfused areas, such as ischemic regions, through functional anastomoses. Anatomical variations that result in the absence or dysfunction of these primary collaterals can be particularly problematic during interrupted perfusion, when local ATP and glucose deprivation exacerbate the outcome of acute AIS.16,17
The C57BL/6 laboratory mouse strain is well-suited to study the effects of vascular anatomical variation on AIS outcome due to its incomplete circle of Willis. 18 This is caused by variable patency of the P1 segment of the posterior cerebral artery, which is absent or hypoplastic in approximately 80% of the mouse population. 18 In our study, unilateral or bilateral absence or hypoplasia of the P1 segment was observed. 10 The absence of the P1 segment following AIS (experimental common carotid artery occlusion) caused a marked reduction in global perfusion of the posterior circulation in the ipsilateral hemisphere, leading to progressive flow reduction in the posterior parietal, auditory, and visual cortices of mice (Figure 2(a), (b1) and (b2)). Although the P1 segment primarily supplies the midbrain and thalamus, its absence indirectly and invariably affects these areas. The poorly perfused posterior parietal cortex (CBF threshold of SD < 25%) is the typical focal area for the development of SD (Figure 2(b1) and (b2)). Therefore, it is suggested that there is a causal relationship between the absence of the P1 segment, marked flow reduction and the occurrence of SD. Previous experimental observations suggest that an incomplete circle of Willis can promote ischemic infarct volume expansion due to insufficient perfusion of structures outside the middle cerebral artery territory. It has been confirmed that an incomplete circle of Willis is detrimental due to the profound reduction in perfusion of the ipsilateral hemisphere caused by AIS (Figure 2(b1) and (c2)). This, in turn, triggers the development of SD and futile reperfusion as a consequence.
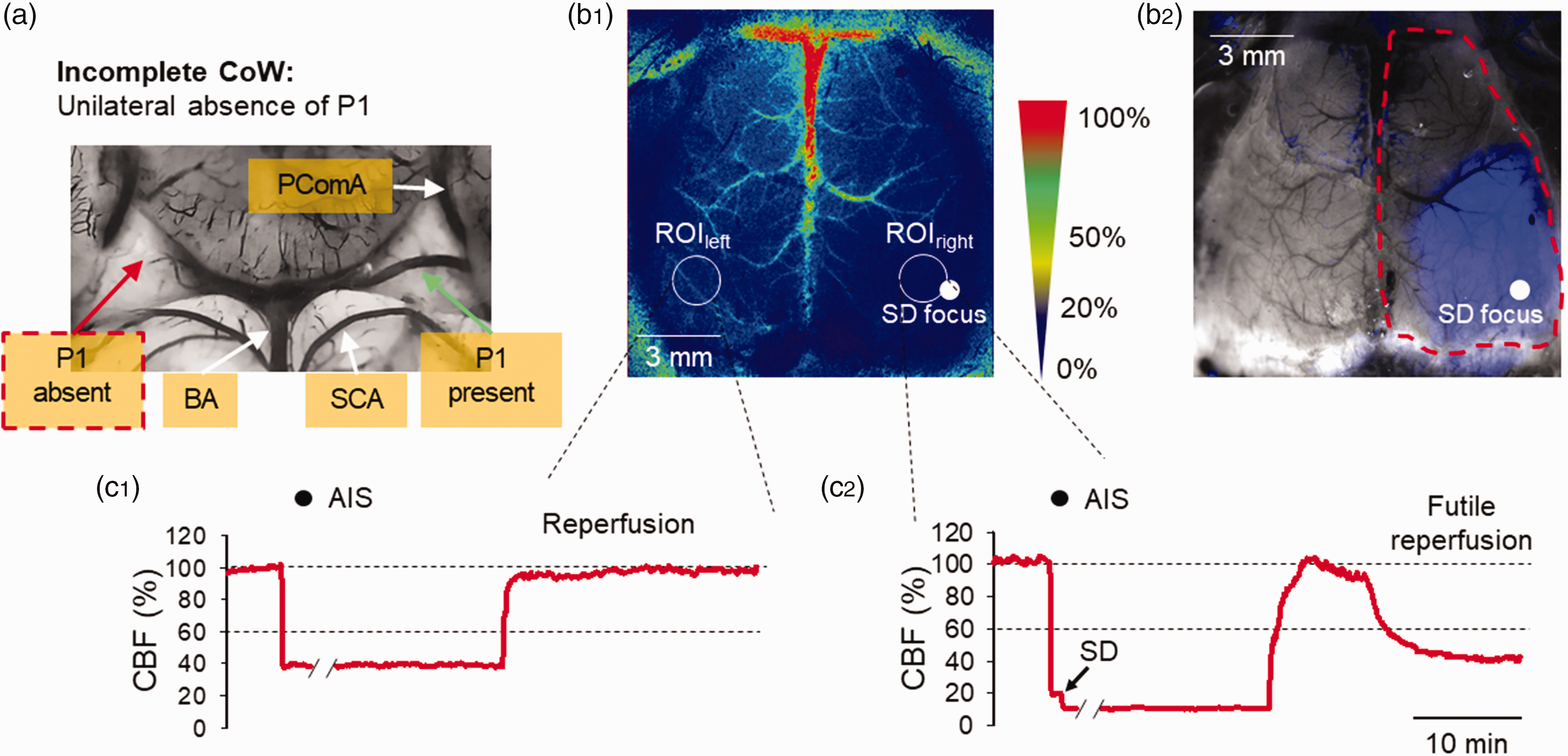
Low perfusion (<30%) develops after AIS and triggers unilateral SD in a mouse with incomplete circle of Willis (CoW). (a) A representative image of the mouse CoW labeled with intraluminal carbon black ink. Note the unilateral absence (right side) of the P1 segment (red arrow). (b1) Representative color-coded CBF map of the same preparation displaying the heterogeneity of perfusion drop after AIS. Note that the low CBF region (CBF < 30%) in the right hemisphere develops ipsilaterally to the absence of the P1 segment and triggers SD (SD focus: white full sphere). In contrast, the CoW is complete on the left side and the CBF drop is less profound (CBF > 30%) in the left cortical hemisphere. White dashed circles are regions of interest (ROIs) placed on both hemispheres to extract CBF traces. (b2) Image of the same preparation under green light illumination, the low CBF region calculated from CBF maps is superimposed on the image (blue area). Importantly, SD (white full sphere representing SD focus) readily occurs in the poorly perfused region of the cortical hemisphere. (c1) A representative CBF trace taken from ROIleft demonstrates that the CBF threshold for SD elicitation was not reached, and reperfusion was optimal after AIS. (c2) A representative CBF trace taken from ROIright displays a profound perfusion drop after AIS promoting SD and futile reperfusion. Note that diagonal black symbols on the x axes indicate axis break after 16 minutes of AIS. Black spheres over the traces indicate the time points where the pictures in B were acquired.
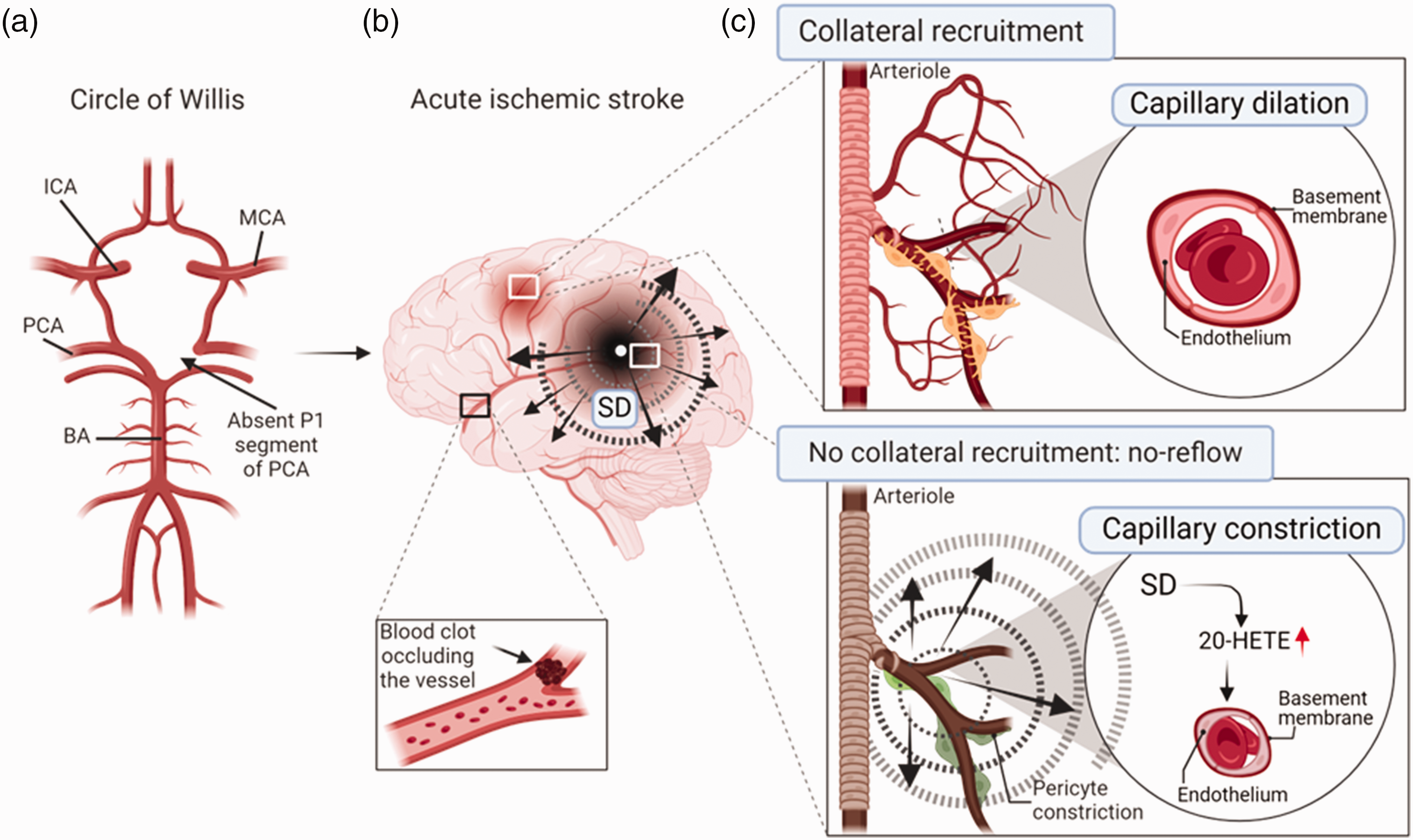
Concept: An incomplete circle of Willis determines the outcome of AIS after middle cerebral artery occlusion in the human brain. (a), Schematic illustration of the absence of the left P1 segment of the posterior cerebral artery, which is manifested with a ∼50% probability in the human circle of Willis. Abbreviations: ICA: internal carotid artery, PCA: posterior cerebral artery, BA: basilar artery, MCA: middle cerebral artery. (b) Schematic illustration of the heterogeneity of perfusion changes in AIS. Unilateral absence of primary collaterals (such as the P1 segment of the PCA) causes a marked CBF drop after AIS in the parietal region of the cortex, where consequently SD develops. In contrast, a rostral region is also shown, where the CBF drop is less severe due to the better collateralization of the territory, and no SD is observed. (c) Overview of the pathology of the no-reflow phenomenon at the cellular level. The upper box presents the normal collateral recruitment, and capillary dilation, that occurs in the rostral cortical region, where the AIS-related perfusion drop remains above the threshold for SD generation. The lower box shows no collateral recruitment and the no-reflow phenomenon. The no-reflow phenomenon is mediated by SDs, which induce the synthesis and accumulation of 20-hydroxieicosatetranoic acid (20-HETE) in the injured region. The accumulation of 20-HETE causes a tonic capillary constriction, which contributes significantly to the no-reflow phenomenon.
Anatomical anomalies of the primary collaterals are common in the human population. In fact, the circle of Willis is incomplete in more than 50% of healthy adults, 19 and the number of missing segments positively correlates with adverse neurological outcomes after AIS. 20 Clinical studies show that, like the C57BL/6 mouse strain, anomalies of the human circle of Willis frequently affect (∼50%) the posterior circulation of the brain.21,22 The most common cases involve the hypoplasia or absence of the posterior communicating artery or the P1 segment of the posterior cerebral artery.21,23 While posterior hypoplasia refers to the underdeveloped (smaller than normal) posterior communicating or posterior cerebral artery P1 segment, the complete absence of these vessels is usually referred as a fetal variation of the circle of Willis. The fetal type P1 or posterior communicating artery results from the embryonic derivation of the posterior cerebral artery from the internal carotid artery. 24 People with fetal-type posterior cerebral artery are at an increased risk of vascular insufficiency in both the anterior and posterior cerebral circulation during an ischemic challenge. 24 Taken together, the absence of collateral flow through the circle of Willis or flow through only one unilateral posterior communicating artery indicates poorer cerebral perfusion.20,25 In contrast, confirmation of a complete circle of Willis predicts better outcomes from ischemia and AIS. 20
SD occurrence implies capillary no-reflow phenomenon
The term ‘no-reflow' refers to microvascular constriction that triggers the development of futile reperfusion despite successful reopening of the occluded vessel. This phenomenon has been confirmed both experimentally and clinically, and typically develops 30–40 minutes after restoration of perfusion.5,26 It is usually preceded by a brief period of hyperperfusion (Figure 2), then the prolonged hypoperfusion that follows is due to capillary constriction resulting from decreased oxygen concentration and severe ischemic damage. 1 Our original observation showed that futile reperfusion (CBF < 60% of baseline CBF) invariably develops in the hemispheres affected by SD (Figure 2). 10 Therefore, SD should be considered as a major contributor to the no-reflow phenomenon. SD causes prolonged vasoconstriction, known as spreading oligemia, which affects arterial divisions from the pial arteries to the capillary beds. 27 Spreading oligemia develops 1–2 minutes after the depolarization wave and persists for 1–3 hours in preclinical models. 28 We propose that spreading oligemia associated with SD extends into the phase of reperfusion and mediates the no-reflow phenomenon of the SD-affected hemisphere. The vasoconstrictor 20-hydroxyeicosatetranoic acid (20-HETE) is considered to be the primary modulator to maintain oligemia after SD. 28 The metabolic, biochemical conversion of 20-HETE from arachidonic acid is restricted to pathological conditions because the CYP450 ω-hydroxylase enzymes that synthesize 20-HETE are inactive in healthy tissues. 29 Reducing the synthesis of 20-HETE effectively limited spreading oligemia after SD evoked in the otherwise optimally perfused cortex. 28 Furthermore, the accumulation of 20-HETE 24 hours after middle cerebral artery occlusion was found to cause pericyte constriction and capillary occlusion in the mouse cortex. 29 Additional data indicate that elevated levels of 20-HETE in cerebrospinal fluid and plasma are predictive of adverse clinical outcomes in AIS patients.30 –32 More direct evidence suggests that GPR75, a newly discovered receptor for 20-HETE, is highly expressed in the human brain. 33 Garcia et al. demonstrated that the G-protein receptor 75 (GPR75) serves as a specific target of activation for 20-HETE in human endothelial cells.33,34
Taken together, an increased acute accumulation of 20-HETE may have contributed significantly to our original observation, since the release of this vasoconstrictor, has been shown to be stimulated by SDs.28,35 Hypothetically, vascular anatomical variations could favor similar scenarios in the human brain, however, the mechanistic background of the SD-related CBF response is more complex. One obvious limitation in the translation of our hypothesis is that while spreading oligemia is regularly observed in mouse AIS models, 36 it is rarely seen in stroke patients. 37 In fact, the CBF response to SDs in patients suffering from malignant hemispheric stroke, subarachnoid hemorrhage, or traumatic brain injury are usually started with an ischemic episode of marked vasoconstriction, known as spreading ischemia.38,39 The confirmed mediators of spreading ischemia are the decreased availability of cortical NO combined with an increase in extracellular potassium. 11 On the other hand, in the ischemic brain, where the prominent SD-related vasoconstriction dominates the response, spreading oligemia cannot be isolated in any species, but mediators of oligemia can be produced. Indeed, SD clusters are routinely monitored by surface or depth electrodes in the brains of patients with acute brain injury. 14 Although cortical folding or gyrification and extensive vascular arborization might prevent entire cortical hemispheres from being affected by SD in AIS, 40 smaller, specialized regions may still be at greater risk than others due to an incomplete circle of Willis. For example, inadequate posterior circulation could lead to poor collateral recruitment and the development of SD in posterior cortical regions after AIS. In contrast, the primary collaterals of the circle of Willis supplying this brain region may recruit blood flow to the less ischemic territory.
Another limiting factor for the translation of the current hypothesis is that the clinical detection of the no-reflow phenomenon is challenging. Although experimental studies confirmed the existence of no-reflow almost 50 years ago, clinical observations have yielded heterogeneous results. 41 Because some diagnosed no-reflow cases simply represented residual vessel obstruction after recanalization, it remained unclear whether no-reflow was an epiphenomenon of lesion progression or the cause of infarction. 41 Despite its diagnosis, the lack of reliable measurements makes the diagnosis of no-reflow a significant clinical challenge. The different assessment of reperfusion using the Thrombolysis in Cerebral Infarction (TICI) or modified TICI (mTICI) scale results in different characterizations of no-reflow, which further complicates standardization among clinicians. 1 There is motivation to standardize the protocol for neuroimaging no-reflow identification, but this initiative requires more multicenter studies for reproducibility. 42
Conclusions
Our original observations classify SD as a major contributor to futile reperfusion in AIS. In concert, SD occurrence obviously fosters the progression of some already known pathologies in the background of the no-reflow phenomenon, such like pericyte constriction 43 or capillary stall formation. 6 We also note that the occurrence of SD is organically related to the lack of collateral recruitment, which is usually due to anatomical variance or the absence of collaterals. Indeed, our results suggest that the SD-related oligemia may extend beyond recanalization of the injured territory and may profoundly reduce CBF after AIS. Consistent with this, the prolonged accumulation of 20-HETE after SD episodes may serve as an important regulator of the no-reflow phenomenon leading to futile reperfusion of the brain, and adverse outcome of AIS. In conclusion, further understanding of the role of SD and 20-HETE in the no-reflow phenomenon provides valuable insights into the pathophysiology of AIS and opens potential avenues for new therapeutic interventions.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: the EU’s Horizon 2020 research and innovation program under grant agreement No. 739593; grants from the National Research, Development and Innovation Office of Hungary (No. FK142218, K134377 and K134334); the Ministry of Innovation and Technology of Hungary and the National Research, Development and Innovation Fund (No. TKP2021-EGA-28 financed under the TKP2021-EGA funding scheme); the Ministry of Human Capacities of Hungary (ÚNKP-21-3-SZTE-78), and the National Brain Research Program 3.0 of the Hungarian Academy of Sciences and the University of Szeged Open Access Fund, Grant ID: 7157.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.