
Abstract
Periventricular white matter lesions (WMLs) are common MRI findings in migraine with aura (MA). Although hemodynamic disadvantages of vascular supply to this region create vulnerability, the pathophysiological mechanisms causing WMLs are unclear. We hypothesize that prolonged oligemia, a consequence of cortical spreading depolarization (CSD) underlying migraine aura, may lead to ischemia/hypoxia at hemodynamically vulnerable watershed zones fed by long penetrating arteries (PAs). For this, we subjected mice to KCl-triggered single or multiple CSDs. We found that post-CSD oligemia was significantly deeper at medial compared to lateral cortical areas, which induced ischemic/hypoxic changes at watershed areas between the MCA/ACA, PCA/anterior choroidal and at the tip of superficial and deep PAs, as detected by histological and MRI examination of brains 2–4 weeks after CSD. BALB-C mice, in which MCA occlusion causes large infarcts due to deficient collaterals, exhibited more profound CSD-induced oligemia and were more vulnerable compared to Swiss mice such that a single CSD was sufficient to induce ischemic lesions at the tip of PAs. In conclusion, CSD-induced prolonged oligemia has potential to cause ischemic/hypoxic injury at hemodynamically vulnerable brain areas, which may be one of the mechanisms underlying WMLs located at the tip of medullary arteries seen in MA patients.
Keywords
Introduction
Demyelination of the periventricular white matter is seen in several neurological disorders. Hemodynamic disadvantages of the vascular supply to this region are thought to be one of the factors creating this vulnerability.1–3 Long and thin penetrating arteries (PAs) originating from the pial arteries at the cortical surface feed this area. PAs do not have anastomoses, making blood flow through them vulnerable to changes in perfusion pressure. Furthermore, they supply oxygen to the capillary-free tissue surrounding them (Krogh’s cylinder),4,5 hence, steeply lose their oxygen as they descend into deep periventricular areas. 6 Accordingly, a reduction in flux rate caused by, for example, cortical spreading depolarization (CSD)-induced constriction of these vessels and the terminal microvascular bed may put the deep periventricular areas at risk of hypoxia. 7 Thus, oligodendrocytes, which are vulnerable to hypoxia may be injured, leading to demyelination. 8
Indeed, T2-hyperintense magnetic resonance imaging (MRI) lesions are more prevalent in patients with migraine with aura (MA) compared to the general population.9–12 In general, they are presumed to have an ischemic/hypoxic origin 13 although the pathological nature and underlying etiology of MRI lesions in migraineurs are unclear. In addition to cerebral microembolism,14,15 one of the proposed causes is the prolonged oligemia seen at the wake of CSD,12,16 the putative cause of migraine aura.17–19 This oligemia is due in part to active vasoconstriction caused by 20-hydroxyeicosatetraenoic acid derived from arachidonic acid released during CSD. 20 Although post-CSD oligemia is generally considered a benign phenomenon with no residual tissue damage in experimental animals, 21 it is likely that it can potentially put the tissue in hemodynamically vulnerable watershed areas supplied by PAs at risk of ischemia/hypoxia,4,5 which may especially be significant in subcortical areas of the human brain fed by long medullary arteries.2,3 MRI lesions located in the distal vascular territories supplied by medullary arteries suggest such a hemodynamic mechanism.22,23 CSD-induced vasoconstriction of the pial and PAs, while causing oligemia in the cortex, can lead to ischemia/hypoxia in the deep subcortical areas supplied by the same long medullary arteries, especially in the presence of hemodynamically unfavorable conditions. Indeed, perfusion-weighted MRI scans performed soon or several hours after aura consistently detected oligemic areas contralateral to the patients’ aura symptoms. Oligemic areas were only observed during MA attacks and disappeared in follow-up scans, suggesting that they correspond to post-CSD oligemia observed in experimental animals.24–28 Endothelial dysfunction observed in at least a group of migraineurs may also predispose to stronger vasoconstriction and make tissue vulnerable to deep white matter lesions (WMLs). 29 Supporting this view, CADASIL patients, who have dysfunctional PAs and suffer from frequent MA attacks (that is, CSDs), develop multiple WMLs as the vascular dysfunction progresses. 30
We have tested these possibilities in the intact mouse brain. In addition to single CSDs, we also triggered repeated CSDs to disclose any potential vascular vulnerability by intensifying CSD-induced hemodynamic stress, because the length of PAs are much shorter in mice compared to humans and rodent models of CADASIL or hypertension are known to be resistant to formation of deep WMLs unlike patients.31,32 Additionally, we subjected BALB-C mice known to develop large MCA infarcts due to deficient collaterals33–35 to single CSD, considering that a single CSD in these mice might be sufficient to disclose the hemodynamic vulnerability. Here, we report that the post-CSD oligemia is more pronounced at medial areas of the MCA territory and can induce ischemic histological changes at the cortical and deep watershed areas between the MCA, ACA, PCA and anterior choroidal arteries as well as at the tip of the medial deep and superficial PAs of BALB-C mice as detected by MRI as well as histological examination of brain sections 2–4 weeks after CSDs.
Materials and methods
Experimental animals and protocol
Male Swiss albino (from Hacettepe University Experimental Animal Facility-originally from Refik Saydam Public Health Institute, Ankara, Turkey) and BALB-C (from Kobay DHL A.Ş., Ankara, Turkey) adult mice weighing 25–35 g were housed under diurnal lighting conditions. Animal housing, care, and application of experimental procedures were carried out in accordance with Hacettepe University, Laboratory Animals Research and Application Centre guidelines. All procedures were approved by Hacettepe University Ethics Committee (2017/46). The experiments have been reported in compliance with the ARRIVE Guidelines. Animals (n = 48) were anesthetized with isoflurane (1.5–2%) or ketamine (100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.). Body temperature was kept at 37.0 ± 0.2 °C by a homoeothermic blanket control unit. Pulse rate and tissue oxygen saturation were monitored by an oxymeter using a mini Y clip attached to one lower extremity. The tissue O2 saturation was kept above 95% with oxygen supplement at 2 l/min. In two separate groups of naïve mice, the arterial blood pressure was noninvasively measured from the tail.
Mice were placed in a stereotaxic frame. After a midline incision, a 3x3 mm of skull between the bregma and lambda on the right side was thinned with a drill under a stereomicroscope. The area was continuously irrigated with artificial cerebrospinal fluid to prevent heating during drilling. CSDs were evoked by placing a cotton ball soaked with 1 M KCl on the dura 1.5 mm anterior and 1.5 mm lateral to Bregma and, in 2 mice, over the parietal bone through a burr-hole. The cotton ball was removed after the first CSD or one hour later in the case of multiple CSDs and any remaining KCl on the dura was rinsed with saline irrigation. Laser speckle contrast (LSC) imaging was used to monitor the CSDs. At the end of 60 minutes of monitoring, the head skin was sewed, and the mice were kept alive for 2–4 weeks.
Histological analyses
To investigate whether CSD-induced oligemia can cause ischemic lesions at watershed areas, Swiss mice were subjected to repeated CSDs under isoflurane (n = 9) or ketamine (n = 6) anesthesia or to a single CSD under isoflurane (n = 5). Fifteen BALB-C mice were subjected to a single CSD under isoflurane anesthesia. Five Swiss mice were subjected to sham surgery and saline-soaked cotton ball application for one hour under isoflurane. One mouse from ketamine group and 7 mice from BALB-C group did not survive for 2 weeks. Surviving mice and sham animals were transcardially perfused 2–4 weeks after the experiment with heparinized saline followed by 4% paraformaldehyde under chloral hydrate anesthesia. Brains were postfixed in 4% paraformaldehyde for 24 hours with intact skull to minimize dark neuron formation. The skull was then removed, and the brains were harvested to prepare 5 µm-thick paraffin sections. Paraffin sections were then stained with luxol-fast blue or hematoxylin and eosin. Sections were evaluated by at least two blinded investigators (F.S., T.D., M.Y. and B.D-D.).
Laser speckle contrast imaging
Cortical blood flow (CBF) was monitored for one hour with Laser Speckle Contrast (LSC) imaging in all Swiss and BALB-C mice subjected to single or recurrent CSDs. A CCD camera (Basler 602 F or acA1300, Basler Vision Technologies, Ahrensburg, Germany) attached to a stereomicroscope (Nikon SMZ 1000, Nikon) and custom-developed software (courtesy of AK Dunn of Texas University) was used for LSC imaging. A laser diode (785 nm wavelength, Thorlabs) was used to diffusely illuminate the cortical surface through thinned skull. LSC images were captured every 10 seconds during 1-hour experiment and processed as described by Dunn AK et al, 2001 36 to construct CBF maps. To compare the CBF changes induced by recurrent CSDs in selected regions of cortex, LSC images were later imported to ImageJ v1.42q NIH as image sequences and then saved in tiff format for further analysis. In order to transform contrast values of each image within the saved sequence to CBF values, inverse correlation time (ICT) images were obtained with MATLAB software by taking into consideration the camera exposure time. A baseline image was constructed by averaging several images before the first CSD for each saved ICT image sequence. Subsequent ICT images were differentially divided by the baseline image and then all calculated values for each pixel were averaged. The results were constructed as a single image and pseudo-colored to illustrate the average percent flow change for each pixel, from which ROIs were selected in order to compare CBF changes in different cortical areas. The mean and standard deviation of each ROI were calculated and histograms were generated with MATLAB software by an investigator blinded to the anesthetic used (G.U.). Adobe Photoshop was used for photo editing and image creation with the images taken by LSC imaging system.
Magnetic resonance imaging
Two mice whose right hemisphere were subjected to KCl-induced repetitive CSDs for one hour under isoflurane, were sacrificed 4 weeks later by transcardial perfusion with heparinized saline followed by 4% paraformaldehyde under chloral hydrate anesthesia. The brains were then extracted as a whole and preserved in PFA until MR imaging. Prior to imaging, PFA-fixed mouse brains were placed in a 2 ml-volume syringe filled with Fluorinert FC-77 liquid (3 M™ Electronic Liquids, Saint Paul, USA), a proton-free fluid with low water solubility and similar magnetic susceptibility to the tissue. This greatly reduces the background noise and artifacts from the surrounding medium during image acquisition. MRI acquisitions were performed on a horizontal 7 T BRUKER Biospec MRI system (Bruker Biospin MRI, Ettlingen, Germany) equipped with a set of gradients of 400 mT/m and controlled via Bruker ParaVision 5.1 workstation. A 2 ml-volume syringe filled with Fluorinert containing the mouse brain sample was placed on a dedicated holder and positioned at the center of the magnetic field system. A Bruker birdcage volume coil (outer diameter = 112 mm and inner diameter = 72 mm) was used for signal transmission, and a Bruker single loop surface coil (15 mm diameter) was used for signal reception and positioned in order to target the mouse brain. Axial oriented MR images were obtained using a 3 D T2-weighted fat saturated sequence based on rapid acquisition with relaxation enhanced (RARE) method. The ex-vivo acquisition parameters were as follow: Echo Time (TE) 80 ms, Repetition Time (TR) 2800 ms, RARE factor = 8 and a number of 4 averages. A field of view (FOV) of 1.50 × 0.75 × 1.50 cm3, and a matrix size of 256 × 128 × 64 reconstructed by interpolation 256 × 128 × 256 provided an isotropic resolution of 78 × 78 × 78 microns, for a total scan time of 12h45 minutes.
After the ex vivo MRI acquisition, the brain samples were preserved in PFA until histological processing. Coronal brain sections were then cut and stained with luxol-fast and H&E. Coronal sections corresponding to the MRI sections exhibiting signal asymmetry were histologically evaluated to assess specificity of the MRI lesion.
Statistics
The mean values for the diameter of the ischemic column in the borderzone, arterial blood pressure, mean changes in CBF and ICT values are given with their standard errors. In addition to the descriptive statistics mentioned above, the normality of each dataset was assessed by Shapiro-Wilk test and then, the mean changes in CBF in the medial and lateral ROIs under the two anesthetics were compared using the Mann-Whitney U test. Uninterrupted LSC imaging data throughout the one-hour recording period was available for 6 Swiss mice from each of isoflurane and ketamine groups. The power analysis for the sample size indicated that 6 mice were satisfactory based on variation in the CBF values (power 90% and α = 0.05). CBF values were available from 15 BALB-C because 7 of these mice died within 2 days after CSD induction, requiring additional mice for histological evaluation performed 2 weeks later.
Results
CSD induces delayed histological lesions at hemodynamically vulnerable brain areas
To specifically test whether the CSD-induced vasoconstriction could lead to ischemia at hemodynamically susceptible tissue fed by PAs, we first examined the coronal brain sections obtained from Swiss albino mice subjected to KCl-induced repeated CSDs for one hour. On average 9.2 ± 0.4 (under isoflurane, n = 9 mice) or 6.0 ± 0.5 (under ketamine, n = 6 mice) CSDs were evoked in an hour by application of KCl on the dura 1.5 mm anterior and 1.5 mm lateral to Bregma. Coronal sections 1.75–2 mm posterior to the KCl application site were studied 2 weeks after CSD. These mice did not have a cranial window except a burr hole in the prefrontal area for KCl application; therefore, they did not show any histological abnormalities related to cranial surgery performed a day before the experiment similar to the sham controls subjected to saline application without CSD induction (n = 5 mice). Interestingly, however, we observed neuropilic edema (loosening), degenerating neurons, swollen peri-neuronal processes (possibly astrocyte endfeet) of varying intensity in a vertical column of cortical tissue around the long PAs located at the frontal cortical watershed areas between ACA and MCA in 5 out of 9 Swiss mice anesthetized with isoflurane (Figure 1(a)). The diameter of the ischemic column was 171 ± 19 µm (n = 5 mice), consistent with the size of the capillary-free tissue oxygenated by passive diffusion from the PA in the mouse (Krogh’s cylinder).4,5 MRI performed before histological examination detected the ischemic watershed lesions (subsequently verified histologically) as asymmetrical (compared to the contralateral site) hyperintense signal area in T2-weighted images at posterior border zone (Figure 1(b)). Prompted by MRI findings, we disclosed a similar lesion at the border zone between the MCA and PCA in 2 mice when we also examined a series of consecutive coronal sections from the posterior brain areas (Figure 1(b)).
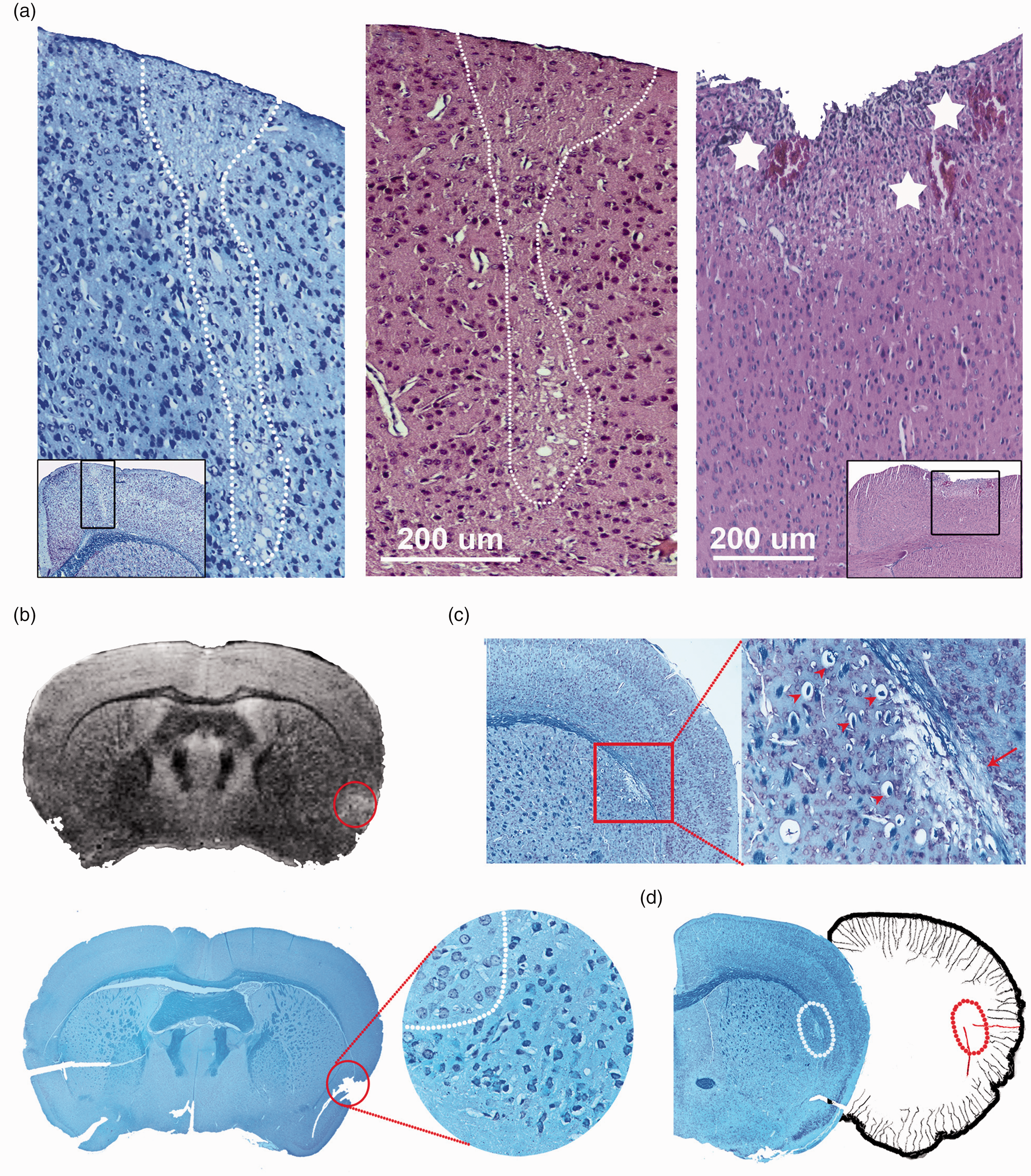
Recurrent CSDs can induce an ischemic lesion at hemodynamically vulnerable cortical watershed areas. (a) Illustrates the ischemic changes at the frontal border zone between the anterior and middle cerebral artery. Reduced Nissl staining, cellular and neuropilic swelling are visible along a narrow strip possibly fed by a long penetrating artery (boxed area in the inset). The adjacent coronal section stained with hematoxylin and eosin. The KCl-soaked cotton ball that was placed more lateral and anteriorly to the hemodynamic lesion for one hour, directly caused a superficial cortical necrosis unlike the ischemic changes induced at hemodynamically vulnerable medial border zone, which extend deep into the cortex along a narrow strip (hematoxylin and eosin stain). Immediately under the cotton ball, some of the dural and pial vessels were thrombosed. *Depicts thrombosed vessels. Scale bar: 200 μm. In contrast, the dura and pia as well as the superficial cortex were intact on coronal sections involving the watershed lesions. (b) CSDs also caused lesions at the MCA-PCA and MCA-choroidal watershed areas. An abnormal signal at the border zone between the MCA and PCA on T2 MRI 4 weeks after recurrent CSDs for one hour (red circle), which was confirmed to be an ischemic lesion by examination of the same brain section with Luxol-fast staining (bottom image). The horizontal cut on the left marks the contralateral normal hemisphere. The softened ischemic tissue was torn during histological processing. The area within the circle exhibits swollen as well as shrunken cells with condensed nuclei neighboring the normal tissue on the left upper quadrant at 400X magnification. Continued.(c) illustrates a small subcortical lesion involving the corpus callosum and the tissue immediately below, corresponding to the border zone (watershed) between the anterior choroidal artery and a long penetrating artery. The magnified panel shows demyelinated axon bundles in corpus callosum (loss of blue staining with Luxol-fast, arrow) as well as spongiform neuropil, swollen and shrunken cells with condensed nuclei (arrowheads). (d) The hemisphere on the left illustrates the CSD-induced white matter lesion detected at the anterior choroidal-MCA watershed zone with Luxol-fast staining (white circle). The hemisphere on the right is modified from 75 to illustrate the long penetrating and anterior choroidal arteries forming the internal watershed zone (marked with red).
We did not observe similar border zone histological lesions in 5 mice subjected to a single CSD under isoflurane anesthesia or in 4 out of 5 mice subjected to recurrent CSDs for one hour under ketamine anesthesia. However, in one mouse in the group subjected to multiple CSDs under ketamine, we detected a demyelinating ischemic lesion involving the corpus callosum and the tissue immediately underneath, which corresponds to the watershed area between the anterior choroidal and MCA (Figure 1(c) and (d)). This lesion was characterized by reduced Luxol-fast staining and swelling of myelin in the corpus callosum, indicating demyelination as well as loosening of the neuropil underneath, which exhibited several shrunken and swollen cells.
Although we irrigated the burr hole after removing the cotton ball to terminate CSD induction, local cortical injury (Figure 1(a)) and strong depolarization at the site of KCl application might have triggered secondary events over a prolonged time course after the 1-hour CSD monitoring period. Contribution of this to the formation of hemodynamic lesions is difficult to investigate, but seems unlikely, as suggested by the absence of anterior border zone lesions in mice subjected to one hour of KCl application under ketamine anesthesia, which also caused a similar lesion at the application site. Unlike a brief application of KCl to induce a single CSD, keeping KCl on the dura for one hour consistently caused discoloration and indentation of the underlying cortex and thrombosed vessels that could be seen macroscopically. Accordingly, coronal sections were cut starting from posterior to this region, except 2 mice from each group in which the lesion area was also sectioned for histological verification. Ketamine, which has a more powerful inhibitory action on CSD generation, but less significant cerebrovascular effects compared to isoflurane,37–40 lowered the number of CSDs ignited during one-hour KCl induction and reduced the intensity of oligemia, implying that the main driver of the hemodynamic vulnerability was the magnitude of spreading oligemia accompanying recurrent CSDs (see next section). Moreover, the presence of lesions in the posterior and internal watershed areas that are distant and disconnected from the frontal KCl application supports the idea that the anterior watershed lesions 1.5 to 2 mm away from the KCl application site were unrelated to the KCl-induced focal necrosis.
CSDs induce deeper oligemia at watershed areas
We next examined the CSD-induced blood flow changes at the MCA-ACA watershed that can be readily monitored with LSC imaging through thinned skull in additional sets of mice (Figure 2(a)). LSC images recorded during recurrent CSDs disclosed that the post-CSD oligemia was most profound in the medial cortex encompassing the border-zone compared to the other areas of the cortex (Figure 2). Unlike the prolonged monophasic oligemia observed following a single CSD, during recurrent CSDs, the CBF returned to the baseline level (or slightly exceeded it) during the passage of each CSD. However, these hyperemic responses were either absent or of much shorter duration at the medial cortical areas under isoflurane anesthesia (n = 6 mice), illustrating that the medial areas are repeatedly exposed to the CSD-induced metabolic challenge without associated blood flow increase. Because isoflurane may compromise cerebral hemodynamics by lowering systemic arterial pressure concomitantly with inducing cerebral vasodilation, which can limit cerebrovascular reactivity (e.g. loss of autoregulation) 37 and cause microvascular shunting, we also tested the effect of multiple CSDs on watershed CBF under ketamine/xylazine anesthesia in another 6 mice, since ketamine has relatively milder unfavorable hemodynamic effects.37,41,42 To confirm that CSD-induced changes in CBF were indeed different between the two anesthetics, we developed an algorithm to calculate the mean change in CBF from baseline for each LSC imaged pixel throughout the one-hour recording period, which allowed us to compare changes in CBF in entire cortex region without selection bias in picking up an ROI, in mice under isoflurane or ketamine anesthesia (Figure 2(b)). The mean CBF change during recurrent CSDs was −24 ± 2% in the medial border zone, whereas it was −15 ± 3% in the lateral parietal area under isoflurane anesthesia (n = 6). Under ketamine/xylazine (n = 6), the CBF changes were −13 ± 2% and −17 ± 2% in the medial and lateral areas, respectively. The mean changes in the medial areas under the two anesthetics were significantly different (p = 0.006). As in the isoflurane anesthesia, the hyperemic responses during CSDs in the medial areas were irregular with some failures, unlike the lateral areas, where a hyperemic response was routinely observed with each CSD wave under ketamine anesthesia, although total oligemia was less severe compared to the isoflurane group (Figure 2(c)). These observations conform with the view that both the depth of oligemia and the hyperemic responses are significantly modified by perfusion pressure, 43 which is reduced toward the distal MCA territory feeding the medial areas and may have been further compromised by relatively lower arterial blood pressure and cerebrovascular dilation under isoflurane. 37
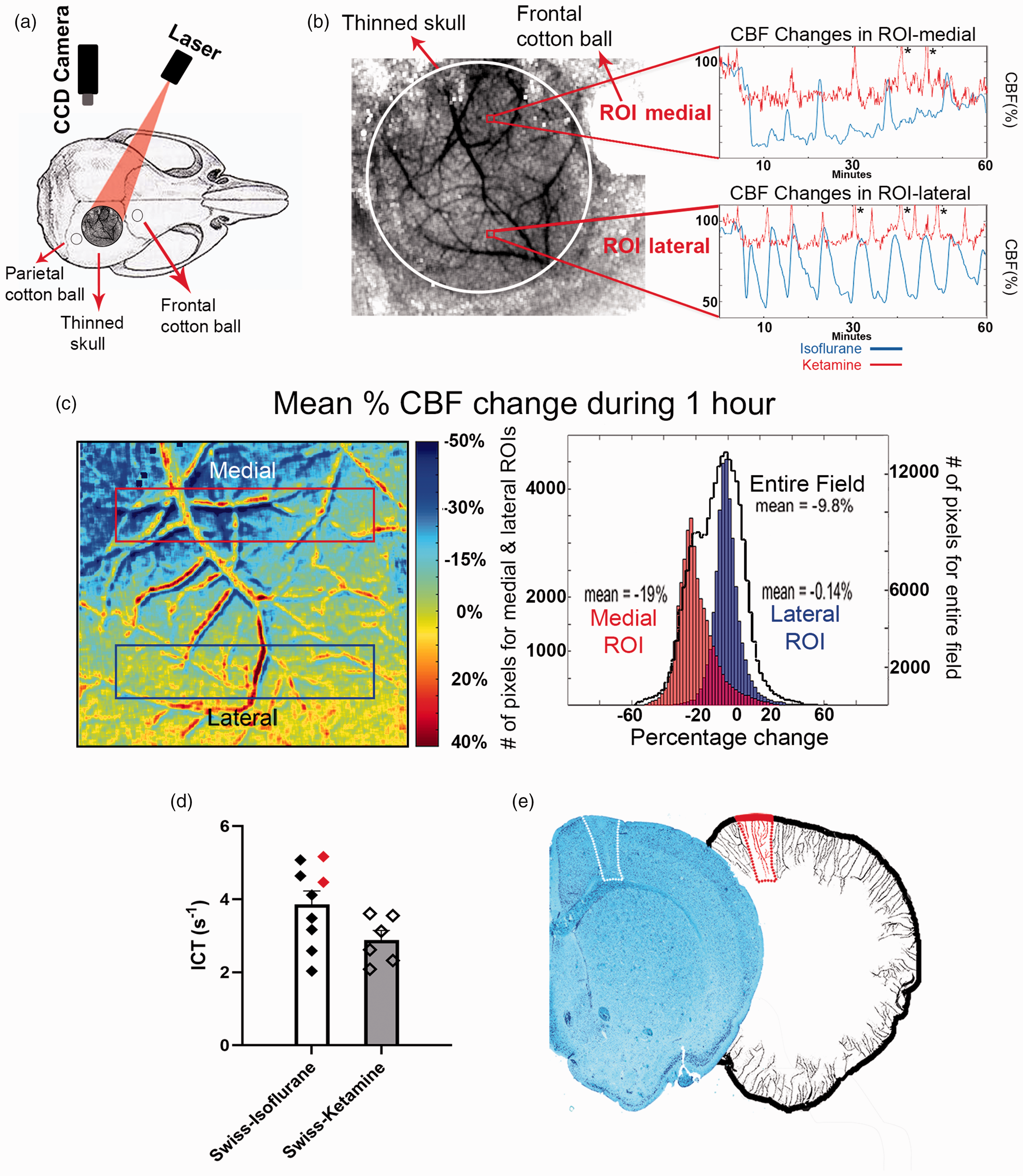
The oligemia induced by recurrent CSDs was more profound in the ACA-MCA border zone compared to lateral areas, predisposing to watershed ischemia. (a) Illustrates the sites where KCl was applied (cotton ball) and blood flow changes were recorded with LSC imaging through the intact thinned mouse skull. (b) Shows the view of cortex and pial vessels imaged and the cotton ball. The relative blood flow changes recorded with LSC throughout one-hour period during KCl-induced CSDs from regions of interests (ROI) over the border zone between the ACA and MCA and, a lateral area within the MCA territory from a mouse under isoflurane anesthesia were shown as plots. Note that hyperemic episodes during each CSD are of shorter duration and, hence, the total oligemia is more pronounced in the medial ROI. The oligemia was less pronounced under ketamine anesthesia (red trace recorded from another mouse); *denotes the CBF responses that were above 110% (c) The pseudocolored image shows the average CBF changes during one-hour period of recurrent CSDs for each pixel as detected by LSC imaging (cold colors illustrate reduction in the CBF compared to baseline). Under isoflurane, the oligemia is deepest at the medial border zone, whereas it becomes less severe toward periphery. The double vessel trace is caused by pulsations and minimal brain displacement that is unavoidable during Continued.CSD-induced blood volume changes but does not significantly affect the measurements obtained from large ROIs. Histograms obtained from the same animal illustrate the distribution of average CBF changes for each pixel in the medial (red rectangle) and lateral (purple rectangle) areas. Black line illustrates all pixels in the entire field imaged, the expanded scale of which is given on y-axis on the right. (d) Graphs illustrate the basal ICT values in Swiss mice under isoflurane or ketamine anesthesia. Red diamonds represent the ICT values from 2 mice in whom CSD was triggered with KCl application through the parietal burr hole. In all other mice, cotton ball was applied through the frontal burr hole. (e) The hemisphere on the left illustrates the CSD-induced lesion detected at the frontal watershed zone with Luxol-fast staining (white box). The hemisphere on the right was modified from 75 to illustrate the long cortical penetrating arteries at the frontal watershed zone (marked with red).
LSC imaging provides only semiquantitative data; therefore, we used relative values as generally practiced. However, this approach eliminates any information related to baseline blood flow, which should be taken into account when comparing the isoflurane and ketamine groups because isoflurane increases the basal CBF.37,41,42 Although it is not possible to assess absolute perfusion without independent calibration by other methods, ICT is correlated with perfusion over a considerable range. 42 In line with the previous reports, the baseline ICT was 34% higher in Swiss mice under isoflurane compared to ketamine37,41,42 (Figure 2(d)). For BALB-C mice under isoflurane, baseline ICT was comparable to Swiss under isoflurane (mean±SE; 3.86 ± 0.37 vs. 4.53 ± 0.31l S−1, p > 0.05). Despite higher baseline level with isoflurane, which suggests that relatively deeper looking oligemia under isoflurane may in fact be corresponding to higher CBF values compared to ketamine group, ischemic injury emerged in isoflurane but not ketamine group. Strongly supporting the presence of a significant post-CSD oligemia, oxyhemoglobin recordings during multiple CSDs in mice under isoflurane closely parallel around 50% decrease in CBF recorded with LSC imaging. 44 This is not surprising given that CBF increase due to vasodilation in the absence of a triggering metabolic demand (i.e., without neurovascular coupling) causes shunting of blood by way of throughfare channels rather than flowing through all capillaries, which cannot be differentiated with LSC imaging. Indeed, this has been clearly documented for hypercapnia-induced vasodilation, where blood quickly shunts from the arteriolar to the venular circulation along with sporadic capillary constrictions to maintain optimal tissue oxygenation. 45
We did not continuously monitor the intra-arterial blood pressure during anesthesia to avoid invasive procedures in these mice that can induce confounding hemodynamic factors in small animals and negatively impact their survival for the following 2 weeks. Repeated non-invasive blood pressure measurements from the tail in this small rodent unfortunately confounded laser-speckle recordings by introducing motion artifacts. Considering that we were not comparing groups based on changes in blood pressure but trying to unambiguously document the hemodynamic vulnerability caused by CSDs, we, instead, monitored the blood pressure in two separate groups of naïve mice for one hour, and found that the blood pressure was within normal range under both isoflurane (n = 3) and ketamine/xylazine (n = 3) anesthesia, but was significantly lower under isoflurane (94.5 ± 0.4 and 110.8 ± 3.4 mmHg respectively, p = 0.050) consistent with the previous experience of our laboratory with this mouse strain and anesthetics.
A single CSD induces hemodynamic lesions when collaterals are insufficient
To disclose the potential vulnerability created by a single CSD at the watershed regions, we also studied its effect in BALB-C mice known to have insufficient collaterals between MCA and ACA, and an incomplete circle of Willis.33–35 As we anticipated, these mice exhibited deeper flow reduction post-CSD than did Swiss mice (p < 0.001). The mean CBF change after single CSD under isoflurane was diffusely profound; −45 ± 4% in the medial and −46 ± 3% lateral MCA areas (n = 15), possibly due to insufficient ACA collateral support in BALB-C in response to flow reduction in the MCA territory (Figure 3(c) and (d)). Corresponding values after single CSD in Swiss mice under isoflurane were −31 ± 4% and −34 ± 4% in medial and lateral areas, respectively. Importantly, in 7 out of 8 BALB-C mice, a single CSD induced mild ischemic changes (neuropilic or astrocyte endfeet swelling) in the medial septal area as well as myelin swelling of the axon bundles within the medial corpus callosum or passing through the basal ganglia (Figure 3(a) and (b)), whereas no necrosis was observed at the KCl application site, as typical in experiments when KCl was immediately rinsed after the first CSD. Seven BALB-C mice were found to be dead (so no histological examination was possible) within the 2 days after completing the CSD experiment. We presume that this might have been caused by hypothalamic ischemia due to hemodynamic vulnerability of deep penetrating arterioles feeding this area, which originate either from the incomplete circle of Willis or proximal segments of its branches. In contrast, no such a lesion in Swiss mice subjected to single CSD was observed. Swiss mice may also have an anomaly in one of the posterior communicating arteries, however, this only causes oligemia in the hippocampus during occlusion of the MCA if a thick filament completely filling the carotid lumen is used. 46
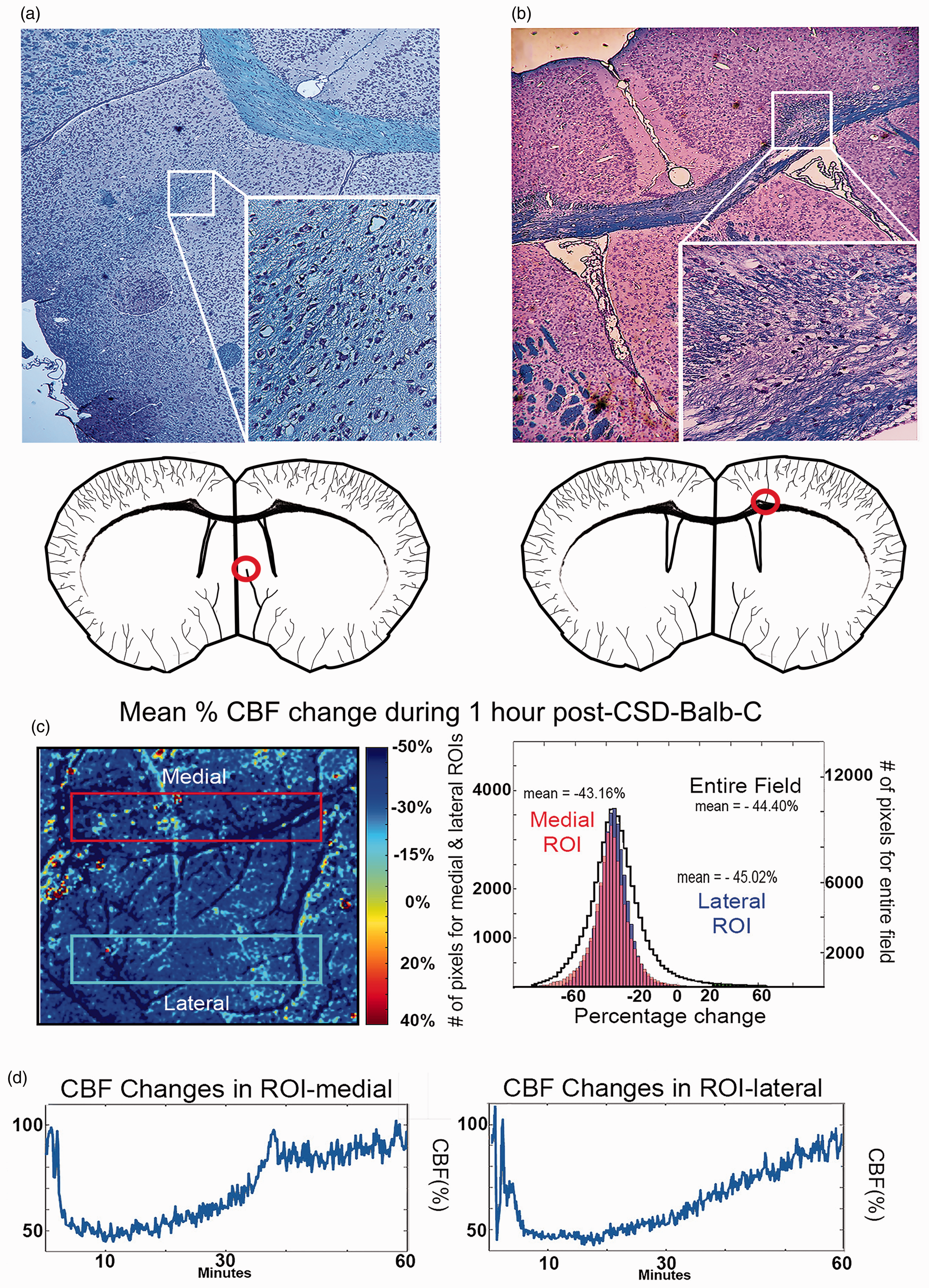
A single CSD can induce an ischemic lesion at hemodynamically vulnerable subcortical watershed areas in BALB-C mice. Top row illustrates the ischemic changes (neuropilic or astrocyte endfeet swelling) induced by a single CSD in the medial septal area Continued.(a) and myelin swelling of the axon bundles within the medial corpus callosum (b). Insets show the boxed lesions at 400X magnification. Both brain sections were stained with luxol-fast and, cresyl violet (a) or nuclear-fast red (b). The hemisphere drawings below were modified from 75 to illustrate the long deep (a) and superficial (b) penetrating arteries affected, and the lesions formed at the hemodynamically vulnerable zones at their tips (marked with red circles). (c) The pseudocolored image shows the average CBF changes during one-hour period following a single CSD for each pixel as detected by LSC imaging (cold colors illustrate reduction in the CBF compared to baseline). The CSD-induced oligemia was equally deep at the medial and lateral cortical areas in a BALB-C mouse with incomplete MCA-ACA collaterals. The histogram obtained from the same animal illustrates the distribution of average CBF changes for each pixel in the medial (red rectangle) and lateral (purple rectangle) areas. Black line illustrates all pixels in the entire field imaged, the expanded scale of which is given on y-axis on the right. (d) The relative blood flow changes recorded with LSC imaging throughout one-hour period during a KCl-induced CSD from regions of interests (ROI) over the border zone between the ACA and MCA and, a lateral area within the MCA territory from a mouse under isoflurane anesthesia were shown as plots.
Finally, because the resolution of vasculature was low in LSC images, we obtained high-resolution vascular images for one hour after KCl-induced single CSD in two mice, which exhibited parallel triphasic arterial diameter changes to the CBF variations observed with LSC imaging (Figure 4(a) and (b)). These images clearly illustrated that pial arteries in the cortex under the thinned skull, including the MCA-ACA border zone, remained partially constricted following the early hyperemic phase after CSD for at least 30 minutes (Figure 4(a) and (b)). The constrictions could be traced down to the point that arterioles dived into the cortex, indicating that a single CSD has the potential to reduce the flux rate in PAs, hence, putting the tissue around and at the tip of them at risk of hypoxia (as convincingly illustrated with optical coherence tomography 7 ) especially in the presence of factors that further compromise cerebral hemodynamics such as hypotension or deficient collaterals.
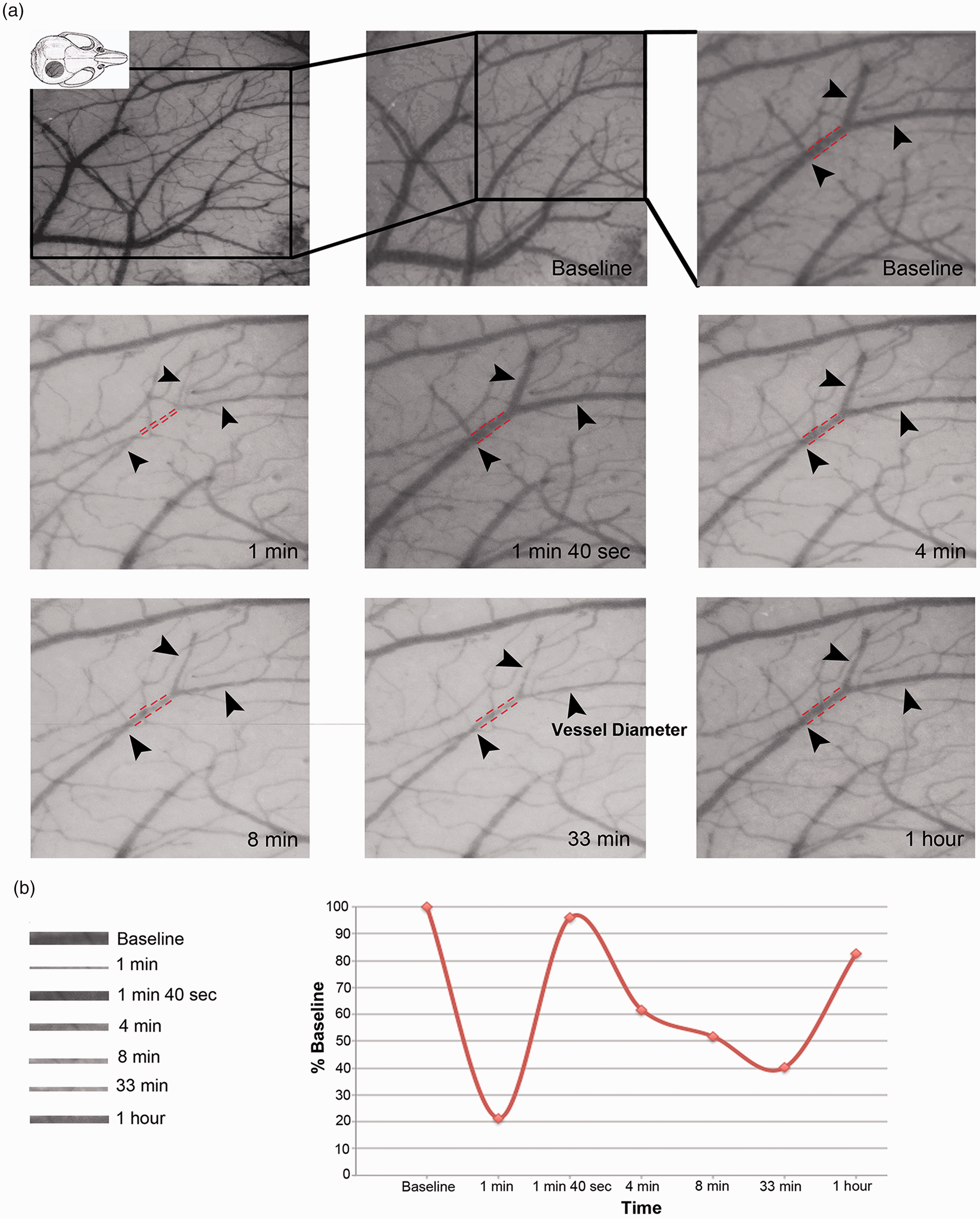
A single CSD induces significant pial vessel constriction. (a) Pial vessels were imaged through an intact thinned skull during and one-hour after a KCl-induced single CSD in a mouse under isoflurane anesthesia. Distal branches of the middle cerebral artery including the ones abutting the border zone are illustrated (arrowheads). A brief but severe vasoconstriction within the first minute (1 min) was followed by vasodilation (1 min 40 sec), which was replaced by a moderate but at least an hour lasting vasoconstriction (8, 33 minutes and 1 hour) and (b) The images of the vessel segment demarcated with broken red lines are consecutively displayed at the bottom for easier comparison. The diameter changes were graphically illustrated at the bottom right (time displayed on x axis is not linear). Note that the cortical light reflectance parallels the blood volume changes.
Discussion
CSD can induce ischemia at watershed areas
A novel finding of this study is that CSD has the potential to cause ischemic/hypoxic injury in hemodynamically vulnerable areas of the brain, although the rest of the cortex and subcortical areas remain intact except for scattered dark neurons and injured cells. This finding may be relevant to some of the WMLs located at the tip of medullary arteries seen in migraine patients, considering that long medullary arteries of humans are particularly vulnerable to hemodynamic perfusion changes. The special conditions (i.e. insufficient MCA-ACA collaterals in BALB-C or recurrent CSDs under mildly hypotensive anesthesia in Swiss mice) gave the opportunity to disclose the significant hemodynamic vulnerability that CSD can create, which may emerge in MA patients at the wake of a single CSD. Post-CSD oligemia after a single event seems to last longer in humans, unlike CSDs associated with oligemia lasting about an hour in rodents and other species.47,48 Lauritzen and Olesen detected hypoperfused brain areas up to 4–6 hours after aura during spontaneous MA attacks with SPECT following xenon inhalation in their seminal studies. 26 Perfusion MRI studies later also found hypoperfused areas in MA patients more than 2 hours after the beginning of aura.25,27,28 In agreement with these imaging findings, the typical duration of migraine aura symptoms has recently been reported to be longer than an hour in 6–10%, 14–27%, 17–60% of patients with visual, sensory and aphasic aura, respectively. 49 Importantly, a significant association was observed between WMLs and the aura duration as well as attack frequency.50–52 A recent PET/MRI study detected widespread parenchymal inflammation, encompassing the subcortical areas in patients having frequent MA attacks, which might aggravate CSD-induced transient hypoxic injury at the tip of PAs and cause demyelination. 53 Prolonged oligemia in humans can therefore predispose the watershed areas to injury when associated with conditions that unfavorably affect cerebral hemodynamics. For example, leukocytosis and cytokines released during a systemic infection 43 or orthostatic hypotension induced by hastily standing up while at bed rest may further compromise CBF in oligemic watershed areas. Supporting the latter possibility, several clinical studies have demonstrated a higher prevalence of syncope and orthostatic/sympathetic insufficiency in migraineurs than controls.54,55 Endothelial dysfunction reported at least in a group of MA patients can also aggravate vasoconstrictions during CSD, causing deeper post-CSD oligemia. 29 Therefore, in translating findings obtained from mouse to patients, the species differences predisposing to hemodynamic vulnerability (e.g. recurrent CSDs in the mouse vs. single CSD but long PAs plus prolonged oligemia in humans) should be taken into account, considering that at least some of the MRI lesions observed in migraineurs are highly suggestive of a hemodynamic etiology. 56
Although our study convincingly demonstrates that post-CSD oligemia has the potential to lead to ischemia by hemodynamic mechanisms, there are two caveats that we need to clarify for translating the observations in the mouse to the MRI lesions seen in MA patients. First, the WMLs are subcortical, whereas some of the ischemic lesions that we observed in Swiss mice were mainly cortical. However, this discrepancy can be explained by the differences between the ratio of the cortical vs. subcortical tissues as well as vasculature of the two regions in the mouse and human. Compared to humans, the mouse brain has a very narrow deep white matter area; the ratio of gray to white matter ratio is 90:10 in the mouse, whereas it is 40:60 in humans. 57 Consequently, the mouse does not have long medullary arteries; PAs in the mouse are generally less than 1–2 mm, whereas human PAs can be as long as a few centimeters, which can make humans particularly vulnerable to prolonged oligemia associated even with a single CSD, unlike the Swiss mouse in which multiple CSDs were required to disclose the potential vulnerability. Indeed, occipital rCBF decreases as large as 53% and persisting up to 2.5 h into the headache phase were detected by perfusion MRI in migraineurs with visual aura.28,58 Therefore, once a single CSD is initiated, prolonged post-CSD oligemia can increase the probability of ischemic episodes in the internal watershed areas fed by long medullary arteries in humans. In line with this, we found that the profound cortical oligemia in BALB-C mouse triggered by single CSD caused focal demyelinated lesions in corpus callosum fed by the longest PAs. Further supporting this view, unlike the common observation in CADASIL patients, the lacunes could not also be faithfully reproduced in mouse models of CADASIL despite overt degenerative changes in PAs possibly because of the above cerebrovascular differences in two species. 31 Similarly, unlike hypertensive patients, deep WMLs are not observed in aged spontaneously hypertensive rats despite overt cerebrovascular degeneration. 32
The second caveat is that most of the WMLs in migraineurs are located in the frontal and parietal lobes, whereas CSDs generally occur in the occipital lobe. It is known that relatively shorter PAs in the posterior regions are more resistant to hemodynamic perturbations than long frontoparietal and insular medullary PAs. 3 However, there is convincing imaging and clinical evidence showing that oligemia can spread up to the frontal regions of the brain in humans.24,26,59–61 In fact, sensory and aphasic auras (possibly caused by CSDs in the cortical areas anterior to the occipital lobe) tend to be longer than an hour as noted above. 49 As CSD propagates roughly at a speed of 3 mm/min, it can travel 18 cm away from the occipital pole in an hour to reach the frontal lobe. Importantly, considering that WMLs are relatively rare compared to countless number of MA attacks a patient suffers throughout his lifetime, coincidence of the above-discussed conditions compromising medullary PA perfusion during aura-associated oligemia is expected to be uncommon.
Mouse model
Use of the mouse that responds to CSD with a more profound oligemia compared to other experimental animal species 62 might have enabled the demonstration of such a hemodynamic vulnerability caused by CSD-induced oligemia in the present study. CSDs in rats, swine, cats and humans induce a predominant hyperperfusion overshooting the baseline level in naïve tissue, which is variably followed by a mild oligemia, whereas the mouse rCBF response to CSD is biased toward vasoconstriction and therefore may create more hemodynamic vulnerability, especially during recurrent CSDs. 62 On the continuum of rCBF responses between normal and pathological conditions, the normal rCBF response to CSD in mice is partly reminiscent of the pathological inverse rCBF response in rats, swine, and humans. 63 This variable CBF response in the normal brain to increased metabolic demand during CSD is likely to be mediated by a neurovascular coupling attempt to maintain tissue oxygenation under increased capillary transient time heterogeneity (CTH), which prompts an increase or decrease in CBF depending on the severity of heterogeneity to optimize oxygen extraction from blood as suggested by Ostergaard et al. 43 In contrast to post-CSD oligemia in normal brain, the fall in CBF during the inverse neurovascular response to CSD in the ischemic brain is much more pronounced and is associated with disrupted transmembrane ionic gradients. Therefore, post-CSD oligemia conceivably reflects an adjustment to promote O2 extraction by reducing CBF in order to compensate for prolonged capillary flow disturbance.64,65 Consistent with this view, CBF responses to CO2 inhalation (including during migraine with aura attacks), 66 neural stimulation and vasoactive substances are also reduced during post-CSD oligemia. Laser speckle blood flow data showed that the vasodilatory action of isoflurane 38 was not able to prevent the post-CSD oligemia although the hyperemic responses overriding oligemia was more pronounced under isoflurane compared to ketamine. These findings suggest that the vasodilatory effect of isoflurane was not sufficient to restore microvascular constriction at rest during post-CSD oligemia as shown by Anzabi et al. 7 in mice under isoflurane but was able to promote some increase in blood flow in response to the high metabolic demand induced by the repolarization phase of CSD. 7 In other words, while increased CTH favors CBF reduction during post-CSD oligemia to enhance resting O2 delivery from the constricted microvessels by lowering transit time, enhanced metabolic activity during CSD mandates dilation of at least some of the upstream capillaries and precapillary arterioles to increase O2 supply as previously illustrated in mice under isoflurane.43,44 Although hyperemic responses remained below the basal CBF level, O2 delivery was sufficient to prevent deep hypoxia and cellular injury, as clearly demonstrated in oxyhemoglobin recordings during the second KCl-induced CSD by Yuzawa et al. 44 Even though basal CBF reading with LSC imaging was less under ketamine compared to isoflurane, its stronger inhibitory action on both the number and duration of CSDs must have reduced the metabolic load (hence, needing smaller hyperemic response) and post-CSD capillary dysfunction (hence, smaller oligemic response), 67 suggesting that, rather than the basal CBF values, the neurovascular response to tissue metabolic demand was more critical in maintaining the effective tissue O2 levels. 43
Importantly, however, hyperemic responses were incomplete or absent in the medial watershed area, indicating that the perfusion pressure in the distal region of the pial branches of MCA was not sufficient to drive the required increase in blood flow during the metabolically demanding repolarization phase. Supporting this idea, a previous study clearly documented that a reduced perfusion pressure due to systemic arterial hypotension diminished the hyperemic response to CSD. 68 The post-CSD oligemia is known to lead to mild hypoxia in the mouse cortex,44,69 accordingly, this additional supply-demand mismatch at the border zones may cause irreversible hypoxic/ischemic injury at areas that are most vulnerable to perfusion deficit even when the CBF values are within oligemic range. 43 Consistent with our hypothesis, multiparametric recordings of mice under isoflurane illustrated that metabolic compromise was more pronounced in the medial areas of the cortex after the first CSD. 44 The reduced perfusion pressure due to arterial vasodilation37,41,42 and relatively lower blood pressure during isoflurane anesthesia may explain the deeper oligemia observed under isoflurane anesthesia compared to ketamine. 37 The lower number of CSDs under ketamine (9.2 vs. 6.0 CSDs during one-hour KCl application) may also have reduced the metabolic burden on tissue by decreasing the episodes of metabolic-hemodynamic uncoupling at the border zone. Ketamine may also have prevented the cortical hypoxic/ischemic injury by its NMDA receptor antagonistic action.
The hemodynamically induced, spatially very limited and delayed ischemic lesions may have previously gone unnoticed as the brains were processed too soon after the injury or the attention was more focused on dark neurons and sporadic selective neuronal necrosis across the cortex.70,71 In the case of endothelin-1-induced CSD, the vasoconstriction was too severe and quickly induced ischemic neuronal injury within 24 hours. 72 However, several laboratories have reported signs of CSD-induced cellular stress.73,74 These pathways can be further aggravated by transient hypoxia induced by PA constriction and, therefore, may be responsible for the lesions that we observed 2 weeks after CSD.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study is supported by grants by The Turkish Academy of Sciences to TD and Hacettepe University Research Fund THD-2018-16387).
Acknowledgements
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Buket Dönmez-Demir performed the experiments, contributed to data analysis, design of the study and drafting the manuscript and prepared the figures. Muge Yemisci supervised the experiments, contributed to data analysis, the design of the study and drafting the manuscript and figures. Gökhan Uruk contributed to ICT image calculations, generating histograms and preparation of the figures. Figen Söylemezoğlu contributed to histological examinations. Radu Bolbos performed and analyzed MRI images. Shams Kazmi contributed to ICT image calculations and generating histograms. Turgay Dalkara conceived the hypothesis, contributed to the design of the study, supervised the experiments and data analysis and wrote the manuscript. All authors discussed the results and commented on the manuscript.