
Abstract
Several studies have shown that an abnormal vascular-immunity link could increase Alzheimer’s disease (AD) risk; however, the mechanism is unclear. CD31, also named platelet endothelial cell adhesion molecule (PECAM), is a surface membrane protein of both endothelial and immune cells and plays important roles in the interaction between the vascular and immune systems. In this review, we focus on research regarding CD31 biological actions in the pathological process that may contribute to AD based on the following rationales. First, endothelial, leukocyte and soluble forms of CD31 play multi-roles in regulating transendothelial migration, increasing blood–brain barrier (BBB) permeability and resulting in neuroinflammation. Second, CD31 expressed by endothelial and immune cells dynamically modulates numbers of signaling pathways, including Src family kinases, selected G proteins, and β-catenin which in turn affect cell-matrix and cell–cell attachment, activation, permeability, survival, and ultimately neuronal cell injury. In endothelia and immune cells, these diverse CD31-mediated pathways act as a critical regulator in the immunity-endothelia-brain axis, thereby mediating AD pathogenesis in ApoE4 carriers, which is the major genetic risk factor for AD. This evidence suggests a novel mechanism and potential drug target for CD31 in the background of genetic vulnerabilities and peripheral inflammation for AD development and progression.
Introduction
Alzheimer's disease (AD) is a chronic neurodegenerative disorder characterized by the pathological accumulation of amyloid beta (Aβ) peptides, neurofibrillary tangles containing hyperphosphorylated neuronal Tau (MAPT) protein and neuroinflammation. Both peripheral chronic inflammation and peripheral vascular disease increase AD risk. 1 However, for most circulating inflammatory factors, it is unclear and how they may be involved in AD pathogenesis by entering the central nervous system from the blood. Generally, the blood–brain barrier (BBB) refers to the vascular barrier from the blood to the brain parenchyma formed by microvascular endothelial cells of the capillary wall, pericytes embedded in the capillary basement membrane, and astrocyte end-feet sheathing the capillary. 2 BBB protects the brain from some detrimental factors present in the systemic circulation. BBB dysfunction includes increased BBB permeability and degenerative endothelial cells and pericytes. Recent studies further support endothelial cells and associated Wnt/β-catenin signaling involved in barrier maintenance and transporter regulation.3–5 BBB breakdown facilitates entry of neurotoxic blood-derived products into the brain, enhancing the neuroinflammatory response 6 and AD pathogenesis.7,8
During peripheral chronic inflammation, some circulating inflammatory factors, such as C-reactive protein (CRP), have the potential to act on the blood-facing endothelia in the brain, facilitating or causing AD pathology. CD31 (platelet endothelial cell adhesion molecular PECAM-1) is an endothelial cell adhesion molecule and signaling receptor comprised of six extracellular immunoglobulin-like homology domains and a cytoplasmic domain that becomes serine and tyrosine phosphorylated upon cellular activation. Moreover, BBB is essential to prevent some detrimental factors entry into the brain and maintain central nervous system (CNS) homeostasis. In this review, we summarize the cellular and molecular functions of CD31 in regulating BBB function and propose that CD31 and its dynamic phosphorylation in brain endothelia putatively contribute to the pathogenesis of AD development.
Overview of CD31: structure and expression
CD31 is a 130 kDa type I, alternatively spliced, transmembrane glycoprotein. Specifically, CD31 is composed of six extracellular immunoglobulin-like homologous domains and a cytoplasmic domain that becomes serine and tyrosine phosphorylated upon cellular activation. 9 The extracellular domains play an important role in the adhesion cascade, where the first two structural domains (2 amino-terminal immunoglobulin homology) determine hemophilic binding10,11 and are involved in leukocyte-endothelial interaction, 12 leukocyte transendothelial migration 13 and stabilization of endothelial cell–cell junctions. 11
CD31 is predominantly expressed in endothelial cells, 14 platelets,9,15 different subpopulations of leukocytes (neutrophils, monocytes, eosinophils, and several lymphocyte subsets),16,17 hematopoietic progenitor cells, 18 and megakaryocytes. 19 In endothelia, CD31 is located just adjacent to tight junction complexes, which have been reported to be regulated by the expression of CD44 and comprise the vascular permeability barrier20–23 (Figure 1). Additionally, CD31 also functions as a cell adhesion molecule when the cytoplasmic domain acts as a dynamic scaffold, mediating binding (and activation) of selected cytoskeletal elements (β-&γ-catenin),24,25 STAT family members (STAT3 & 5), 26 src family members, 27 and small G proteins (Gαi2) 28 in an exon-specific manner (Figure 2).
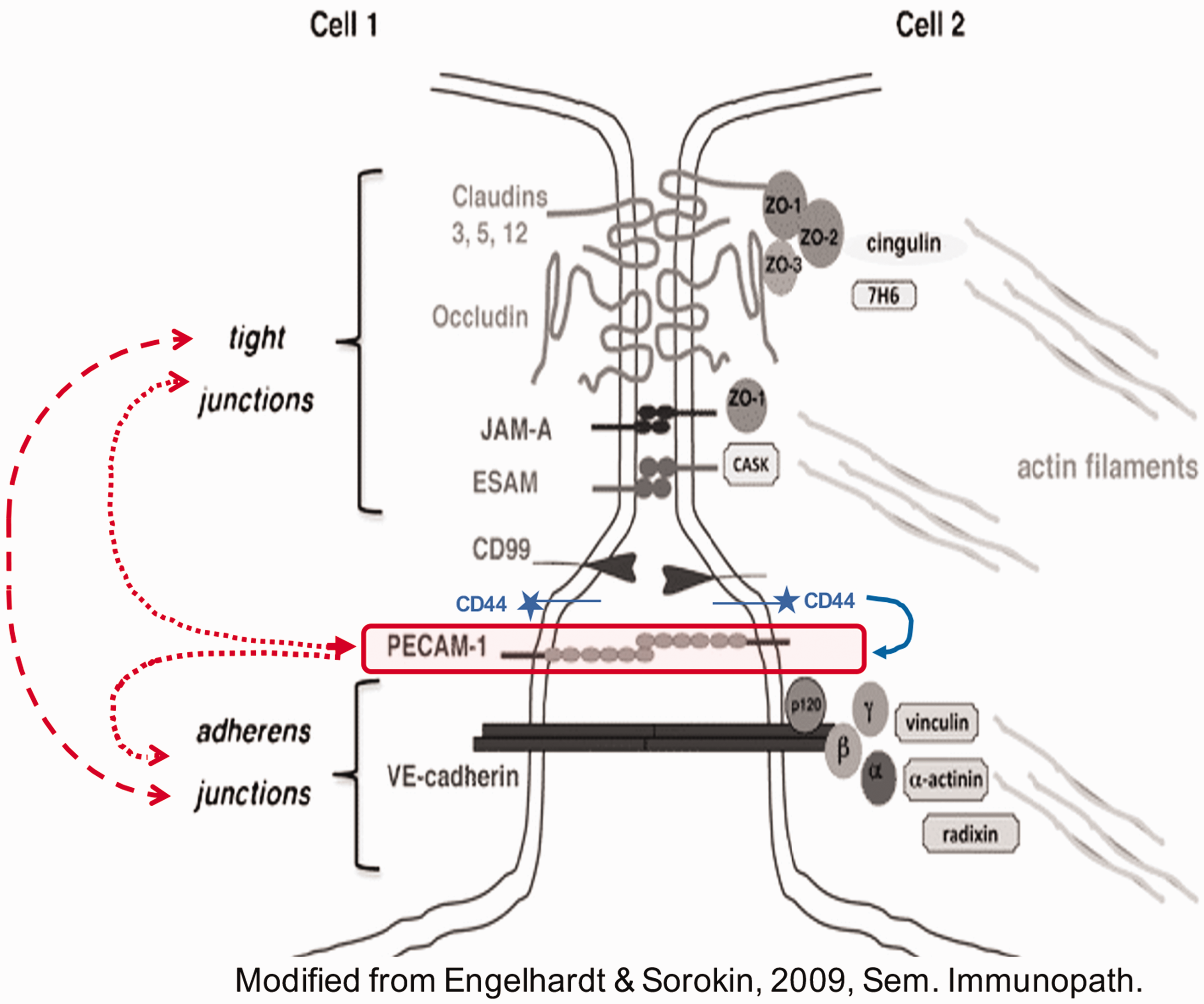
Endothelial cell–cell junctions: the relationship of CD31 to tight and adherens junctions (adapted from Engelhardt and Sorokin, Seminars in Immunopathology, 2009). While CD31 (PECAM-1) is in close proximity to both adherents and tight junctional complexes, as is CD99, both molecules are outside of these two organized structures. Studies have shown that CD31 may function as a modulator of VE-cadherin and adherens and tight junction formation as well as endothelial cell survival, proliferation and apoptosis. In addition, CD31 expression has been shown to be modulated by CD44, 25 and CD31 expression, localization and phosphorylation can be affected by the dynamics of adherens and tight junctions. 26
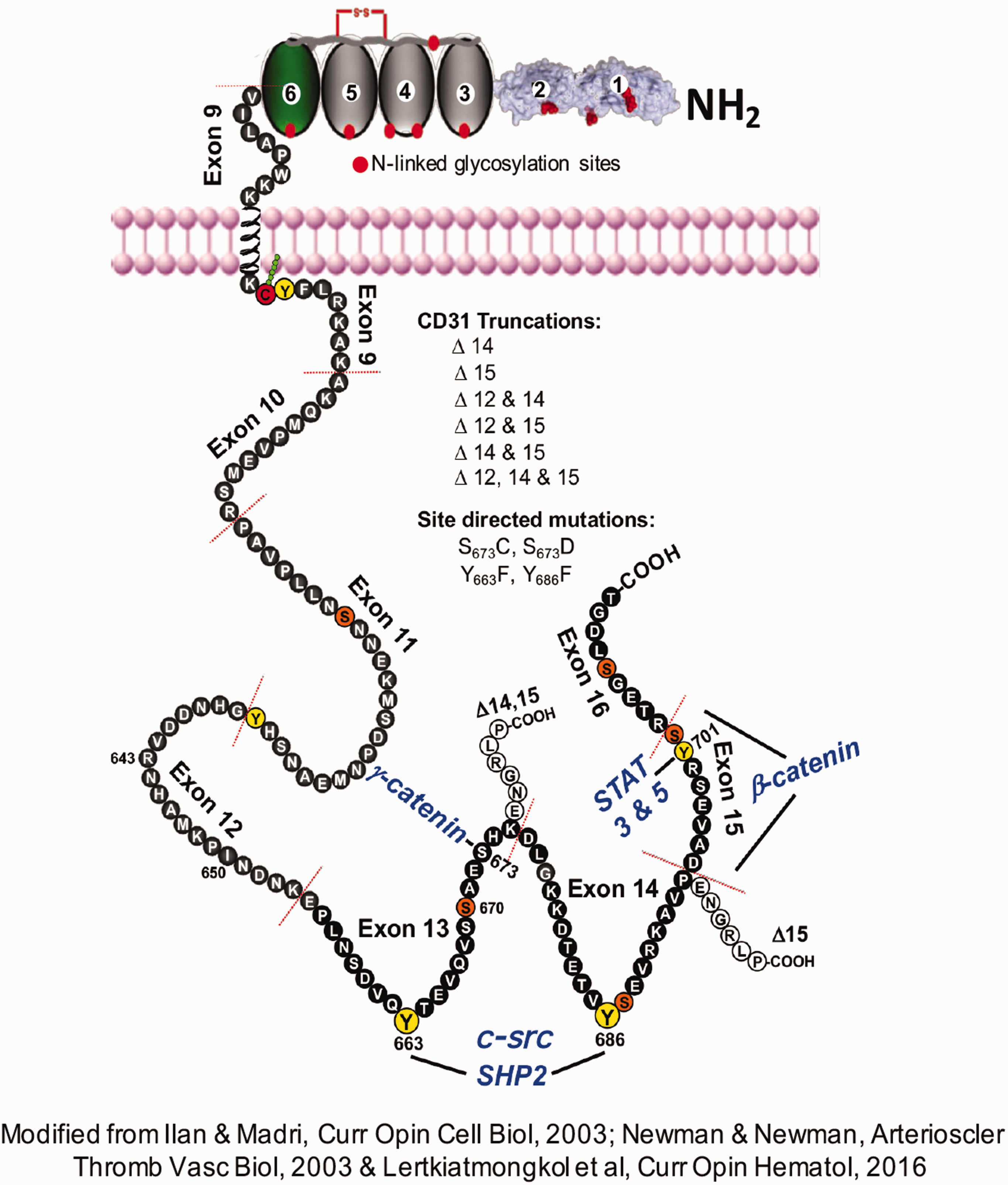
Schematic of the structure and selected binding properties of CD31 (PECAM-1) (adapted from Ilan & Madri, Curr Opin Cell Biol, 2003; Newman & Newman, Arterioscler Thromb Vasc Biol, 2003 & Lertkiatmongkol et al., Curr Opin Hematol, 2016). The molecules found to bind to CD31 (β-catenin, γ-catenin, SHP-2 and STAT3 & STAT-5) are denoted in bold blue type. The CD31 truncations and the site-directed mutations listed were utilized to determine the binding specificities of the binding molecules noted. CD31 functions as a dynamic modulator of cellular behaviors driven in part by differential Y & S phosphorylation, regulating downstream signaling cascades to affect several biological processes, including vasculogenesis, angiogenesis, megakaryocytopoiesis, osteoclastogenesis, permeability and modulation of gene expression (MMP-2, tissue factor and thrombosis) and endothelial cell (migration and cushion formation) and PMN migration. The phosphorylation of Y663 and Y686 mediates the binding and activation of SHP-2 and increases c-src binding. Together, these residues mediate an increase in c-src binding and modulate the protein tyrosine phosphatase SHP-2 binding and activation. The phosphorylation of S673 mediates a decrease in γ-catenin and could also affect SHP-2 binding and its kinetics. When β catenin is tyrosine-phosphorylated, its binding affinity to exon 15 is increased. Y701 mediates STAT family member binding, while Y686 mediates STAT family member Y phosphorylation; PECAM-1 has also been shown to modulate Rho activation via Gai2 binding, affecting single-cell mobility and directed migration.
The formation of the CD31/SHP-2 complex is thought to inhibit ITAM-mediated cellular activation events in circulating blood cells. 29 CD31 modulates several distinct downstream signaling cascades, which, in turn, affect a diverse range of cellular functions, including permeability, apoptosis, coagulation, MMP expression, angiogenesis, cytokine expression, motility, migration and homing. Among these, several signaling pathways activated after chronic inflammation may be involved in subsequent BBB disruption and neuroinflammation, which is integral to the development and progression of AD. 30
CD31 can undergo alternative splicing to yield several different isoforms that affect its function. Interestingly, the exons involved in scaffold function (exons 12–15) are known to undergo alternative splicing during development. Currently, the biological effects of CD31 alternative splicing are mostly unknown. However, the alternatively spliced CD31 isoforms have proven to be useful in studies focused on identifying exon-specific binding of kinases, phosphatases, transcription factors, and cytoplasmic proteins that bind CD31, as illustrated in Figure 2.
CD31 protein phosphorylation for cellular function
Phosphorylation of proteins is known to change cellular behavior and plays an important role in both physiological and pathological pathways. Scr family kinases are involved in tyrosine/serine phosphorylation of CD31. The cytoplasmic domains of CD31, as determined by 2-dimensional NMR, undergo sequential phosphorylation of serine and ITIM tyrosine residues that regulate the recruitment of the protein-tyrosine phosphatase SHP-2 upon cellular activation. In particular, phosphorylation on Ser-729 may initiate the dissociation of the membrane-interacting segment (residues 709–729) from the cell membrane, allowing for sequential phosphorylation of Tyr-713 and Tyr-690, Try663 and 686. 31
The YSEI motif in CD31 containing tyrosine residue 713 has been identified as the Scr phosphorylation binding site. 32 Tyr-663 was found to be required for efficient trafficking of CD31 to and from the lateral border recycling compartment (LBRC) and is necessary for the LBRC membrane to be targeted around migrating leukocytes. 33 The tyrosine residues 663 and 686 located in the ITIM domain are required for the recruitment of SHP-2, which then dephosphorylates β-catenin, thereby allowing for it to re-associate with VE-cadherin and re-stabilize the endothelial junction. 34 Phosphorylation of serine at position 702 located on the plasma membrane is increased in AD brains 11 and becomes necessary for the tail to detach from the membrane, which engages the phosphorylation of C-terminal ITIM and sequential phosphorylation of the N-terminal ITIM. When CD31 is phosphorylated via its ITAM domain to recruit and activate SH2 tyrosine/inositol phosphatases and downstream signaling molecules, it leads to modulation of cellular behavior, including endothelial cell adhesion, permeability, and leucocyte cell migration, which may contribute to AD pathogenesis.
In addition to phosphorylation/dephosphorylation, proteolysis, and alternative splicing, the role of CD31 is modulated by various single nucleotide polymorphisms (SNPs). CD31 polymorphisms are involved in aspects of vascular and angiogenic processes, endothelial survival, proliferation, migration, permeability, epithelial to mesenchymal transformation (EMT), differentiation, hematopoiesis, PMN-directed migration, B and T lymphocyte activation, cytokine expression and monocyte adhesion. In particular, the CD31 polymorphism rs1131012 (670 R/G) (residue 643 in the mature protein) has been found to affect CD31 tyrosine phosphorylation and mononuclear cell behavior;35,36 thus, it could potentially play a role in influencing AD development and progression.
The following cellular behaviors of CD31 are summarized in Figure 3:
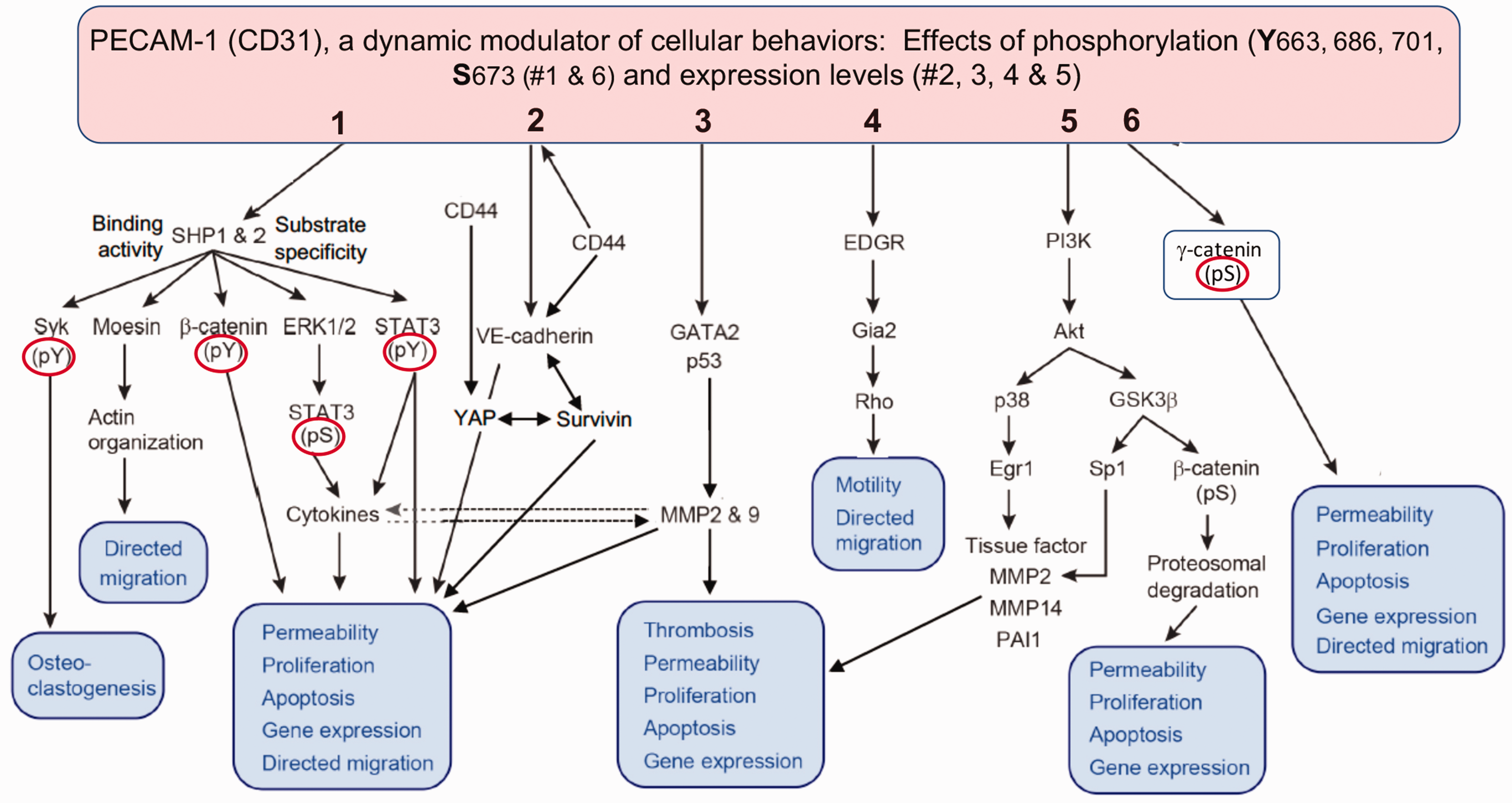
CD31 (PECAM-1) is a dynamic modulator of the cellular behaviors of endothelia and immune cells. CD31 (PECAM-1) mediates the cellular activities in endothelia and immune cells as illustrated: (1) The binding of SHP-1 & -2 to differentially phosphorylated CD31 (Y663 & Y686) results in distinct binding to CD31 and substrate specificities. CD31/SHP-1 & 2 interactions have been shown to modulate moesin phosphorylation, affecting the directed migration of neutrophils and megakaryocytes; the tyrosine phosphorylation state of β-catenin and FAK, vascular permeability, proliferation, apoptosis, gene expression and migration; the activation state of ERK1/2, affecting STAT phosphorylation and cytokine responsiveness; and the tyrosine phosphorylation state of STAT3, affecting cytokine induction. (2) The dynamic interactions of CD31 with CD44 and VE-cadherin decrease the levels of CD31 and are associated with the loss of adherens and tight junction integrity, altering the levels of YAP and survivin and resulting in decreased endothelial apoptosis. (3) CD31 on the surface of endothelial cells modulates the expression of MMP-2 and MMP-9 via induction and nuclear targeting of GATA2 and p53 transcription factors. (4) CD31 interacts with Gαi2, affecting Rho activation, cell motility and migration. (5) CD31's interactions with PI3K modulate Akt activity, which in turn regulates Egr-1 expression via p38 activation, leading to blunting of tissue factor induction, as well as MMP-14 and PAI-1 induction, reducing thrombosis, permeability and apoptosis in endothelial cells. (6) CD31/PI3K interactions also regulate GSK-3β activity via Akt phosphorylation, resulting in blunting of β-catenin serine phosphorylation and reducing its proteosomal degradation. Tyrosine phosphorylated β-catenin binding to CD31 results in sequestration of β-catenin, rendering it incapable of binding to VE-cadherin, affecting junction formation; the binding of γ-catenin to exon 13 of CD31 is dependent upon CD31 residue S673. Effects of CD31 phosphorylation (#1 & 6, Y663, 686, 701, S673) and CD31 expression levels (#2, 3, 4 & 5).
Clinical evidence of the involvement of CD31 in cerebrovascular pathology in Alzheimer’s disease
Several clinical and preclinical studies have demonstrated that CD31 may be involved in affecting endothelial and immune cell function and implicated in AD pathology. During endothelial apoptosis, metalloproteinase-dependent cleavage of CD31 results in the shedding of the soluble form of CD31, 41 which may serve as a surrogate marker for endothelial or immune cell pathology. In fact, soluble CD31 levels were higher in the serum of AD patients than in controls. 42 Consistently, another study found that CD31-positive endothelial microparticles were increased in the plasma of AD patients, which was associated with cognitive decline. 43
Several neuropathological studies have shown an association between CD31 expression in the cerebrovasculature and AD pathology; namely, the mRNA levels of endothelial CD31 in the cerebrovasculature of AD brains were lower than those in controls. Decreased expression of CD31 in AD brains with cerebral amyloid angiopathy (CAA), compared with brains suffering from AD but without CAA, possibly implicates CD31 in BBB breakdown in capillary CAA. 44 As the cerebellum controls movement and AD patients at a late stage can have gait dysfunction, one study with a small sample size showed that the number of CD31-positive vessels was found to be increased, albeit with a reduction in the ratio between PDGFRβ of CD31, suggesting hyper-vascularity with a relative failure of perivascular investiture potentially contributes to gait dysfunction and cognitive decline. In addition, colocalization of RAGE and CD31 has been shown in the microvasculature of the human AD hippocampus and is thought to be involved in the increased Aβ-mediated transendothelial migration of monocytes. 45 In the AD cerebrovasculature, the increased deposition of Aβ40/42 is thought to ignite intracellular signaling that can promote transmigration of HL-60 cells, a monocyte cell line, from the apical to basolateral direction of brain endothelial CD31 cells. 46
Preclinical evidence of CD31 involvement in Alzheimer’s disease
The evidence from preclinical studies supporting the involvement of CD31 in AD is complex and may reflect some aspects of AD pathology in humans. A significant increase in CD31-positive vessels and neuroinflammation was observed in the brains of mice injected with AAV-expressing tau. 47 Aβ, another component of AD pathology, induces HL-60 monocyte cell line migration across brain endothelium via CD31 and is thus inhibited by anti-CD31 antibodies.45,46
While CD31 plays an important function in neurovascular protection, it has a double-edged role during inflammation. For example, when transplanting neural stem cells into the AD animal models APP/PS1 brain, CD31 expression and neurovasculature were found to be increased in the cerebral cortex, and the cognitive function was improved. 48 Similarly, using a transgenic mouse model expressing mutant human amyloid precursor protein (APP) under the Thy1 promoter, vascular fitness was evaluated using CD31. After neurotrophic peptide mixture Cerebrolysin treatment CD31 was found to be upregulated in the treated mouse brains, suggesting preservation of the cerebrovasculature. 49 Additionally, CD31-deficient mice exhibit profound defects in leukocyte transmigration,50,51 which further supports the involvement of CD31 in the trans-basement membrane migration of neutrophils. 52 In different CD31-deficient strains of mice, the role of CD31 in the development of T-cell 53 and B-cell immunity 54 was shown to be related to the early onset of clinical symptoms during experimental autoimmune encephalomyelitis. In demyelinating disorders of the CNS, the absence of endothelial CD31 influences the cellular route of T-cell diapedesis across the BBB, which is repaired by upregulated expression of CD31 to restore brain vascular integrity and cell migration.55,56 Recently, the CD31 receptor was found to induce a metabolic response via activation of Akt or AMPK pathways to sustain junction reprogramming, which is required for endothelial barrier recovery in brain inflammatory conditions. 57 These studies suggest that CD31’s involvement in vascular integrity and its antagonists might have either beneficial or detrimental effects in AD progression mediated by binding directly to CD31 or its ligands.
CD31 as a possible gatekeeper protein of blood-brain in peripheral chronic low-grade inflammation
Although multiple studies have shown that peripheral chronic inflammation increases AD risk,58,59 the mediating key factors in the pathological process are largely unclear. Several studies have shown that chronic reactive protein CRP is associated with an increased risk of AD.60,61 CRP is one of the key elements in peripheral chronic low-grade inflammation and plays a role in the immune response to toxins or injuries in peripheral inflammation. 62 There are two forms presented in the body: 63 1) native pentameric CRP (pCRP) oligoprotein that is produced during active inflammatory reactions and 2) monomeric CRP (mCRP) that is produced during and after the acute phase by the irreversible dissociation of pCRP. Notably, CRP is also reported to mediate toxicity in vascular injuries/diseases through microvascular endothelial cells. 64 ApoE4 has been implicated in numerous processes including crosslink with Aβ and lipid metabolism to affect BBB permeability in inflammatory responses, due to loss of protective function or due to gain of toxic function. 65 Our recent study revealed that elevated CRP exhibits a strongly additive risk with apolipoprotein E4 (ApoE4) but not with ApoE3 or ApoE2 58 and was linked with AD biomarkers in cerebral spinal fluid (CSF) in ApoE4/4 carriers. 66 In support of this notion, our research revealed robust binding of mCRP to cerebral vasculature after intraperitoneal injection (i.p.) into different ApoE mouse models. 67 We identified CD31 as one of the receptors for peripheral mCRP, the binding of which can induce phosphorylation of CD31 and decrease its expression. Furthermore, mCRP and ApoE protein (ApoE2 > ApoE3 > ApoE4) competitively bind to CD31. Our research demonstrates the ApoE-mCRP-CD31 axis as a novel pathological pathway in the blood-facing endothelia that differentially affects AD risk in cerebrovasculature in an ApoE allele-specific manner. Elevated peripheral mCRP decreases endothelial CD31 expression and increases the phosphorylation of CD31 (pCD31) in the brains of ApoE4 mice. In parallel, AD brains have higher levels of mCRP-CD31 binding and pCD31 but lower levels of endothelial CD31 and ApoE-CD31 binding than control brains in humans. These findings suggest that CD31 is a possible gate-keeper receptor in the inflammatory blood–brain axis, which was differentially modulated in ApoE manner.
Putative mechanisms of CD31 action in Alzheimer’s disease development
Based on published literature and our own findings, we propose the following mechanisms for the role of CD31 in promoting the pathogenesis of AD (Summarized in Table 1).
CD31 involvement in pathways during different biological processes in the development of Alzheimer’s disease.

The role of CD31 in the regulation of leukocyte transendothelial migration
CD31 could mediate the transendothelial migration of leukocytes through the cerebrovascular wall into the brain, a critical step for BBB permeability leading to AD. 68 This process may be regulated by microvascular endothelial CD31, 44 leukocyte CD31 69 or the soluble form of CD31.42,70 CD31 signaling in endothelial cells is activated by homophilic interactions between leukocytes and endothelial cells under inflammatory conditions (such as those induced by IL-1β).
Features of transendothelial migration regulation in different types of leukocytes include CD31 homophilic interactions between monocytes and endothelial cells, which appear to be responsible for the early stages of transmigration. 71 Although neutrophils and eosinophils are not known to be involved in AD, homophilic interactions of neutrophil-endothelial CD31 induced by IL-1β exposure could stimulate the enhanced expression of alpha-6/beta-1 integrin on the cell surface, facilitating neutrophil migration across the BBB.72–75 Similarly, the heterophilic interaction of CD31 on eosinophils with alpha-V/beta-3 integrin on endothelial cells could promote adhesion of eosinophils to endothelial cells. 69 Integrins, as extracellular matrix proteins (ECMs), mediate cell-ECM and cell–cell adhesion events that determine whether vascular remodeling by angiogenic vessels or increased vascular density occurs in pathologically diagnosed AD postmortem brain tissue. 76
CD31 induces leukocyte-endothelial adhesion through homophilic interactions between leukocyte CD31 and endothelial CD31. This biological process activates Kinesin1 molecular motors 77 and increases endothelial Ca('2+) cytosol concentration in a TRPC6-dependent manner. 78 This in turn triggers the mobilization of lateral border recycling compartment (LBRC) vesicles to the junctional plasma membrane of endothelial cells, increasing the membrane surface area at such sites.79,80 LBRC is an endothelial intracellular vesicle compartment that forms a membrane network just below the plasma membrane at regions of cell contact. Trafficking of LBRC vesicles to the cell contacts is initially triggered by leukocytes through CD31 on leukocytes and, after initiating paracellular or transcellular diapedesis, a second wave of vesicle trafficking is triggered by MIC2.81,82 LBRC mobilization to the junctional plasma membrane is accompanied by the expression and structural exchanges of junction elements such as VE-cadherin, Claudin-5 and CD31.83,84
The AD pathological components Aβ40 and Aβ42 interact with RAGE on the basolateral surface of brain endothelial cells to induce cellular signaling, leading to transmigration of HL-60 monocytes from the apical to basolateral direction of the endothelium and across the BBB in a CD31-dependent manner. 46 Leukocyte CD31 acts a critical process in traversing the basal lamina of the perivascular basement membrane, triggering leukocyte extravasation. It is possible that the soluble form of CD31 (which is increased in AD) retains the ability to bind to its counter receptors (e.g., lymphocyte activation marker CD38 85 or neutrophil-specific NB1 86 ) and thus may activate circulating leukocytes, increasing their adhesion capacity with microvascular endothelial cells to impact BBB integrity.
CD31 as a mechanoresponsive cell-surface receptor
In the AD brain, capillaries (at the BBB) may undergo progressive degeneration caused by amyloid deposits, thickened basement membrane, cerebral atrophy, reduced vessel elasticity, or genetic predisposition. 44 When these structural abnormalities of the brain microvasculature begin to interfere with fluid dynamics, the consequent morphological compromise will result in increased cerebral capillary resistance, high blood viscosity, abnormal flow patterns, and changes in shear stress and shear rate in vessel walls. 87
CD31 is known to act as a direct mechanical force sensor and transmitter, mediating the endothelial response to mechanical stimuli. When endothelial cells are stimulated by fluid shear stress, the cytoplasmic domain of CD31 is phosphorylated by c‐Src on its cytoplasmic domain residue 71332,88 or by p120-catenin that is bound with Fer tyrosine kinase at cell contacts. 89 Activated CD31 can recruit c-Src to phosphorylate GAB1 on Y627, thereby inducing translocation of SHP-2 and GAB1 from the cytoplasm to the junction sites.90,91 These events together may lead to activation of the H-Ras/c-Raf-1/MEK1/2/ERK1/2 signaling cascade, which is dependent on CD31 tyrosine phosphorylation.92,93 ERK1/2 stimulation results in actin cytoskeleton remodeling, which promotes the transendothelial migration of leukocytes and elongation of endothelial cells; this process is considered one of the major histologic changes that occur in the aging population. 94
In addition, tyrosine phosphorylation of CD31 can enhance the association with functional eNOS (endothelial nitric oxide synthase). Under fluid shear stress, GAB1/SHP-2 signaling stimulates the activity of the phosphatidylinositol 3-kinase (PI3K) complex by phosphorylating PtdIns (4,5)-P2 into PtdIns (3,4,5)-P3, which in turn binds to and phosphorylates AKT (protein kinase B); activated AKT can further induce phosphorylation and activation of downstream eNOS phosphorylation.95,96 Endothelial dysfunction is characterized by impaired activity of eNOS, resulting in reduced production of nitric oxide (NO). NO is one of the most important vasoactive signaling molecules in cardiovascular homeostasis; it has multiple physiological effects, including vasodilation and neuroprotection, as an anti-AD factor.97,98 Together, these studies suggest that CD31 is a mechanotransducer that responds to mechanical forces (such as fluid shear stress imposed by blood flow) via its phosphorylation in the endothelial cells of the cerebrovasculature in the AD brain.
CD31 as an anti-apoptotic factor of endothelia
CD31-mediated anti-apoptotic effects have been shown to require both homophilic binding and intracellular signaling via its immunoreceptor tyrosine-based inhibitory motif (ITIM) domains. 99 Homophilic interactions between monocytes and endothelial cells can reduce endothelial cell death after serum deprivation via c-Src-dependent activation of protective molecules A20/A1 100 and induction of anti-apoptotic protein BFL1.101,102
Apoptosis, including of endothelial cells, is increasingly recognized as an important neurodegenerative factor in AD pathogenesis. Various factors and diverse pathways are known to provoke multiple deleterious events in mitochondria and the endoplasmic reticulum, leading to cell death. For example, CD31-mediated upregulation of the anti-apoptotic protein BFL1 is likely to contribute to anti-AD signaling. It is conceivable that its anti-apoptotic action may explain the CD31-driven accelerated restoration of the endothelial cell barrier in some acute and chronic inflammatory diseases. 103 Our previous study suggested that mCRP can increase the pathway of oxidative phosphorylation and lead to increased levels of phosphorylated CD31 in the presence of ApoE4 and in human AD brains. It is possible that phosphorylated CD31, unlike CD31, does not function as a potent suppressor of apoptosis in cerebrovascular cells, which is known to cause AD.
CD31 as a modulator of the T-lymphocyte response
T-cell accumulation and activation were observed in the brains of patients with AD. 104 The T-cell population, particularly CD8+ effector memory CD45RA+ T (TEMRA) cells, appear to be negatively associated with cognition and has been frequently observed in the cerebrospinal fluid of patients with AD. 105 TEMRA cells are well known to be involved in immunological memory and secrete proinflammatory and cytotoxic molecules. 106 In addition, exposure to infection (e.g., respiratory infections) can promote T-cell infiltration and Aβ deposition, which are known to accelerate cognitive decline in AD animal models 107 and AD patients. 108 These studies suggest that infection is a critical factor, especially in elderly individuals with neurodegeneration, potentially causing accelerated cognitive decline.
CD31 performs a dual function in regulating T cells. On the one hand, in naive T cells, T-lymphocyte-anchored CD31 is transhomophilically linked to either the cell-anchored CD31 of the antigen-presenting cells or to a soluble form of CD31. T-lymphocyte-anchored CD31 coclustered with MHC class II-stimulated TCR α/β molecules can recruit Scr homology 2 (SH2)-domain-containing protein-tyrosine phosphatases SHP-1/2 or activate its cytoplasmic tail via ITIMS; this ultimately leads to dephosphorylation of tyrosine kinases (e.g., CD3-activated ZAP70) known to be involved in T lymphocyte activation. These studies point toward a role for CD31 in the suppression of T-lymphocyte activity and differentiation and the induction of T-lymphocyte survival and tolerance.109–111 On the other hand, in activated or memory T cells, extracellular cleavage of CD31 leads to the loss of the transhomophilic sequence, attenuates the SHP-1-dependent cascade, and induces proinflammatory T-cell migration. 112
Peripheral inflammation with an increased number of terminally differentiated T cells may trigger TCR α/β-dependent cleavage of CD31 and increase phosphorylation of CD31, resulting in the release of proinflammatory molecules and active cytotoxic T cells. Although mCRP induces the phosphorylation of the tyrosine residue at position 701 of CD31 and the level of this pCD31 is high in AD brains, this specific CD31 phosphorylation should not relate to the tyrosine residues that reside in the CD31 ITIMs, which are at positions 663 and 686. 34 These hyperactivated T cells can contribute to neuroinflammation through the release of inflammatory molecules or via direct contact with the neurons themselves, 113 exacerbating chronic activation of the adaptive immune system in AD.
Dual role of CD31 in platelet activation
Several studies have demonstrated that enhanced platelet activation is observed in both patients with AD and animal models.114,115 Platelets have been considered the principal source of amyloid precursor protein (APP) and Aβ in human peripheral blood.116,117 The hyperactivated state of circulating platelets could be caused by potential factors such as damaged cerebral endothelial cells or membrane abnormalities.
In collagen-stimulated platelets, CD31 mainly functions as a negative regulator of collagen I/glycoprotein VI/Fc gamma RII alpha signaling. CD31 stimulates AKT(PKB)/GSK3 beta/Dinamin-1 118 and α-IIb/β-3 integrin-mediated internalization of GP-IB alpha; 119 this leads to the inhibition of von Willebrand factor/GP-IB alpha-dependent platelet activation. Elevated von Willebrand factor is a biomarker of vascular injury. The cerebrovascular von Willebrand factor is increased through the binding of mCRP with CD31 during peripheral chronic inflammation. 67 Thus, as platelet activation and adhesion on the vascular wall is a key step of vascular inflammation, platelet CD31-dependent activation could be linked with cerebrovascular risk factors and AD pathogenesis.
Possible therapeutic strategies targeting the CD31-mediated pathways for Alzheimer’s disease
The potential of CD31 as a therapeutic target in peripheral diseases, including atherosclerosis and inflammation, has been considered over several years. For example, in the healing of atherothrombotic arteries, preclinical studies suggest that the immobilization of a CD31 homotypic peptide on coronary stents could improve the integration of the metallic device to improve vascular homeostasis and arterial wall healing. 120 In vivo and in vitro CD31-targeting antibodies suppress leukocyte transmigration, resulting in a reduction in immune response. CD31 agonists can accelerate the resolution of the inflammatory phase, which is essential for wound healing in the vascular–blood interface homeostasis process.9,121
Based on our published study, we speculate that the use of a CD31 antagonist holds promise to block mCRP binding and counteract the inflammatory process of neurodegeneration, particularly in ApoE4 carriers, to prevent AD pathogenesis. In addition, our previous findings suggested that peripheral mCRP binds with extracellular CD31 via phosphorylation, probably by Src kinase, to activate mitochondrial and inflammatory pathways and suppress vascular pathogenesis. An agonistic effect of CD31, targeting molecular Scr family tyrosine kinases, could play key roles in regulating cell surface CD31 signal transduction in the context of a variety of cellular environments. As a target for new pharmaceutical drugs, the Scr family has been investigated for the treatment of pathologies in AD. The src family kinase inhibitor saracatinib (vAZD0530) is the first effort to target Fyn, a member of the Scr family kinase, with a specific inhibitor, which was assessed favorably in a small phase II clinical trial in AD 122 and is currently in a phase III AD clinical trial. 123 It may provide a novel sight for regulating pathological vascular-blood barrier recovery. Meanwhile, a healthy BBB is important for therapeutic drug delivery and decrease the initiated AD onset.124,125
Conclusion
The blood- brain barrier during peripheral chronic inflammation are probably mediated through responding cascades at the interface of blood factors and microvascular endothelia in the brain. This dynamic process is likely modulated by patient genetic vulnerabilities, the states of inflammation (acute or chronic), and the relevant treatments. Furthermore, dysfunction of endothelial barrier can amplify neuroinflammation and act as a key element in the development of neuroinflammatory diseases including AD. In this review, we discuss and focus on studies regarding the dynamic regulators of CD31 in mediating important biological processes involved in vascular neuroinflammation. There are a series of potential molecular mechanisms that may explain the relationship between CD31 and AD (Figure 4). CD31 has a key role in suppressing the immune response and sustaining endothelial vascular barrier function. As a leukocyte- and vascular cell-specific surface molecule, CD31 is involved in the transendothelial migration of leukocytes and integrin regulation and activation, especially T lymphocytes. By establishing homophilic/hemophilic interactions, CD31 acts a critical process in the immune response and sustains endothelial vascular barrier function by recruiting phosphatase activation. Interruption of these pathways could increase risk factors, including BBB disruption, amyloid plaque formation and neurofibrillary tangle formation, to induce neuroinflammation. Given that neuroinflammation is enhanced in AD pathogenesis in the brain, dynamic changes in the relative levels of CD31 could be a key element in the pathological process. Future studies are warranted to investigate whether anti-CD31 or inhibiting CD31 phosphorylation drugs can be developed and are efficacious for preventing or mitigating AD.
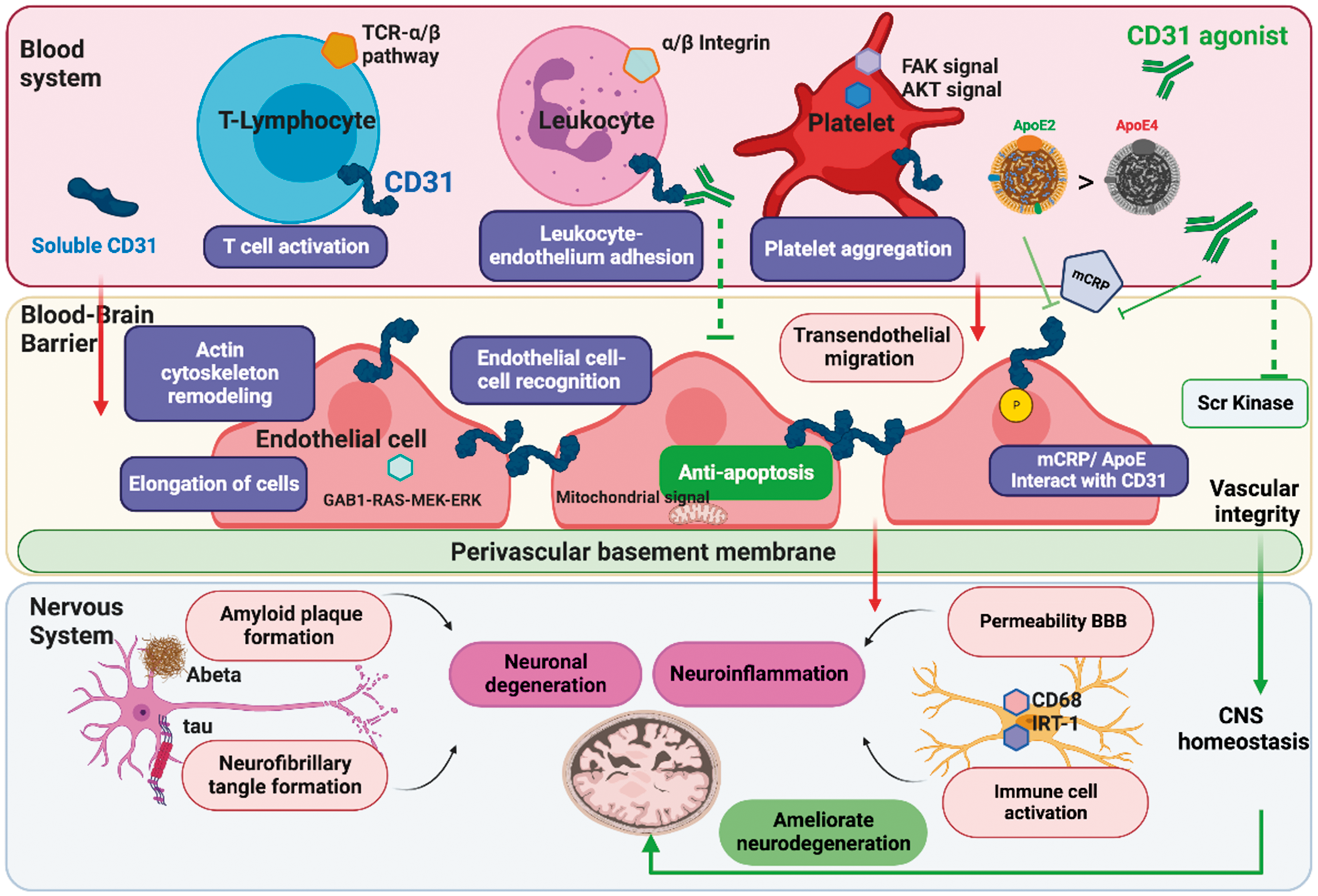
Map of CD31 involvement in pathological/protective molecular mechanisms in AD development. As a leukocyte- and vascular cell-specific surface molecule, CD31 is a probable gate-keeping receptor in the inflammatory blood–brain axis. It has been shown to be involved in risk pathways for AD, including transendothelial migration of leukocytes, integrin regulation, T-cell function and competitive binding of ApoE vs. mCRP to the CD31 receptor in regulating the permeability of the BBB and inducing neuroinflammation and neurodegeneration in the brain. Additionally, CD31 mediates anti-AD signaling pathways by suppressing apoptosis and the function of platelets to accelerate the restoration of the BBB. CD31 agonists have the potential to inhibit Scr kinase in the mCRP induced detrimental process, and then by accelerating the resolution of the inflammatory phase to maintain the BBB homeostasis process.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the grant from NIA, R01 AG054546 (W.Q.Q).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Z.Z. and W.Q. designed the article outline; J.M. contributed to manuscript’s suggestion and revision; Q.G, J. H. and Q. T. provided relative references; Z.Z. and W.Q. were responsible for manuscript preparation and writing.