
Abstract
Acute ischemic stroke (AIS) afflicts millions of individuals worldwide. Despite the advancements in thrombolysis and thrombectomy facilitating proximal large artery recanalization, the resultant distal hypoperfusion, referred to “no-reflow” phenomenon, often impedes the neurological function restoration in patients. Over half a century of scientific inquiry has validated the existence of cerebral “no-reflow” in both animal models and human subjects. Furthermore, the correlation between “no-reflow” and adverse clinical outcomes underscores the necessity to address this phenomenon as a pivotal strategy for enhancing AIS prognoses. The underlying mechanisms of “no-reflow” are multifaceted, encompassing the formation of microemboli, microvascular compression and contraction. Moreover, a myriad of complex mechanisms warrant further investigation. Insights gleaned from mechanistic exploration have prompted advancements in “no-reflow” treatment, including microthrombosis therapy, which has demonstrated clinical efficacy in improving patient prognoses. The stagnation in current “no-reflow” diagnostic methods imposes limitations on the timely application of combined therapy on “no-reflow” post-recanalization. This narrative review will traverse the historical journey of the “no-reflow” phenomenon, delve into its underpinnings in AIS, and elucidate potential therapeutic and diagnostic strategies. Our aim is to equip readers with a swift comprehension of the “no-reflow” phenomenon and highlight critical points for future research endeavors.
Introduction
Acute ischemic stroke (AIS) is a significant global health concern, accounting for substantial morbidity and mortality rates.1 –3 Even though thrombolytic therapy and thrombectomy have gained widespread acceptance, improving outcomes for numerous patients,4 –7 a considerable proportion (>50%) of those treated continue to exhibit poor prognoses due to a range of adverse events. 8 A potentially overlooked aspect of post-stroke pathophysiology is the diminished microvascular reperfusion subsequent to complete recanalization, otherwise known as the “no-reflow” phenomenon. This primarily affects the precapillary arteriole, capillary, and postcapillary venule (Figure 1).9 –12 These microvessels measure between 6–20 μm and are chiefly composed of peripheral vessel cells, including endothelial cells, astrocytes, microglia, neurons, and pericytes.13,14
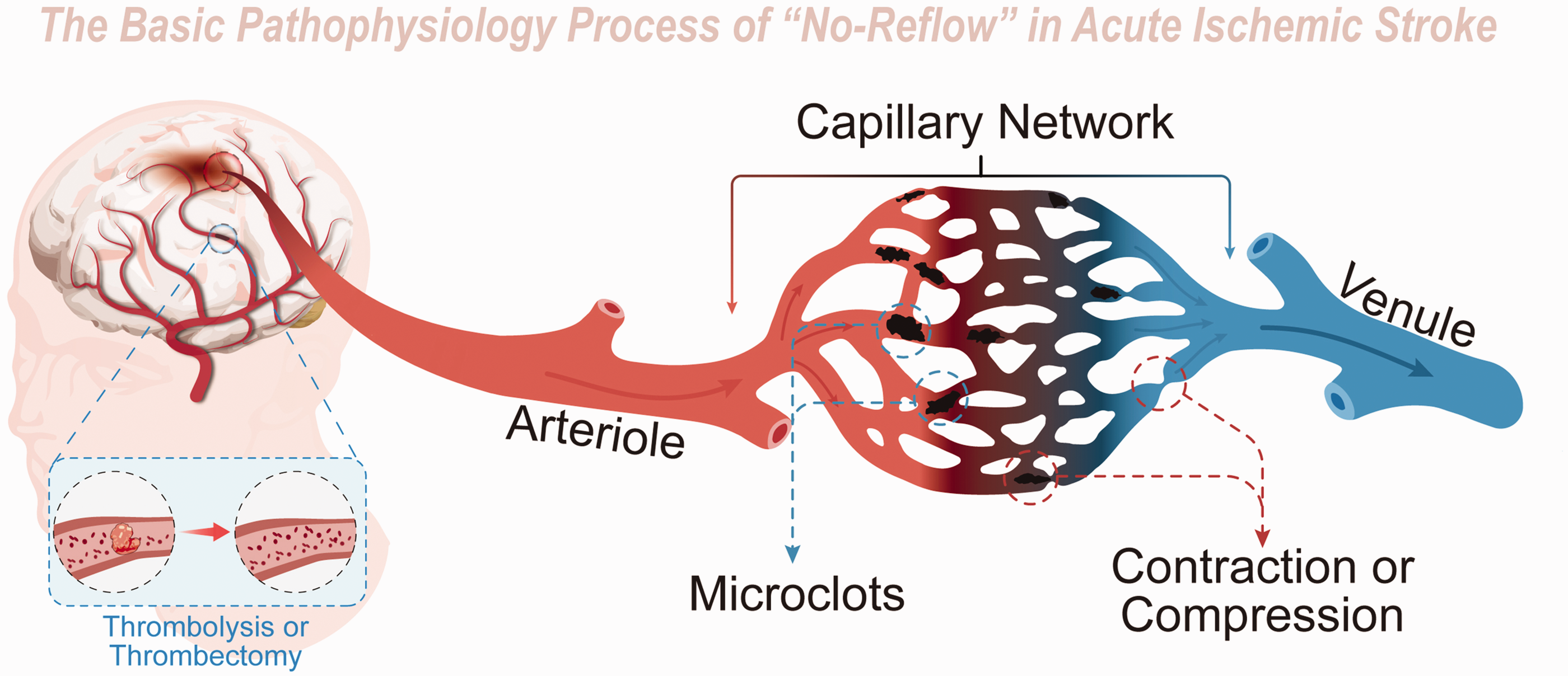
Schematic diagram of Basic Pathophysiology of “no-reflow” in acute ischemic stroke. Though proximal macrovessels are recanalized after thrombolysis or thrombectomy in acute ischemic stroke, distal microvessels are still under poor perfusion due to microclots and microvessel compression and contraction.
Recent literature has revealed that the supplemental administration of alteplase (0.225 mg/kg) to patients exhibiting an expanded Treatment in Cerebral Ischemia (eTICI) score of ≥2b50 15 after thrombectomy, intending to dissolve microcirculation thrombi, culminated in superior clinical outcomes. 16 Furthermore, a pooled analysis of three prospective international multicenter thrombectomy randomized controlled trials (RCTs) disclosed that cerebral “no-reflow” was prevalent in humans and was correlated with adverse events and long-term prognosis. 17 These emerging insights suggest a close association between enhanced microcirculation post-stroke treatment and neuroprotection. 14 Thus, there is a pressing need to advance microvascular reperfusion and minimize the “no-reflow” phenomenon.
To mitigate the “no-reflow” phenomenon effectively, a comprehensive understanding of its research history, currently identified pathophysiological mechanisms, the treatment modalities that have been implemented or are anticipated to be clinically relevant based on the mechanism, and the latest clinical diagnostic tools is imperative. These areas of focus form the foundation of this review.
The history of “no-reflow”
The historical progression of the “no-reflow” concept is elegantly depicted in Figure 2. Initial explorations into cerebral “no-reflow” originated from groundbreaking studies conducted by Ames et al. in 1968, unearthing this phenomenon in the rabbit brain. 18 The researchers deployed a cuff, inflated to 350 mmHg, around the rabbit's neck. This approach obstructed blood flow in both common carotid arteries and basilar artery for a minimum of 5 minutes, instigating global cerebral ischemia. Then the colloidal solution was injected into the carotid arteries to stain perfused brain regions. However, parts of the brain did not regain perfusion after proximal large artery flow was restored and the size of perfusion deficits increased with longer periods of ischemia. Additionally, by utilizing a Ringer solution to clear intravascular blood and benzene staining for trapped blood, Ames et al. found that areas of trapped blood were synonymous with regions of perfusion deficits. These observations led to the inference that, following an ischemic injury, blood components were trapped within the capillaries, potentially causing hypoperfusion post-proximal reperfusion. To further understand this, Ames et al. flushed the cerebral vessel with a Ringer solution, followed by 15 minutes of bloodless ischemia. Interestingly, the restoration of flow did not present the “no-reflow” phenomenon, which highlighted the crucial role of blood components in “no-reflow”. Furthermore, red blood cells were once again trapped following blood reintroduction post-bloodless ischemia. This suggested that besides alterations in blood composition following an ischemic event, the microvascular unit also suffered damage that inhibited reflow. This pioneering research essentially laid the groundwork for understanding the “no-reflow” phenomenon, paving the way for a plethora of subsequent basic research on cerebral “no-reflow”.
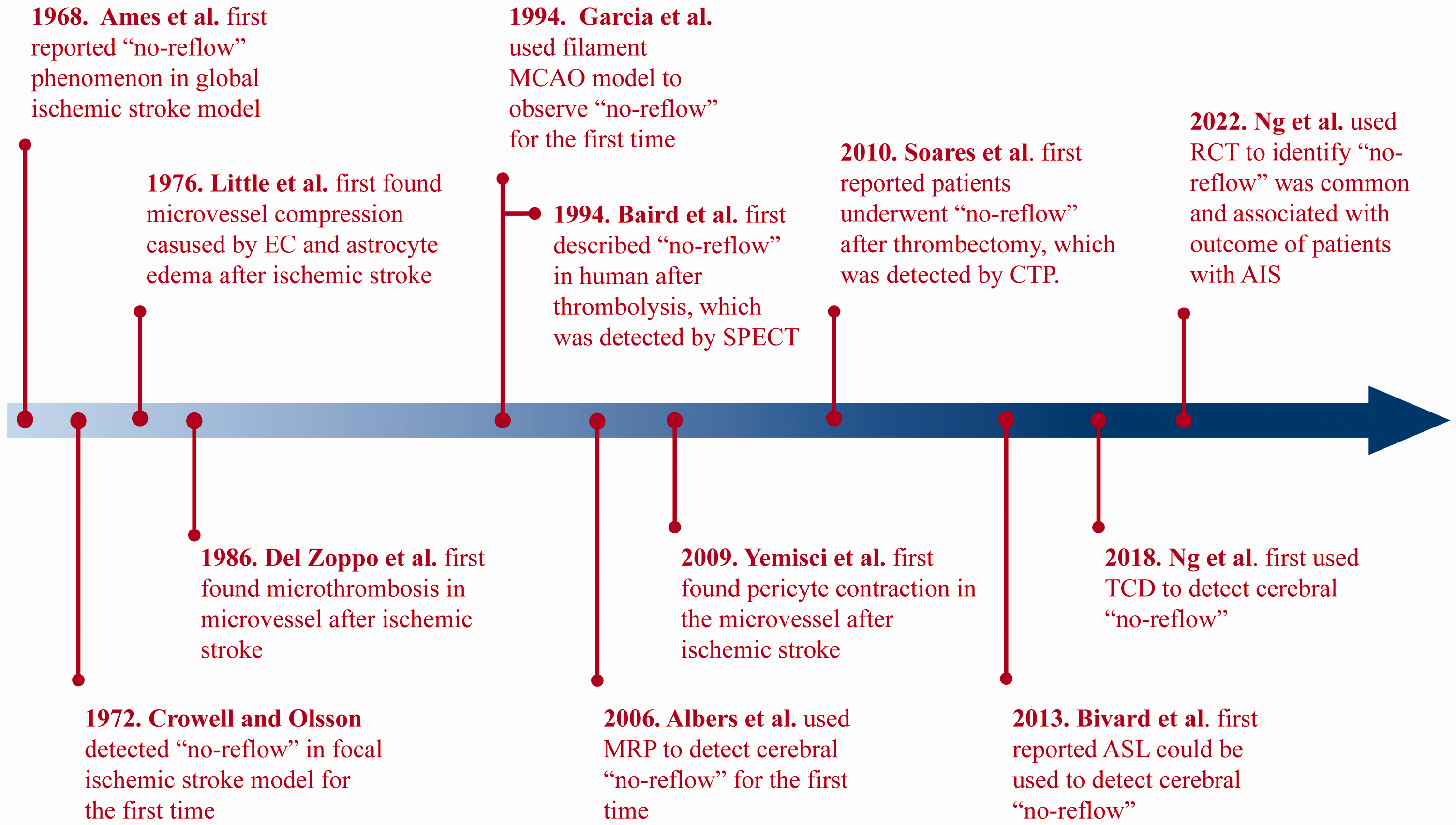
History of “No-reflow” Phenomenon. EC: endothelial cell; MCAO: middle cerebral artery occlusion; SPECT: single photon emission computed tomography; MRP: magnetic resonance perfusion; CTP: computer tomography perfusion; ASL: arterial spin labeling; TCD: transcranial doppler; RCT: randomized controlled trial; AIS: acute ischemic stroke.
Contrasting with global ischemic stroke models, which emulate cardiac arrest and resuscitation,18,19 the focal ischemic model predominantly simulates intracranial arterial embolism. In 1972, Crowell and Olsson were the first to mimic transient focal cerebral ischemia in monkeys, thereby detecting microvascular filling. 20 After inducing a 4-hour temporary middle cerebral artery occlusion (MCAO) using an aneurysm clip and reperfusing with a carbon-black suspension, the researchers observed impaired vascular filling in the focal ischemic area. Notably, treatment with low-molecular-weight dextran, heparin, or hyperventilation could attenuate this impairment. Furthermore, in 1976, Little et al., employing an electron microscope, delved deeper into the causes of “no-reflow” in monkeys. They discovered that swollen endothelial cells (ECs) and astrocyte endfeet could constrict microvessels following proximal large artery occlusion. 21 Moreover, in 1986, Del Zoppo et al. first confirmed the presence of thrombotic material occluding microcirculatory branches in ischemic region. 22 Although these studies used non-human primate (NHP), to find the “no-reflow” phenomenon in their brain, from which the evidence was far higher than other mammals, the widespread use of NHPs as a research tool is hampered by their exorbitant costs and extended breeding cycles.23,24
Ever since the introduction of the reversible endovascular filament MCAO methodology by Koizumi et al. in 1985, later refined by Longa et al. in 1989, 25 rodents have become the most employed animal models to study focal MCAO.26,27 Leveraging this model, the exploration of the “no-reflow” phenomenon's mechanisms and potential treatments has broadened substantially. In 1994, Garcia et al. reported the “no-reflow” phenomenon in a rat filament MCAO model, revealing, in addition to red blood cells, leukocytes and platelets were also detected to be trapped in the microvessels (capillaries and venules) of the ischemic hemisphere. 28 A foundational finding regarding the “no-reflow” phenomenon came from Yemisci et al. in 2009, who noted that, in transient MCAO mice, pericytes—regulators of vessel diameter—could contract in response to oxidative-nitrative stress, despite the successful opening of an occluded cerebral artery, thereby hampering capillary reflow. 29 Based on these pioneering studies, the fundamental pathophysiological alterations associated with “no-reflow” have been identified: microemboli obstructing microvessels, and both active contraction and passive compression of microvessels reducing their diameter (Figure 1).
Although the identification of the “no-reflow” phenomenon in the brain has spurred research into its presence in systemic organs, such as the heart and kidney, clinical discovery of cerebral “no-reflow” seems to lag behind basic research. This may be attributable to the relatively recent introduction of stroke treatments utilizing reperfusion, such as thrombolysis and thrombectomy. The first clinical evidence for cerebral “no-reflow” surfaced in 1994, when Baird et al. employed single-photon emission computed tomography (SPECT) in the case of a patient treated with streptokinase for thrombolysis. They observed a mismatch between successful large artery recanalization and failure of reperfusion. 30 Furthermore, in 2006, Albers et al. pioneered the application of magnetic resonance perfusion (MRP) technology to identify the occurrence of “no-reflow” phenomenon in patients following thrombolysis. 31 Comparatively, the progression of thrombectomy technology has been slower, primarily due to the gradual advancement of intracranial interventional instruments. It was only in 2010 that Soares et al. utilized the computed tomography perfusion (CTP) to report patients experiencing “no-reflow” following thrombectomy. 32 Since then, numerous clinical studies have underscored the significance of “no-reflow”.33 –35 For instance, Bivard et al. were among the first to report the utility of arterial spin labeling (ASL) for detecting “no-reflow” in 2013, 36 while Ng et al. played a pioneering role in advocating the use of transcranial doppler (TCD) to identify this condition in 2018. 37 Nonetheless, owing to patient variability, treatment modalities, and evaluation criteria, the reported incidence of “no-reflow” varies considerably among studies. Ter Schiphorst et al. even proposed that the “no-reflow” phenomenon is relatively rare and should not be a major therapeutic target. 38 Contrastingly, a multi-center randomized controlled trial (RCT) study published in 2022 reported “no-reflow” as a common occurrence with a prevalence of 25.3% and highlighted its strong association with patient outcomes. 17
In summary, the “no-reflow” phenomenon has been verified in both small and large animals, as well as humans, in both global and focal ischemia scenarios. This cumulative evidence underscores the “no-reflow” phenomenon as a significant pathophysiological characteristic following ischemia, potentially impeding patient recovery post-recanalization treatment. Therefore, it is imperative to understand the underlying mechanism and devise mechanism-based treatments to mitigate the “no-reflow” phenomenon. Furthermore, effective diagnosis of “no-reflow” is crucial, a topic that will be elaborated upon in the following sections.
Theories and potential treatments of “no-reflow”
Building on the basic pathophysiological characteristics previously detailed—namely, microemboli, and the contraction and compression of microvessels—research has further delved into the intricate mechanisms underlying the “no-reflow” phenomenon, culminating in diverse theories (Figure 3). Furthermore, while some RCT studies have not explicitly assessed the perfusion state, these studies have indeed employed methods aimed at treating specific elements of the pathophysiological changes inherent in “no-reflow”, corroborating the efficacy of these methods. Consequently, mechanisms confirmed by clinical trials and their associated therapeutic approaches appear to be of paramount importance. Nevertheless, we do not dismiss the significance of other mechanisms and their therapeutic progress, which warrant further validation. Existing studies on the treatment of “no-reflow” are summarized in Table 1.
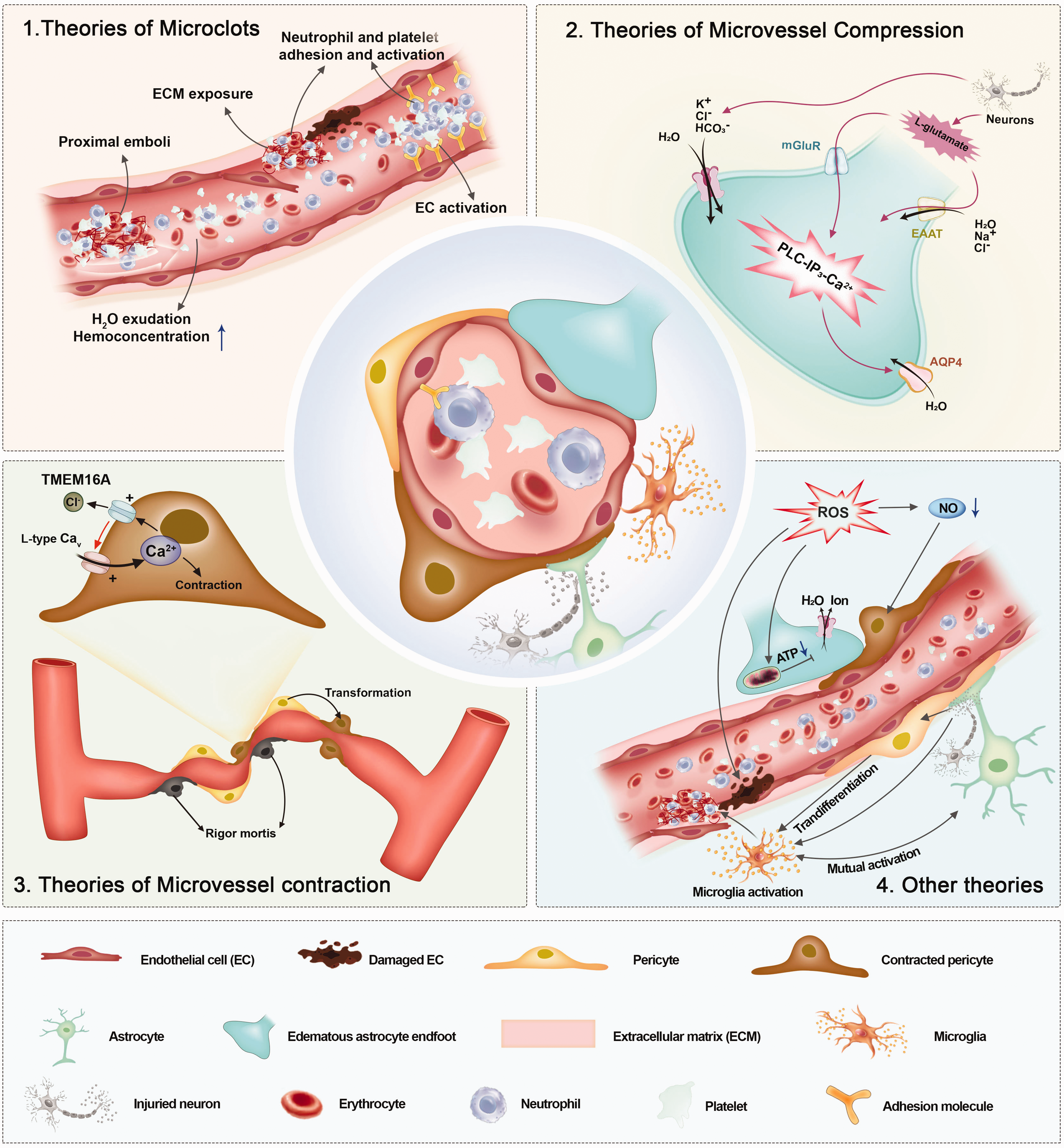
Complex mechanism under basic pathophysiology of “no-reflow”.
Overview of different treatment to “no-reflow” in various animal models.
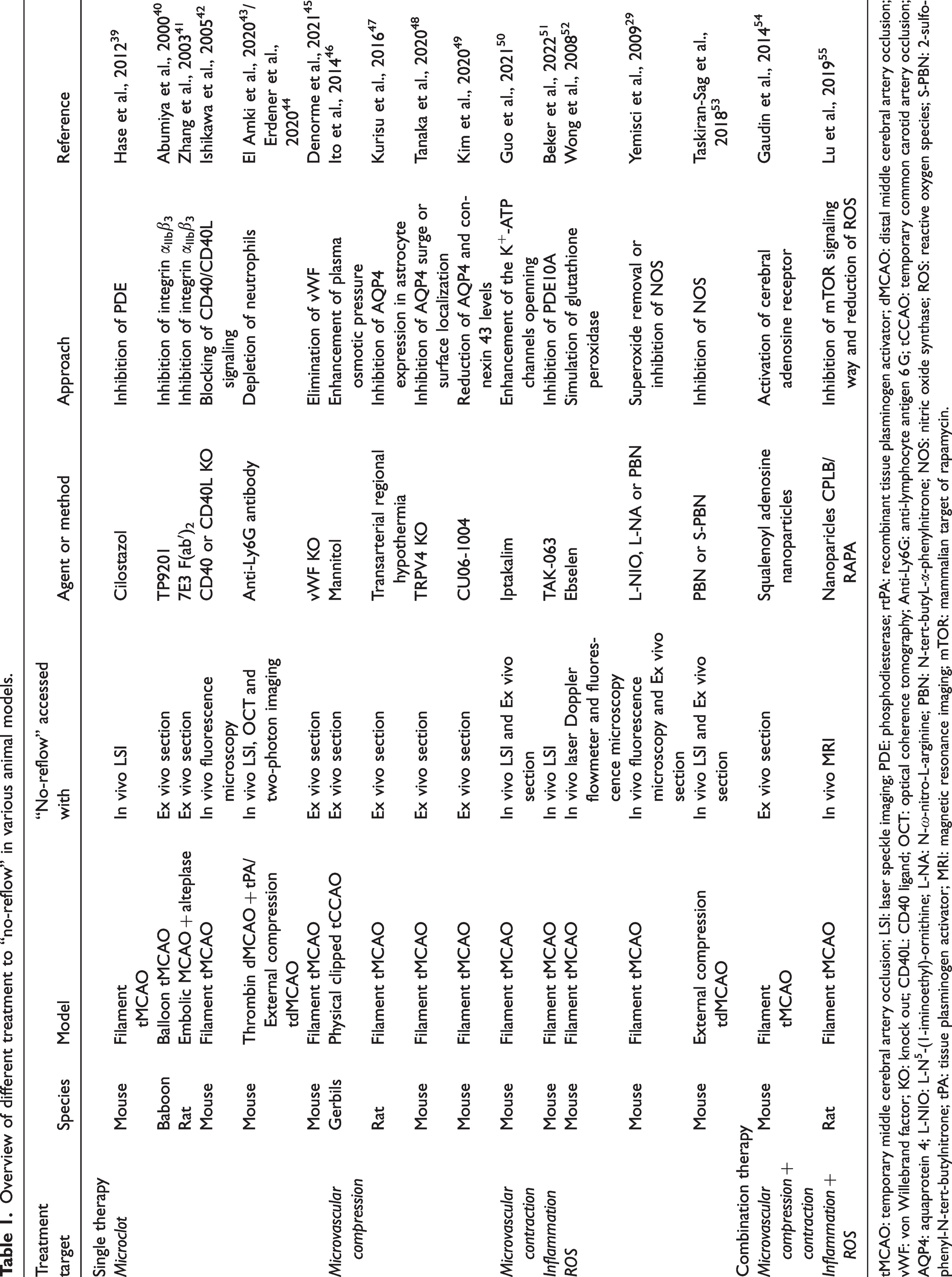
tMCAO: temporary middle cerebral artery occlusion; LSI: laser speckle imaging; PDE: phosphodiesterase; rtPA: recombinant tissue plasminogen activator; dMCAO: distal middle cerebral artery occlusion; vWF: von Willebrand factor; KO: knock out; CD40L: CD40 ligand; OCT: optical coherence tomography; Anti-Ly6G: anti-lymphocyte antigen 6 G; tCCAO: temporary common carotid artery occlusion; AQP4: aquaprotein 4; L-NIO: L-N5-(1-iminoethyl)-ornithine; L-NA: N-ω-nitro-L-arginine; PBN: N-tert-butyL-α-phenylnitrone; NOS: nitric oxide synthase; ROS: reactive oxygen species; S-PBN: 2-sulfo-phenyl-N-tert-butylnitrone; tPA: tissue plasminogen activator; MRI: magnetic resonance imaging; mTOR: mammalian target of rapamycin.
Theories of microclots
Given that microclots represent the earliest identified pathophysiological manifestation of “no-reflow”, they have naturally emerged as a primary research target. Several studies suggest multiple factors may precipitate the formation of microclots, such as exposure to the extracellular matrix (ECM), activation of endothelial cells (ECs), and involvement of blood factors.
Del Zoppo et al. proposed that during ischemia/reperfusion (I/R) injury, vascular ECs undergo extensive damage, revealing the subendothelial ECM components, including collagen and laminin. This exposure triggers the adhesion, spreading, and aggregation of circulating platelets, eventually leading to thrombus formation (mediated by the integrin αIIbβ3 on activated platelets). The thrombus entraps erythrocytes and leukocytes, subsequently impeding microvessel flow.11,22 Additionally, ECs form connections with the ECM via integrins located on the basal lamina side membrane. It has been noted that the expression of integrin α6β1 (a receptor for laminins and type IV collagen in ECM) decreases in the ischemia core due to the effects of tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β), leading to EC detachment from the ECM and subsequent ECM exposure. 56 Another study indicated a reduction in integrin α1β1 on the abluminal endothelial membranes following MCAO, exacerbating EC detachment. 57 Consequently, whether through EC damage or EC detachment, each pathway can lead to ECM exposure and subsequent microclot formation based on the ECM.
Following activation induced by I/R injury, thrombin, and other factors, ECs may express a variety of adhesion molecules on the outer cell membrane. These include selectins (E-, P-, and L-selectins), as well as members of the immunoglobulin superfamily such as the intercellular adhesion molecule (ICAM)-1 and the vascular cell adhesion molecule (VCAM)-1. These molecules facilitate the adhesion of platelets and leukocytes, thereby augmenting microthrombus formation and inciting inflammation. 58 With respect to selectins, although L-selectin can mediate leukocyte transmigration, Yenari et al. found that blocking L-selectin did not reduce ischemic injury in a rabbit model of transient focal cerebral ischemia. This suggests that E- and P-selectins, rather than L-selectin, play crucial roles in regulating leukocyte transmigration and exacerbating stroke injury. 59 Consequently, the expression of adhesion molecules on the EC membrane following EC activation may contribute to microclot formation based on ECs.
In addition to microvascular-related factors, components of the blood may also play a role in the “no-reflow” phenomenon. For instance, the breakdown of the brain-blood barrier (BBB) and plasma exudation can increase blood viscosity through concentration of blood cells, fostering cell aggregation, reducing blood flow velocity, and enhancing the likelihood of microthrombosis. These events have been suggested as primary contributors to cerebral “no-reflow”. 60 In addition, the release of microemboli composed of atherosclerotic debris, blood clots, and platelet plugs into the microcirculation, particularly during the restoration of normal blood flow by thrombolysis, angioplasty, stenting, or other interventional procedures, may contribute to “no-reflow”. These microemboli can be carried distally with the blood flow and obstruct microvessels smaller in diameter than themselves, thereby precipitating “no-reflow”. 9 Recent in vivo studies have also shown that, although recanalization of the proximal artery can be achieved, the stalling of capillaries in the distal vascular network restricts tissue reperfusion. Notably, neutrophils were the primary constituents of the stalled capillaries within the infarct core post-thrombolysis.43,44 Furthermore, systemic inflammatory responses may intensify platelet accumulation. A recent study reported that ischemic stroke could prompt an inflammatory response in circulating neutrophils in both humans and mice, with activated neutrophils forming aggregates with platelets and producing neutrophil extracellular traps, which provide a scaffold for platelet accumulation.61,62
Multiple mechanisms can instigate the formation of microemboli, and although many of these mechanisms remain challenging to treat, the collective body of research underscores the contribution of microemboli to the “no-reflow” phenomenon. Therefore, treatments targeting microemboli could provide promising avenues for addressing “no-reflow”. Thrombosis is a very complex process, and here we believe that targeted treatment of endothelial cells in situ within brain tissue is more important and does not affect other tissues. Thus, in addition to microemboli-targeting treatments, early protection and inactivation of ECs may be vital given their crucial role in “no-reflow”.
However, it is important to note that the sole use of anticoagulants to prevent microthrombosis may elevate the risk of intracerebral hemorrhage (ICH), potentially leading to worsened clinical outcomes.40,64Thus, combining anticoagulants with drugs that promote the integrity and patency of microvessels may present a safer approach for treating “no-reflow”. Cilostazol, a phosphodiesterase (PDE) inhibitor acting as an antiplatelet agent, has shown potential in significantly reducing the endothelial expression of adhesion molecules (P-selectin and ICAM-1), preventing platelet aggregation and leukocyte plugging within microvessels, maintaining microvascular integrity by decreasing MMP-9 expression, and thereby protecting against cerebral ischemic injury by preventing “no-reflow” and hemorrhagic transformation in a transient MCAO mouse model. 39 Furthermore, Zhang et al. demonstrated that co-administration of the integrin αIIbβ3 antagonist [monoclonal antibody, 7E3 F(ab′)2] with tissue plasminogen activator significantly reduced microvascular platelet accumulation and enhanced the patency and integrity of cerebral microvessels. This approach resulted in a reduction of infarct volume, enhanced microvessel patency, inhibited hemorrhage, and improved neurological outcomes. 41 While these combination therapies seem promising, the respective adjuvant drugs are currently not approved for clinical treatment of AIS, thereby necessitating further clinical evidence to validate their efficacy and safety.
In addition to pharmacotherapy, certain antibodies and gene-editing therapies have shown potential. Ishikawa et al. found that knockout of CD40, an inducer of cellular adhesion molecules, and its ligand (CD40L) resulted in decreased adhesion of platelets and white blood cells in microvessels. 42 Besides, based on findings that a majority of stalled capillary segments were due to neutrophil clogging (60-75%), 43 El Amki et al. and Erdener et al. utilized a targeted murine monoclonal antibody, anti-lymphocyte antigen 6 G (Ly6G) to induce neutropenia, thus improving microcirculatory flow, reducing infarct volumes, and enhancing outcomes.43,44 Furthermore, Denorme et al. found in von Willebrand factor (vWF) knockout mice, there were fewer microthrombi in the stroke-affected brain. 45 Although these methods are challenging to apply clinically at present, they do underscore the key roles of specific cells, molecules, or pathways in “no-reflow”, and they may serve as potential therapeutic targets in the future.
Collectively, these data suggest that microthrombosis, one of the primary pathophysiological mechanisms underlying “no-reflow”, involves numerous potential mechanisms that support its formation. Alteplase has been shown to exert a beneficial effect on “no-reflow” and the outcomes of AIS patients following successful recanalization of a proximal large artery. However, its adverse effect of promoting intracerebral hemorrhage transformation needs to be mitigated. Thus, the combination of alteplase with drugs that maintain the integrity of microvessels could be a promising strategy, warranting further exploration in clinical trials.
Theories of microvessel compression
The occurrence of cytotoxic edema surrounding microvessels – inclusive of ECs and astrocytic endfeet – leading to microvessel compression is another crucial pathophysiological aspect of the “no-reflow” phenomenon. 21 EC edema is mainly induced by ionic disorder and water accumulation after ischemia. Compared with ECs, astrocyte endfeet edema are more serious and obvious. 65 Intriguingly, in vitro studies have posited that hypoxia alone cannot induce astrocytic endfeet edema, suggesting the existence of multifaceted mechanisms in vivo. 66 These mechanisms can be broadly categorized into three major groups: the ion channel mechanism, the water channel mechanism, and the glutamate transporters mechanism.
The ion channel represents the first mechanism. Research conducted by Walz et al. established that I/R injury instigates an exodus of neuronal K+ ions into the extracellular space, resulting in significant astrocyte depolarization. This process stimulates the opening of anion channels, thereby permitting the influx of K+, Cl–, and HCO3−, along with water, into the cells. Consequently, water accumulation within astrocytes prompts astrocytic swelling. 67 Given the rapid passive transport function of the ion channel and the abundant presence of K+ ion channels in endfeet compared to soma, astrocytic endfeet edema quickly manifests post-ischemia, at a faster rate than in the soma. 68
The second mechanism involves the water channel, specifically aquaporin 4 (AQP4). As the primary water channel protein expressed in the central nervous system, AQP4 is densely localized within astrocytic endfeet.69,70 A significantly increased expression of AQP4 in astrocytic endfeet, as well as endfeet edema, was confirmed in cell culture models [subjected to oxygen and glucose deprivation (OGD) with or without reoxygenation], animal models (MCAO), and cerebral infarction patients.71 –73 The mitogen-activated protein kinase (MAPK) pathways - comprising three main members: extracellular signal-regulated kinase (ERK), C-Jun N-terminal kinase (JNK), and p38-MAPK - have been implicated in this context. Independent studies demonstrated that AQP4 is upregulated following the activation of ERK and p38-MAPK pathways and the JNK and p38-MAPK pathways in the OGD/reoxygenation model.72,74 Besides an increase in AQP4 expression, AQP4 also necessitates further activation through glutamate induction, as discussed below.
Finally, glutamate transporters present the third mechanism underlying astrocytic endfeet edema. Post-ischemic stimuli cause an increased release of L-glutamate from neurons75,76 into the extracellular space, which, in turn, triggers a high expression of astrocytic excitatory amino acid transporters EAAT2 and EAAT1 (GLT-1, GLAST). These transporters clear L-glutamate from the extracellular space and concurrently cotransport Na+, Cl−, and water into the astrocytes, ultimately leading to astrocytic endfeet swelling.77,78 Furthermore, elevated glutamate concentrations can also activate the metabotropic glutamate receptor (mGluR), contributing to endfeet edema. Specifically, group I mGluR, encompassing mGluR1 and 5, is known to activate phospholipase C, generating inositol triphosphate and prompting the release of Ca2+ from intracellular stores. Subsequently, Ca2+ activates calcium/calmodulin-dependent protein kinase II, which phosphorylates nitric oxide synthase (NOS) and thereby initiates the NO-cGMP-protein kinase G (PKG) signaling pathway. Ultimately, PKG phosphorylates AQP4 to enhance its permeability. 79
The comprehensive structure of microvessels encompasses ECs and astrocytic endfeet. Therefore, edema in ECs and astrocytic endfeet can result in a widespread compression of microvessels. As suggested by the aforementioned mechanisms, although astrocytic endfeet edema is more severe than that in ECs, it could be mitigated through various pathways, which could serve as a focal point for future studies.
Aside from osmotic therapy, several unconventional therapeutic strategies have demonstrated efficacy in mitigating the “no-reflow” phenomenon by inhibiting EC and astrocyte endfeet edema. For instance, Kurisu et al. revealed that a transarterial injection of 10 °C saline to cool the brain could inhibit AQP4 expression and astrocyte endfeet swelling, resulting in significantly improved neurological outcomes. 47 Transient receptor potential vanilloid 4 (TRPV4) knockout may reduce AQP4 surge or surface localization, thereby lessening astrocyte endfeet swelling, preserving the lumen diameter of microvessels, and subsequently leading to milder neurological symptoms in transient MCAO mice. 48 Additionally, CU06-1004 can moderate AQP4 and connexin 43 levels that mediate edema to alleviate astrocyte endfeet swelling, maintain microcirculation, and attenuate cerebral damage in mice subjected to cerebral I/R injury. 49 Furthermore, adenosine has been shown to potentially inhibit the swelling of ECs and astrocyte endfeet. 82 Due to its instability in plasma, Gaudin et al. used squalenoyl adenosine nanoparticles, achieving an extended retention time of adenosine in the blood, reducing ECs and astrocyte endfeet edema, enhancing microvessel perfusion, and providing a neuroprotective effect. 54 While these novel therapeutic approaches show promise in reducing the incidence of the “no-reflow” phenomenon and alleviating the deterioration of neurological function by moderating the edema of ECs and astrocyte edema, these methods require further extensive validation prior to their application in humans.
In summary, edema in ECs and astrocyte endfeet is a crucial mechanism underlying microvessel compression and serves as a promising treatment target for the “no-reflow” phenomenon. Conventionally, osmotic therapy may be employed post-recanalization to prevent “no-reflow” through the inhibition of ECs and astrocyte endfeet swelling. Furthermore, given the greater severity and complexity of astrocyte endfeet edema compared to that in ECs, future research should prioritize the exploration of astrocyte edema, aiming to enhance the “no-reflow” inhibition effect through the attenuation of endfeet edema.
Theories of microvessel contraction
Traditionally, ECs and astrocyte endfeet edema have been solely attributed to the diminution of small vessel diameter. However, the discovery that pericytes bear similarities to smooth muscle cells (SMCs), given their active ability to contract and reduce the diameter of microvessels, has prompted an intensive exploration of their potential role in the development of the “no-reflow” phenomenon. 83 Although SMCs also regulate vascular diameter and are critical to cerebral blood flow, their distribution is mainly restricted to pial arterioles and penetrating arterioles, which are not predominantly involved in the “no-reflow” phenomenon; hence, SMCs will not be discussed in this review of the theory of microvessel contraction.13,84
In a pioneering study utilizing in vitro retinal slices, Peppiatt et al. first suggested that pericytes might govern cerebral capillary diameter under simulated ischemic conditions. 85 Further research by Yemisci et al. posited that in vivo pericyte contraction plays a critical role in “no-reflow” after middle cerebral artery (MCA) recanalization, a phenomenon that can be attenuated by superoxide scavengers and NOS inhibitors. Thus, the authors postulated that oxidative-nitrative stress disrupted pericyte mitochondria, endoplasmic reticulum, and calcium pumps, leading to intracellular calcium overload, a principal factor for pericyte contraction. 29 In addition, Hall et al. reported that constricting pericytes died following ischemic injury, leading to a persistently contracted state (rigor mortis) and a long-lasting reduction of capillary diameter. This death of pericytes was attributed to ischemia-induced lack of adenine nucleoside triphosphate (ATP) molecules, which could prevent the dissociation of myosin and actin. 86 Based on the research above, the importance of pericytes in the regulation of cerebral microvessels, especially in the post-infarction situation, has received more and more attention. Nortley et al. discovered that in Alzheimer's patients, amyloid β oligomers elevated cerebral reactive oxygen species (ROS) concentration, which then prompted the release of endothelin (ET)-1, ultimately resulting in pericyte contraction. 87 Given that ROS is also prevalent in I/R injury, this signaling pathway might contribute to pericyte contraction in “no-reflow”, which warrants further investigation.
Korte et al. examined ion channels as cytoplasmic Ca2+ concentration ([Ca2+]i) in pericytes is an integral regulator of pericyte contraction. They suggested that ischemia triggered a modest increase in [Ca2+]i in pericytes, instigating Cl- efflux via the Ca2+-gated anion channel TMEM16A, thereby depolarizing the cell and opening voltage-gated calcium channels which further amplified pericyte [Ca2+]i rise and capillary constriction. 12 They also highlighted that this [Ca2+]i rise, particularly at the soma of pericytes where the majority of circumferential processes are located, induced significant capillary constriction. 88 However, a more recent study that methodically observed vasodynamics along different segments of the cerebral vasculature found no difference between the altered capillary diameter influenced by the pericyte soma and pericyte process alone following brain ischemia/recanalization. 89 Therefore, the differential influence of pericyte soma and process on vascular diameter necessitates further scrutiny.
Observations within 24 hours post-I/R injury by Zhang et al. indicated an increased contraction of pericytes over time, and the morphology changed from thin-stranded pericytes to mesh pericytes (a transitional phenotype between SMCs and pericytes) which expressed an elevated level of α-smooth muscle actin (α-SMA), a contractile protein integral to the role of pericytes in regulating the diameter of microvessels. 14
Aside from the modification of pericytes themselves, astrocytes could play a significant role in pericyte contraction. 90 Mulligan et al. suggested that increased Ca2+ concentration via mGluR activation in astrocyte endfeet could stimulate Ca2+-sensitive phospholipase A2 (PLA2), leading to increased arachidonic acid (AA) formation from membrane phospholipids. The AA then diffuses to SMCs where it is converted to 20-HETE by CYP4A (a cytochrome P450 enzyme subtype), ultimately resulting in arteriole contraction. 91 Given the functional similarities between SMCs and pericytes, it is posited that the signaling pathways controlling pericyte contraction for capillaries would mirror those of SMCs for arterioles. 90 Recent studies have corroborated the expression of CYP4A 92 and the contractile function of 20-HETE in pericytes under healthy conditions, 86 thus substantiating the above hypothesis. However, further research is warranted to determine if astrocytes mediate pericyte contraction in “no-reflow” via 20-HETE.
In conclusion, compelling evidence demonstrates that pericytes exert a direct effect on vessel contraction, playing an essential role in the formation of the “no-reflow” phenomenon. While some potential mechanisms are extrapolated from other diseases or similar cell types, these hypotheses require further confirmation in the context of pericytes in I/R injury.
In addition to anti-calcium influx therapies, several studies have employed other strategies to inhibit the pericyte contraction response that results in decreased blood flow following recanalization. The research data of Guo et al. suggested that iptakalim could activate K+-ATP channels, thereby reducing intracellular calcium and the secretion of ET-1, a potent vasoconstrictor, consequently inhibiting pericyte contraction and increasing capillary diameter. 50 Additionally, beyond its role in inhibiting the edema of ECs and astrocyte endfeet, adenosine, as previously mentioned, also exhibits an anti-pericyte contraction effect. 97 Researchers observed that squalenoyl adenosine nanoparticles could also reduce pericyte contraction to preserve microvascular diameter. 54
In summary, the recent discovery of pericyte contraction leading to a reduction in microvessel diameter is a fascinating development in the study of the “no-reflow” phenomenon. However, as only a limited number of experimental studies have validated its therapeutic potential, extensive exploration and research are required to develop effective treatments for pericyte contraction in clinical practice. The targeted inhibition of calcium influx in pericytes appears to be a promising therapeutic strategy.
Other theories of “no-reflow”
Beyond basic pathophysiology, inflammation and reactive oxygen species (ROS) have been consistently implicated as potential aggravators of outcomes for patients with AIS.98,99 Recent studies have further substantiated that both inflammation and ROS can also exacerbate the “no-reflow” phenomenon.
In terms of inflammation, microglia - the resident immune cells within the brain that represent the initial line of defense in response to injuries such as stroke100,101 – can undergo phenotypic polarization into a pro-inflammatory type. This state leads to the release of an array of pro-inflammatory cytokines and additional chemokines that recruit peripheral immune cells. These cells can obstruct microvessels and intensify the inflammatory response.102,103 Hence, as initiators of brain tissue inflammation, microglia could indirectly exacerbate “no-reflow”. Additionally, the recruited immune cells can become activated and secrete pro-inflammatory cytokines and chemokines. 104 Apart from traditional inflammatory cells, astrocytes and pericytes also participate in neuroinflammatory reactions. Typically, microglia respond to injury prior to astrocytes, and activated microglia can promote astrocyte activation, which in turn further stimulates distant microglia, amplifying the secretion of characteristic inflammatory cytokines. 105 For instance, microglia can secrete IL-1β to activate astrocytes. 106 Active astrocytes can then secrete elevated levels of factors like lipocalin-2, an acute-phase protein, under hypoxic conditions. This process enhances microglial activity and the production of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α, leading to leukocyte infiltration and microthrombosis. 107 Recent studies have suggested that pericytes are implicated in immune and inflammatory responses. Reports have indicated that pericytes can be activated, undergo transdifferentiation into a microglia-like phenotype, and trigger inflammatory reactions.108,109 Numerous in vitro studies have found that brain pericytes express receptors for damage-associated molecular patterns (DAMPs), including toll-like receptor 4, scavenger receptors, and Fc receptors.108,110 These receptors initiate non-infectious immune responses, leading to the secretion of pro-inflammatory cytokines, including IL-6, and chemokines such as IL-8, CXCL1, CXCL2, CXCL3, and CCL2.110,111 Furthermore, several studies have suggested that various inflammatory conditions can induce pericytes to amplify the pro-inflammatory response. For example, TNF-α and transforming growth factor-beta 1 (TGF-β1) can enhance the expression of the classical pro-inflammatory cytokine, IL-6, in human cerebral pericytes.112,113 TNF-α and IL-1β can stimulate cerebral pericytes to secrete the chemokine IL-8. 114 Under inflammatory conditions, pericytes activated by TNF-α can upregulate the expression of the adhesion molecule ICAM-1 and release chemoattractant to draw in leukocytes. 115 Therefore, pericytes may also contribute to leukocyte infiltration during an ischemic stroke. These results suggest that pericytes may participate in the inflammatory response of ischemic stroke throughout the initial and amplification stages of inflammation. Neuronal death resulting from I/R injury leads to the release of copious amounts of DAMPs, including adenosine, heat shock proteins, high mobility group box 1, and IL-33. 116 Microglia can detect DAMPs through pattern recognition receptors, leading to their activation and secretion of inflammatory cytokines. Some clinical studies have also suggested that the composition of microclots may also contribute to inflammation.117,118 Thus, inflammation is a systemic process involving various cell types, which can recruit peripheral immune cells that block microvessels. Inhibiting systemic neuroinflammation is a plausible strategy to mitigate the “no-reflow” phenomenon.
Overproduction of ROS is another critical mediator in neurological deterioration following I/R injury, and ROS might instigate the “no-reflow” phenomenon by participating in all fundamental pathophysiological processes. For instance, ROS can intensify damage to ECs, exposing the ECM and promoting microglia activation,
119
This process mediates the adhesion and infiltration of leukocytes that obstruct microvessels. Moreover, mitochondrial ROS production forms a self-amplifying process, known as “ROS-induced ROS release”.
120
Hence, ROS overproduction signifies mitochondrial dysfunction, which inhibits ion metabolism in ECs and astrocytes and results in their edema, leading to microvessel compression. Additionally, as previously mentioned, ROS participates in the intracellular calcium overload in pericytes that results in pericyte contraction. Vessel dilation primarily depends on vasoactive mediators, most of which are nitric oxide (NO), produced via neuronal and endothelial NOS (nNOS and eNOS) in neurons and ECs. NO can inhibit the production of 20-hydroxyeicosatetraenoic acid (20-HETE), an essential inducer for pericyte contraction.
90
However, I/R injury causes neuronal and EC dysfunction and an increase in ROS. Consequently, NO is metabolized to peroxynitrite (
Nevertheless, several experimental studies have validated that inflammation and ROS treatment might enhance neurological function by inhibiting the ‘no-reflow' phenomenon. The transformation of microglia into a pro-inflammatory phenotype has been identified as a potential initial trigger for inflammation. Hence, in a study conducted by Lu et al., researchers utilized microthrombus-homing nanoparticle CPLB/RAPA, which contains an ROS response fragment and rapamycin, to successfully induce microglia polarization to an anti-inflammatory phenotype, thereby facilitating the reperfusion of microvessels. 55 TAK-063, a potent and highly selective inhibitor of PDE10A (a dual-substrate specific enzyme of cAMP/cGMP), has been found to attenuate microglial activation via cAMP/PKA signaling. Consequently, Beker et al. utilized TAK-063 to inhibit PDE10A, thereby diminishing cytokine/chemokine expression and enhancing regional microcirculation. 51
The nanoparticles developed by Lu et al. were also found to respond to and scavenge ROS, as well as release rapamycin to safeguard tight junctions and support the reflow of microvessels. 121 Glutathione peroxidase-1 (Gpx1), a protein expressed in the brain, plays a pivotal role in regulating ROS-mediated injury. In a study by Wong et al., they identified ROS as an inducer of MMP-9 and found that the “no-reflow” phenomenon was more prevalent in Gpx1−/− mice undergoing focal cerebral ischemia via MCAO as compared to their wild type counterparts. However, these negative outcomes could be reversed with the pre-emptive use of the anti-oxidant, ebselen. 52 Moreover, a comparative study of the ROS inhibitor, N-tert-butyl-α-phenylnitrone (PBN) and its BBB impermeable analog, 2-sulfo-phenyl-N-tert-butylnitrone (S-PBN), revealed that S-PBN could directly inhibit vessel wall ROS generation and indirectly offer neuroprotection, possibly secondary to improved microcirculation post-recanalization, which may subsequently reduce ROS generation by parenchymal cells. 53 Additionally, oxygen and nitrogen radicals formed in the microvasculature during I/R injury in mice caused persistent pericyte contraction and microvessel perfusion reduction, which could be counteracted by oxidative-nitrative stress suppressing agents, such as L-N5-(1-iminoethyl)-ornithine (L-NIO), N-ω-nitro-L-arginine (L-NA) and N-tert-butyL-α-phenylnitrone (PBN). 29
In summary, neuroinflammation and ROS overproduction serve as significant contributors to the basic pathophysiology of the “no-reflow” phenomenon. Although compelling evidence supporting the effectiveness of anti-inflammatory and anti-ROS treatments on AIS patient prognosis remains elusive, current experimental studies provide valuable insights for future research. Such studies can direct their focus towards subgroups with post-recanalization hypoperfusion to examine whether anti-inflammatory and anti-ROS treatments can aid in the neurological recovery of patients by suppressing the “no-reflow” phenomenon.
Diagnosis of “no-reflow”
In the clinical landscape, the diagnosis of the “no-reflow” phenomenon encounters significant impediments due to constraints in monitoring instruments and methodologies. Consequently, owing to the dearth of persuasive diagnostic findings, a widespread consensus on the definition of “no-reflow” remains elusive. In this context, we examine past and present diagnostic tools used to identify “no-reflow”, analyze their strengths and limitations, and highlight several promising methods that might be employed for diagnosing “no-reflow”, in order to provide a reference framework for future researchers.
Following the original characterization of cerebral “no-reflow” by Ames et al. in 1968, 18 hypoperfusion in the ischemic brain was identified by the absence of black staining in infarcted tissue histological sections, when colloidal carbon was employed as the intravascular contrast agent. 127 In accordance with this definition, perfusion detection with the injection of a contrast agent is still practiced in the clinical setting to ascertain “no-reflow”. Single Photon Emission Computed Tomography (SPECT) perfusion was initially utilized as a diagnostic tool for “no-reflow”,30,33 however, due to the challenges posed by isotope radiation and its implementation in community hospitals, its usage dwindled almost two decades ago. More recently, iodine-enhanced CTP and gadolinium-enhanced MRP have become the primary methods for detecting “no-reflow”.17,31,32,35,128 –130 These techniques primarily incorporate four hemodynamic parameters: cerebral blood volume (CBV), cerebral blood flow (CBF), mean transit time (MTT), and time to maximum (Tmax). Various studies have employed different parameters to define “no-reflow”, leading to an absence of consensus. The current challenge for CTP and MRP, which are software-analyzed, lies in accurately identifying “no-reflow” regions that interpenetrate regions of normal perfusion.
Perfusion imaging techniques that rely on contrast agent injections may elevate the risk of allergic reactions and place additional strain on renal function in patients. ASL, a non-invasive Magnetic Resonance-based technique that generates truly quantitative perfusion maps devoid of contrast agent limitations, has been validated against conventional contrast agent-dependent perfusion methods in AIS patients.131,132 Some recent studies have also employed ASL to monitor reperfusion status post-recanalization treatment and to identify “no-reflow”.38,133 Despite these advancements, the application of ASL in diagnosing “no-reflow” warrants further exploration, due to the limited number of related studies currently available and ASL's yet-to-be-resolved challenges in “no-reflow” diagnosis.
Moreover, the diagnostic examinations discussed above are often conducted hours, or even days, following vascular recanalization treatment due to the frailty of AIS patients. Consequently, even if the “no-reflow” phenomenon is identified in brain tissue, the window for effective treatment may have already passed. Hence, certain prerequisites should be met by diagnostic methods for “no-reflow”: (1) non-invasive methods to reduce patient discomfort; (2) bedside techniques to limit patient transportation; (3) rapid diagnosis to facilitate prompt treatment decisions by clinicians; and (4) high-quality imaging capable of detecting small blood vessels and deep brain tissue. Diagnostic techniques meeting these criteria should be explored to guide early intervention in patients experiencing cerebral tissue “no-reflow” after vascular recanalization treatment for ischemic stroke.
Non-invasive ultrasound, which can be performed bedside, seems to be a promising option. TCD has been adapted for stroke patients who are unfit for moving and for real-time monitoring of reperfusion therapies.134 –136 Elevated cerebral microvascular resistance in the ischemic territory, despite complete recanalization, 137 can be evaluated by TCD. The Gosling pulsatility index (PI) and the Pourcelot resistance index (RI), derived from TCD parameters, are well-established indices of cerebrovascular resistance.138,139 Ng et al. recently utilized TCD to provide the first evidence of post-recanalization acute microvascular dysfunction in humans, which may represent a convenient, real-time, and clinically relevant biomarker of the “no-reflow” phenomenon. 37 However, this study did not compare the roles of TCD and perfusion imaging in evaluating “no-reflow”, which necessitates further verification. Moreover, this method does not directly image cerebral blood vessels but instead converts cerebral blood flow through specific parameters, which may not be sufficiently intuitive.
Recently, Zhang et al. employed contrast-enhanced ultrasound and echo particles to map porcine cerebral microcirculation, demonstrating that blood vessels with diameters of approximately 120 μm could be readily distinguished. 140 This diameter closely mirrors that of the penetrating artery in the human brain. Although there remains a significant gap to the imaging of capillaries (6-7 μm), this study represents a significant stride towards non-invasive imaging of smaller vessels, a requirement for diagnosing “no-reflow”.
In addition to conventional imaging methods, near-infrared imaging instruments are being developed for real-time monitoring of cerebral blood flow in bedside settings. The most promising techniques include near infra-red spectroscopy (NIRS)141 –143 and diffuse correlation spectroscopy (DCS).143,144 However, both techniques exhibit specific strengths and weaknesses. NIRS primarily exploits the low absorption rate of hemoglobin for light (red and infrared) with a wavelength above 650 nm, allowing for simultaneous acquisition of blood, blood flow, and blood oxygen parameters of brain tissue. Nevertheless, NIRS measurements amalgamate intracranial and extracranial blood signals, complicating the separation process. In contrast, DCS signals solely measure the intracranial blood signal, circumventing the issue of signal separation, but the current equipment does not meet the ideal DCS requirements to reduce the signal-to-noise ratio.
Conclusion and perspectives
The body of evidence we have reviewed underscores that, after six decades of meticulous research, the cerebral “no-reflow” phenomenon has been unequivocally confirmed in both animal models and humans. The cardinal pathophysiological alterations in cerebral “no-reflow” comprise microthrombosis, along with microvascular compression and contraction, intertwined with a multitude of complex mechanisms. At present, anti-microthrombosis therapy is the sole clinical strategy with the potential to alleviate “no-reflow”, yet numerous prospective therapeutic schemes warrant comprehensive investigation. Constraints in the precision of existing instrumentation, which impede the detection of intracranial microvessels and their accurate perfusion status, and the lack of expert consensus due to limited clinical research on “no-reflow”, could delineate the future trajectory of “no-reflow” diagnostics. Succinctly, considering the criticality of “no-reflow”, amalgamation of macrovessel and microvessel diagnostics and treatments must be prioritized in future endeavors, to achieve bona fide reperfusion and enhance neural protection.
The “no-reflow” phenomenon was initially identified in the brain and can be explicitly detected in animals. However, due to the specific application of recanalization therapy for AIS, clinical studies on cerebral “no-reflow” have been sparse. With the ongoing advancements in AIS recanalization treatment, the therapeutic window for thrombolytic therapies has been extended, and an increasing number of patients can avail benefits from recanalization treatment. 145 Consequently, we recommend that clinicians should increase their vigilance towards the potential surge in “no-reflow” cases.
While numerous studies have dissected the mechanisms of “no-reflow”, the majority have only focused on the impact of specific intracerebral cells. Recently, the neurovascular unit (NVU), an integrative concept that encompasses all brain cells, has been suggested as a tool to delve deeper into the comprehensive mechanisms of “no-reflow”. 146 Comprising neurons, astrocytes, pericytes, microglia, ECs, and the ECM, the NVU is thought to regulate microvessel flow in both healthy and disease states. Thus, future studies should focus on the effect of comprehensive reaction between NVU on “no-reflow”. Transcriptome databases of concurrently isolated NVU cells from the ischemic brain system could offer invaluable insights. 147 Moreover, NVU co-cultures, including organoids and microfluidic devices, can shed light on neurovascular communication and disease mechanisms, such as “no-reflow”. 148
Furthermore, it has become apparent that the brain is not the insulated entity as it was once perceived, and the NVU communicates with systemic biology in a bidirectional manner. 149 Systemic disorders, such as metabolic abnormalities, can disrupt the NVU regulation, thus conditions like diabetes, hyperlipidemia, and atherosclerosis, which are often indicative of systemic metabolic irregularities, can enhance the probability of NVU disorder following ischemic stroke, 149 and thereby increase the predisposition to “no-reflow”. Moreover, aging can trigger molecular and cellular consequences in all NVU cell types, leading to cellular dysfunction. 150 Consequently, aging may be another factor instigating NVU disruption and inducing the “no-reflow” phenomenon.
SMCs were excluded from the microvessel contraction theory due to the limitations of this article on the vessels involved in “no-reflow”, however, as important vessel diameter control cells, SMCs could also contract vessel after cerebral infaction, thus, proximal emboli could be retained in contracted pial arterioles and penetrating arterioles, which result in “no-reflow”. The influence mechanism of SMCs for “no-reflow” is also worth further exploration.
Although anti-microthrombosis therapy may increase the incidence of intracranial hemorrhage (ICH), 151 thrombolysis therapy, aiming to mitigate “no-reflow” following recanalization, has been adopted clinically and has shown improved outcomes in AIS patients. Thus, future research may focus on integrating anti-microthrombosis therapy with BBB protective therapy.40,41 Experimental studies have also spotlighted other potential treatment targets to curb “no-reflow”, including microvessel compression and contraction, inflammation, and reactive oxygen species (ROS). However, given the absence of clinically approved drugs to tackle these targets, their therapeutic implications require more in-depth exploration. Furthermore, based on our experience, a solitary treatment approach appears to have limited impact on the restoration of neural function following AIS; therefore, coupling microvessel protection with neuronal preservation may yield superior outcomes.
To date, the principal limitation in the context of the “no-reflow” phenomenon pertains to its diagnosis. The absence of universally accepted diagnostic criteria precludes clinical practitioners from definitively ascertaining whether a patient is experiencing “no-reflow”. Nonetheless, as the number of patients receiving recanalization treatment post-AIS increases, the cerebral “no-reflow” phenomenon is becoming more conspicuous. Expert clinicians can leverage perfusion imaging to discern patterns, thereby cultivating a consensual understanding regarding the diagnosis of the “no-reflow” phenomenon. Furthermore, there is an urgent call for a non-invasive, bedside, and accurate method of cerebral blood flow examination for AIS patients who are unfit for transfer. This would enable real-time detection of “no-reflow”, thus guiding physicians to timely intervention. In addition to cultivating direct diagnostic methods, another prospective avenue is the prediction of “no-reflow” through machine learning (ML) applied to big data analytics. In an era where continuous collection of biomedical imaging data, physiological metrics, and electronic medical records are producing more therapeutically relevant data than ever before, the human capacity to evaluate the predictive value of such multidimensional medical data for guiding clinical decision-making is limited, given its volume and complexity. 152 Recent strides in ML have enabled computational analysis of medical imaging on an unprecedented scale, thereby unlocking opportunities to utilize these vast medical datasets to direct medical treatment in various fields, including AIS. 153 For instance, regression random forests have been trained using data obtained from the ischemic lesion, surrounding tissue, and the rest of the brain to forecast modified Rankin Scale (mRS) scores of patients after 90 days. In all 19 cases, their model accurately predicted the 90-day mRS, with an average absolute error of 1.05 ± 0.62. 154 Consequently, using pre- and post-thrombectomy imaging alongside baseline patient characteristics, ML might predict the “no-reflow” phenomenon and guide clinicians to proactively implement preventive measures.
In conclusion, the introduction of the “no-reflow” concept has spurred progress in understanding the failure of neural function following proximal vascular recanalization. However, given the relatively recent advancement of intracerebral recanalization therapy, our current clinical understanding of intracerebral “no-reflow” is still in its nascent stages. It is our hope that this review will pique the interest of an increasing number of researchers in the “no-reflow” phenomenon. An essential objective is to further investigate how different cells directly or indirectly precipitate “no-reflow”, and specifically inhibit crucial influential factors to effectively mitigate “no-reflow” without inducing other side effects. Furthermore, through wider observational studies and more sophisticated examination methods, clinical practitioners can more easily identify “no-reflow” and forge a broad consensus, thereby setting the direction for future “no-reflow” diagnosis. We optimistically anticipate that more and deeper studies focused on treating both macrovessel occlusion and “no-reflow” will render patients suffering from AIS to garner more benefits after macrovessel recanalization.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (82027802, 82071466, 82071468 and 82102220); 2019 Beijing Ten Million Talents Project (2019A36); Beijing Municipal Science and Technology Commission (Z221100007422023); General Projects of Scientific and Technological Plan of Beijing Municipal Education Commission (KM202010025023).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.