
Abstract
Spreading depolarizations (SDs) have been linked to infarct volume expansion following ischemic stroke. Therapeutic hypothermia provides a neuroprotective effect after ischemic stroke. This study aimed to evaluate the effect of hypothermia on the propagation of SDs and infarct volume in an ischemic swine model. Through left orbital exenteration, middle cerebral arteries were surgically occluded (MCAo) in 16 swine. Extensive craniotomy and durotomy were performed. Six hypothermic and five normothermic animals were included in the analysis. An intracranial temperature probe was placed right frontal subdural. One hour after ischemic onset, mild hypothermia was induced and eighteen hours of electrocorticographic (ECoG) and intrinsic optical signal (IOS) recordings were acquired. Postmortem, 4 mm-thick slices were stained with 2,3,5-triphenyltetrazolium chloride to estimate the infarct volume. Compared to normothermia (36.4 ± 0.4°C), hypothermia (32.3 ± 0.2°C) significantly reduced the frequency and expansion of SDs (ECoG: 3.5 ± 2.1, 73.2 ± 5.2% vs. 1.0 ± 0.7, 41.9 ± 21.8%; IOS 3.9 ± 0.4, 87.6 ± 12.0% vs. 1.4 ± 0.7, 67.7 ± 8.3%, respectively). Further, infarct volume among hypothermic animals (23.2 ± 1.8% vs. 32.4 ± 2.5%) was significantly reduced. Therapeutic hypothermia reduces infarct volume and the frequency and expansion of SDs following cerebral ischemia.
Keywords
Introduction
Current therapies for acute ischemic stroke are limited to thrombolysis in the form of either tissue plasminogen activator administration or endovascular thrombectomy, 1 which are currently only available to a small subpopulation of stroke patients due to the narrow therapeutic time window. 2 Infarct volume expansion was associated with peri-infarct depolarizations (PIDs).3–6 PID is a historical term that is no longer used and has been replaced by the SD continuum concept. 7 SDs have long been identified in experimental stroke and in stroke patients 8 and are related to the spread of ischemia. 7 They have also been recently confirmed in patients with subarachnoid hemorrhage.9,10 Therapeutic hypothermia is the single most effective adjunctive treatment for acute ischemic stroke in preclinical animal models. 11 Cooling affects multiple pathways involved in the evolution of ischemic stroke and this multifaceted mechanism is thought to contribute to its strong neuroprotective effect. 12 However, further translational investigations in large species are needed to overcome clinical barriers which seem to preclude the routine use of therapeutic hypothermia in stroke. Given the incredible potential for the use of hypothermia to improve long-term outcomes after ischemia, the paucity of clinical trials opens the possibility of learning how and when to combine hypothermia with standard therapy. In similar scenarios, clinical trials have shown that hypothermia improved clinical outcomes in comatose survivors of out-of-hospital cardiac arrest 13 and neonatal hypoxic-ischemic encephalopathy.13,14
Following the primary injury, patients with ischemic stroke can endure secondary damage and infarct growth from excitotoxicity, blood-brain barrier damage, SDs, and other deleterious processes. These may lead to cerebral edema, mass effects, and worse outcomes despite timely administration of reperfusion therapies. 15 To the best of our knowledge, the effect of hypothermia on SDs in the context of ischemic stroke has not been explored in large species.
In the present study, we used a middle cerebral artery occlusion (MCAo) swine model to evaluate the effects of hypothermia on SDs and infarct volume expansion in a swine model of permanent focal cerebral ischemia.
Materials and methods
Animal preparation
The protocol for the experiments (no. G-74/14) was approved by the Institutional Animal Care and Use Committee in Karlsruhe, Baden-Württemberg, Germany. Experiments were conducted in the Interfaculty Biomedical Research Facility (IBF 347) in accordance with the University of Heidelberg Animal Ethics Policy. The ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines were considered for the study report.
Female German Landrace swine (n = 16), aged 3–4 months, weighing 28–32 kg, were used. Four animals were used for surgical training and establishment of monitoring and 12 were assigned to two groups through simple randomization 16 using a shuffled deck of cards with six cards for each group. As the randomization was performed after MCAo, group assignment was blinded to the surgical procedure and animal preparation, except for the last animal whose assignment was known according to the group sample. All the animals were pre-anesthetized intramuscularly with midazolam (Dormicum®, 0.5–0.7 mg/kg) and azaperone (Stresnil®, 4 mg/kg), followed by a 1 mg IV application of 1% propofol after an overnight fast. After endotracheal intubation, anesthesia and deep sedation were maintained with isoflurane (0.6–1.0%) and midazolam perfusion. All animals were deeply sedated throughout the procedure to minimize animal movement, discomfort, and pain. In the event of shivering or movements due to a temporal decrease of the deep sedation, muscle relaxation (rocuronium) was administered intramuscularly when necessary.
A venous line was placed in the right ear vein and peripheral capillary oxygen saturation (SpO2) and heart rate (HR) were monitored from the left ear. After surgical exposure of the right femoral artery, a 4-Fr catheter was inserted to continuously monitor the mean arterial pressure (MAP) (Raumedic AG, Helmbrechts, Germany). Rectal temperature was measured continuously during the entire experiment using a temperature sensor. In total, the animals were monitored for 18 hours.
Operative procedures
The heads of the animals were firmly held in a stereotactic frame. The technique used to establish MCAo has been previously described in detail. 17 In brief, orbital exenteration was performed to approach the optic foramen and the underlying dura. After the dura incision, the internal carotid artery and middle cerebral arteries (MCAs) were identified. Following an extensive bilateral craniectomy and durotomy, the exposed cortex was protected from drying with a mineral oil layer (about 1–1,5 cm of paraffin oil) that improved the image quality by reducing multilayer light reflexes. During the MCAo, the neurosurgeons were blinded to the group allocation. An intracranial pressure (ICP)/temperature probe (Raumedic AG) was placed over the right frontal lobe under the dura mater to directly measure cortical temperature. In addition, two 5-contact electrocorticography (ECoG) recording strips (Ad-Tech Medical Instrument Corp., Racine, WI, USA) were placed laterally in both cerebral hemispheres. One hour after the onset of ischemia, mild hypothermia was induced in group 2, reducing the temperature from normothermic (36–37°C) to hypothermic (32–33°C), at intervals of 2°C per hour. Wet cooling blankets (textile blankets 80% cotton and 20% polyester) were placed on the animal body and hypothermia was induced under continuous air circulation using a fan (Hantech, Anada GmbH, Karlsruhe, Germany). Body and brain temperature were monitored constantly, and the wet cooling blankets were changed intermittently to maintain the induced hypothermia. The fan was turned off when the target brain temperature was reached.
The animals were euthanized after 18 hours of MCAo with an IV overdose of potassium chloride.
Physiological parameters, ECoG, and intrinsic optical signal monitoring
Cardiovascular and respiratory monitoring was performed as previously described. 17 In brief, the HR was maintained between 60–120 bpm, MAP between 65–100 mmHg, SpO2 at >90%, pCO2 at 35–45 mmHg, and pO2 at >80 mmHg. A crystalloid infusion (Sterofundin ISO®) was used in all experiments to maintain MAP above 65 mmHg and, if necessary, cafedrine/theodrenaline (200 mg/10 mg) was added to the infusion. Failure of the mechanical ventilator and/or anesthesia machine for 2 minutes, a MAP <50 mmHg over 3 h, or a heart rate <170 over 3 h were considered factors for immediate termination of the experiment. ECoG electrodes were connected in a sequential monopolar fashion. The sampling rate of monitoring was set at 1,000 Hz using a near-DC amplifier Powerlab 16/SP analog-to-digital converter. ECoG registration and analysis were performed using LabChart 7 (AD Instruments, Bella Vista, Australia) using standard methods. 18 Intrinsic optical signal (IOS) imaging was executed using a charge-coupled device camera (Smartec GC1621M 8-bit grayscale, 1628 × 1236 pixels, 1/1.8 sensor; MaxxVision GmbH, Stuttgart, Germany) positioned 25–30 cm above the exposed cortex, with a 50 mm lens (Fujinon HF50HA-1B 50 mm Fixed Focal Lens, f = 1.4; MaxxVision GmbH). An optical band-pass filter (564 nm, 14 nm FWHM, Schott, Germany) was positioned in front of the lens, allowing only light within the green spectrum to pass through the lens. One image was acquired per second, in full resolution, as described previously. 19 For the IOS analysis, images were elastically transformed with a registration process. This processing method was used to improve imaging and analysis quality, as previously described.19,20 Two regions of interest of about 5 pixels (0.03–0.12 mm2) were chosen in a radial direction, one close to the ischemic core, and the other distant to it. Expansion in ECoG recordings was defined as the number of electrode channels, expressed in percentage, in which one SD was detected. Expansion in IOS recordings was defined as the number of ROIs, expressed in percentage, in which one SD was detected in the visible area. The researchers who performed the data analysis were blinded to the group allocation.
Infarct volume measurement
Ischemic infarct volume was estimated via 2,3,5-triphenyltetrazolium chloride (TTC) staining. After termination, the brains were rapidly removed, placed in isopentane at −20°C for approximately 10 minutes, and coronally sectioned into 4-mm-thick slices using a custom-made cutting device. Afterward, the brain slices were incubated with 2% TTC in NaCl for 30 minutes at 37°C. TTC-stained brain sections were placed in the frontal to occipital orientation and photographed using a digital camera (Canon, Japan). Afterward, the brain slices were fixed in a 4% formalin solution for 5 days. After fixation, all brain slices were scanned using a flatbed scanner (Canon CanoScan LiDE) on both sides. The unstained area of each brain slice was defined as an infarction. Infarct areas were measured using Image J version 1.44p (National Institutes of Health, Bethesda, MD, USA). The total infarction volume was calculated by multiplying the sum of infarcted areas of all sections by the thickness of the sections (4 mm). 21 The researchers who performed the data analysis were blinded to the group allocation.
Statistical analysis
Continuous variables were assessed for normality using histograms and the Kolmogorov–Smirnov and Shapiro–Wilk tests. The independent samples t-test was used to compare the analyzed parameters between groups with parametric distributions. The Mann–Whitney U test was used to compare the measured parameters between groups with non-parametric distributions. All statistical analyses were performed using values from all variables recorded between the 4th and 14th hours after MCAo since hypothermic stable values (32°C) were obtained at the 4th hour of recording. The results are reported as means ± standard deviations. The statistical analyses were performed using SPSS version 20.0 (IBM Corp., Armonk, NY, USA). Analysis items with p < 0.05 were considered statistically significant.
Results
The data of one animal in the normothermia group were excluded from the analysis because it was confirmed later that a very small MCA infarction occurred (1.5 cm3), presumably due to failure of the MCA clipping technique. The final analysis included five normothermic animals and six animals subjected to therapeutic hypothermia. The temperature target was reached in the 4th hour in the hypothermia group; thus, data from the first three hours were excluded from the statistical analysis. The quality of recordings from both ECoG and IOS was assessed during the first 14 hours. Recordings acquired during the last 4 hours were excluded from the analysis because of large amounts of artifacts and invalid measurements resulting from noise contamination or animal movements potentially resulting from seizures, a decrease in deep sedation, or shivering, as well as due to a change in baseline values and loss of quality in the visualization of the cortex during IOS assessment, secondary to a probable aggregation of fibrin products, brain shift, or swelling after the infarct. The animals were sacrificed, as planned, to perform the infarction size analysis after 18 hours. A schematic of the study design and results obtained is shown in Figure 1.
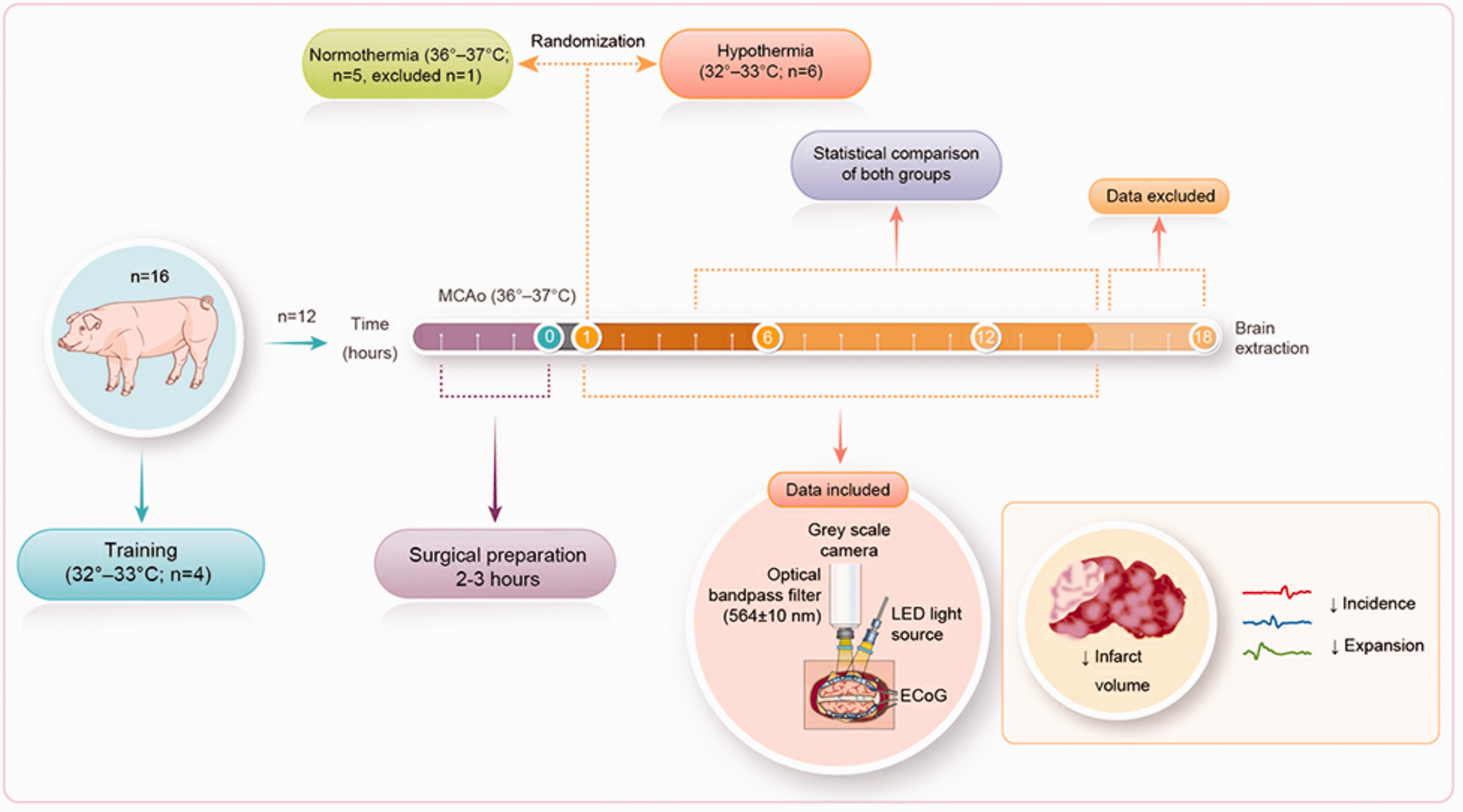
A schematic representation of the study design and results.
Physiological parameters
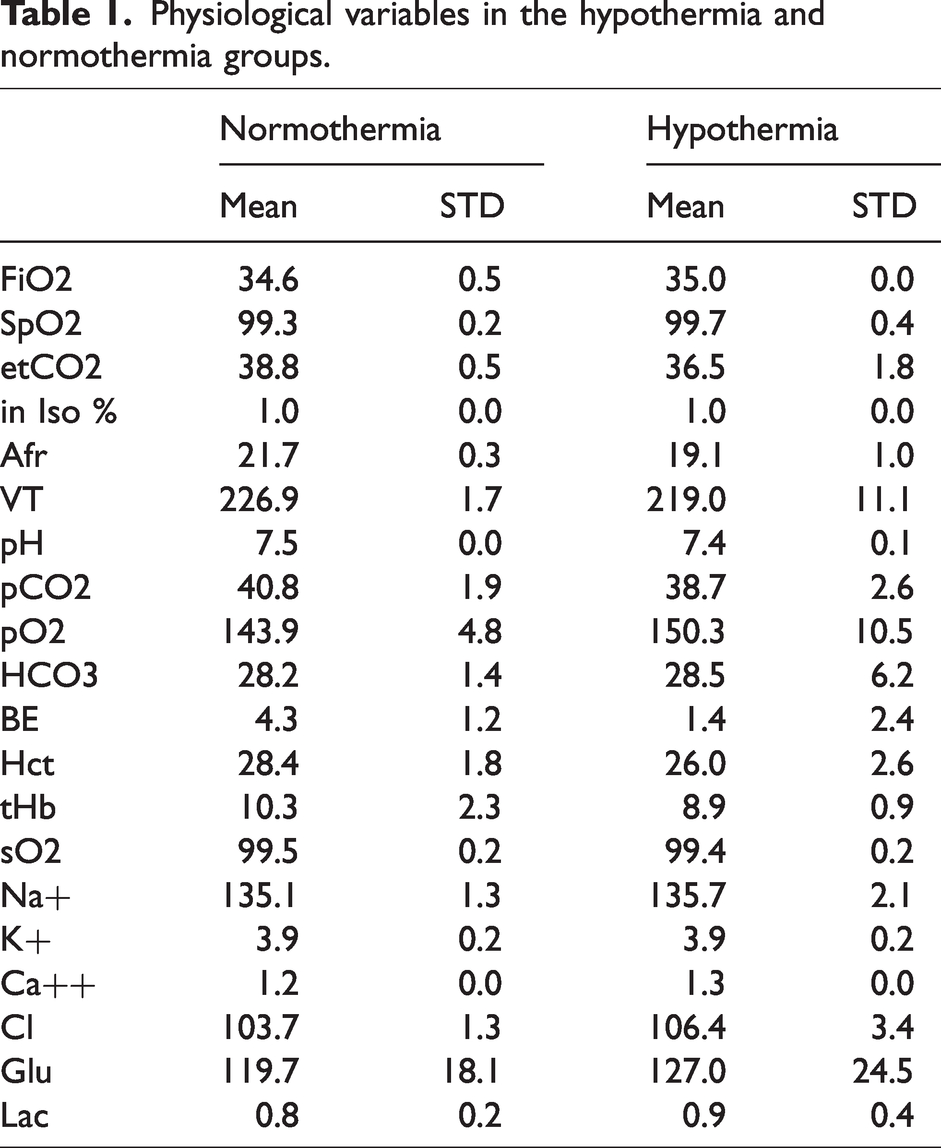
To characterize the physiological response to hypothermia, three parameters were statistically analyzed in all swine: HR, MAP, and body/brain temperatures (Figures 2(a) to (c)). We found no significant difference in HR between normothermic (103.1 ± 11.5 bpm) and hypothermic (93.2 ± 15.3 bpm) swine (p = 0.267). Nonetheless, a significant difference in MAP (p = 0.013) was observed between swine with normothermia (78.0 ± 2.2 mmHg) and hypothermia (72.3 ± 3.7 mmHg), even though interventions were performed to maintain MAP in a normal range in hypothermia (in two animals cafedrine/theodrenaline (200 mg/10 mg) was added to the crystalloids). Body (normothermia: 36.9 ± 0.2°C; hypothermia: 32.6 ± 0.1°C) and brain (normothermia: 36.4 ± 0.4°C; hypothermia: 32.3 ± 0.2°C) temperatures were found to be significantly different between the groups (each p < 0.001). From the beginning of the experiment, hypothermic swine showed constant decreases in body and brain temperature from mean values of 36.9°C to 32.5°C and 36.5°C to 32.1°C, respectively (Figure 2A). The mean and standard deviation of further physiological parameters are presented (Table 1).
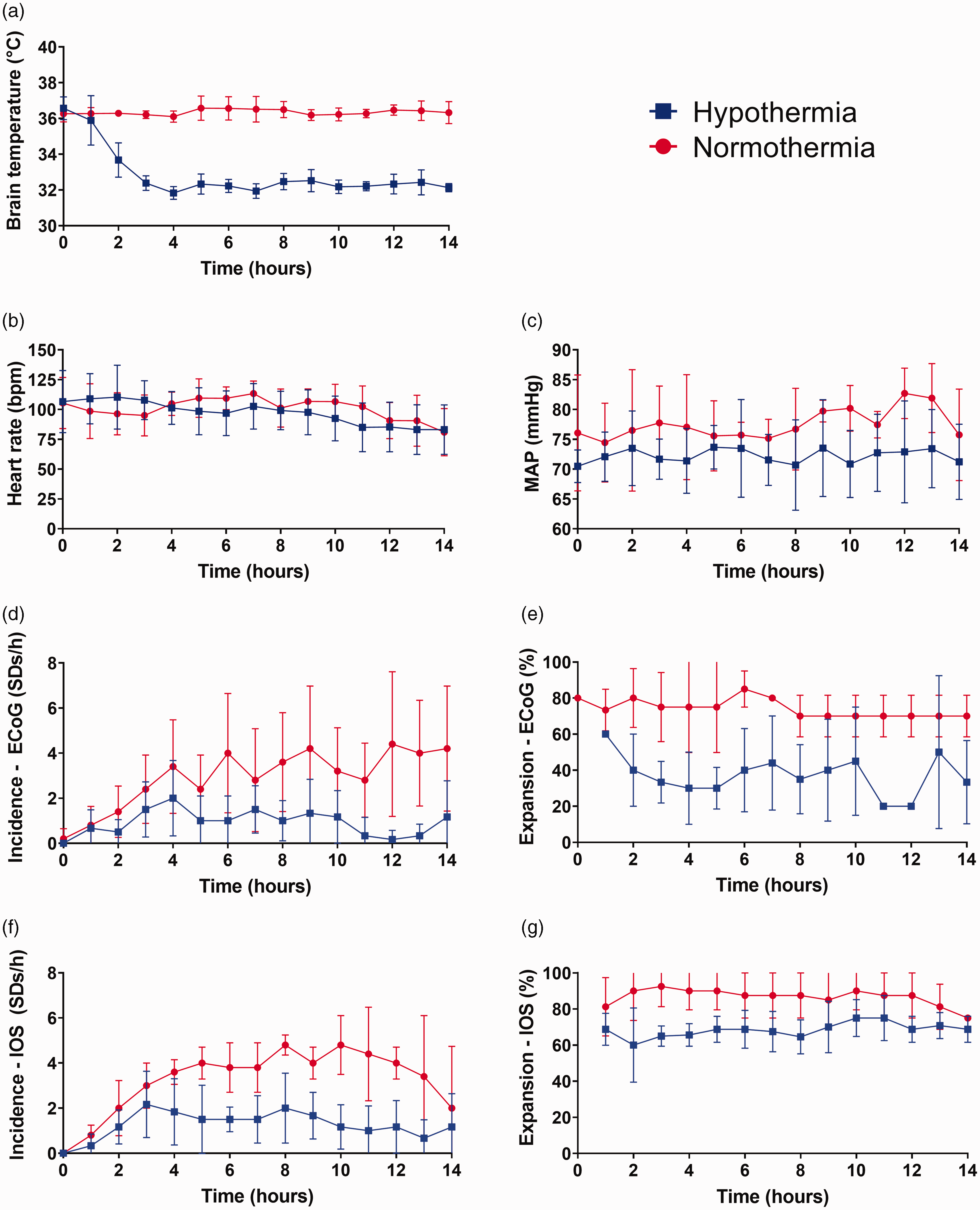
Effect of hypothermia on physiological parameters and SD incidence and expansion. (a) After the implementation of our hypothermia protocol, brain temperature decreased in the hypothermic group at a rate of approximately 2°C/hour until it reached a mean brain temperature of 32.1°C. (b and c) Important physiological parameters, such as heart rate and MAP, were also measured in both groups. Compared to the normothermic group, the hypothermic group showed an important reduction in SD incidence (d and f) and expansion (e and g), over 14 hours of both ECoG and IOS recordings.
Physiological variables in the hypothermia and normothermia groups.
Effect of hypothermia on SD characteristics, incidence, and expansion
Hypothermia showed a significant reductive effect on the number of SDs in both ECoG and IOS recordings, compared to normothermia. During the 14 hours of ECoG recordings of the left cerebral hemisphere of all animals, we found a total of 219 SDs (43.8 ± 25.4 SD/animal) in the normothermic group vs. 82 SDs (13.7 ± 9 SD/animal) in the hypothermic group. Furthermore, we detected a reduction in SD frequency and expansion in the hypothermic group, with a mean frequency of 3.5 ± 2.1 SDs per hour and expansion by a mean of 73.2 ± 5.2% in the normothermic group vs. a mean frequency of 1.0 ± 0.7 SDs per hour and expansion by a mean of 41.9 ± 21.8% in the hypothermic group (incidence: p = 0.020; expansion: p = 0.028; Figures 2(d) and (e)). Similarly, IOS recordings revealed a total of 242 SDs (48.4 ± 5,5 SD/animal) in the normothermic group vs. 113 SDs (18.8 ± 8,5 SD/animal) in the hypothermic group. In these recordings, the mean frequency of SDs in the normothermic group was 3.9 ± 0.4 per hour with expansion by a mean of 87.6 ± 12.0%, vs. 1.4 ± 0.7 SDs per hour with expansion by a mean of 67.7 ± 8.3% in the hypothermic group (incidence: p < 0.001; expansion: p = 0.010; Figures 2(f) and (g)). In terms of amplitude and duration of the near-DC shift, ECoG and IOS recordings revealed no significant differences between the normothermic and hypothermic groups.
Effect of hypothermia on infarct volume
Finally, we aimed to evaluate the impact of hypothermia on infarct volume via TTC staining of postmortem brain tissue. After brain extraction, the average brain volume of all animals was 63.2 ± 3.3 cm3. In terms of infarct volume, we found a significant difference between the normothermic (9.0 ± 0.8 cm3) and hypothermic (6.0 ± 1.0 cm3) groups (p < 0.001), indicating an important reduction in the infarction volume among animals that underwent hypothermia (Figure 3). Similarly, there was a statistically significant difference in the percentage change in infarct volume of the affected cerebral hemisphere between the normothermic (32.4 ± 2.5%) and hypothermic (23.2 ± 1.8%) groups (p < 0.001).
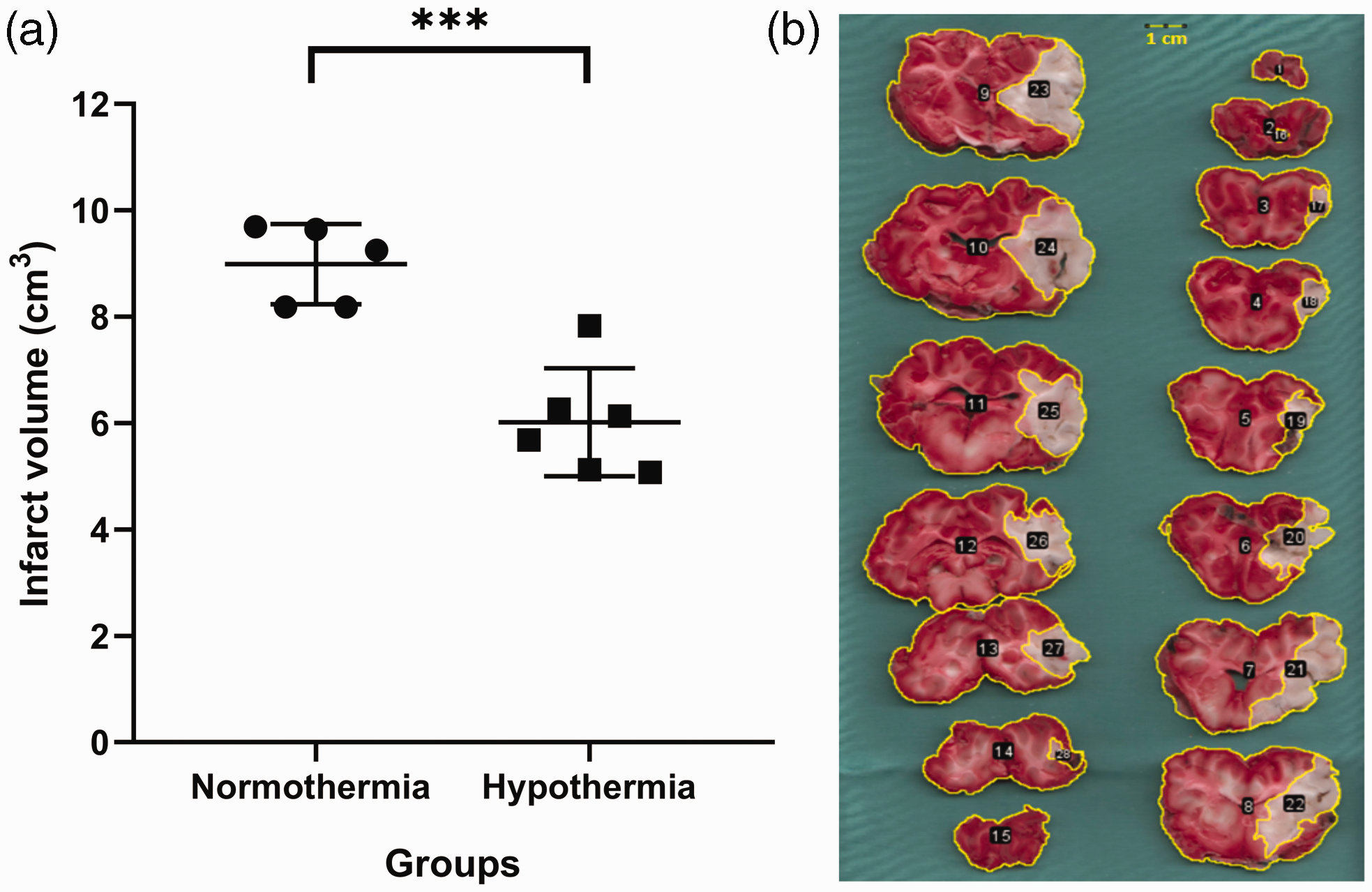
Effect of hypothermia on infarct volume. (a) Following postmortem brain tissue examination, the hypothermic animals showed a significant reduction in infarct volume compared to the normothermic animals and (b) After brain extraction (normothermic brain), all brains were coronally sectioned into 4-mm-thick slices and stained with TTC (2,3,5-triphenyltetrazolium chloride). TTC-stained brain slices (numbers 1–15) are displayed here in a frontal to occipital orientation, allowing us to visualize the infarct tissue stained in white (numbers 16–28).
Discussion
In this study, we demonstrated for the first time that mild hypothermia reduces SD frequency and expansion, as well as infarct volume in the brains of swine. SDs have shown to produce infarct expansion 7 and are therefore markers of ongoing brain damage. To date, four studies investigated hypothermia as a neuroprotective treatment in swine submitted to cardiac arrest or direct damage to the cerebral arteries to generate an ischemic environment in their brains. Different efficacy parameters were used including histological findings from different brain areas, the infarct volume measured by MRI or pathologic analysis, and the neurological outcome.22–24 Combined with rodent-based studies, the results in the swine models were sufficiently effective to implement hypothermia treatment in humans.
On the other hand, the efficacy of hypothermia was debated in clinical trials due to the heterogeneous results within studies. In the studies with fruitful outcomes, two key factors were determined as compulsory for a successful hypothermic therapy: recanalization and good collateral flow. 11 Hypothermia plus recanalization diminishes the infarct zone volume, lowers cerebral edema, prevents hemorrhagic transformation, and enhances the neurological outcome.1,24–26
In DISCHARGE-1 (Depolarisations in ISChaemia after subarachnoid HAemoRrhaGE-1), a recent prospective, observational, multicenter, cohort, diagnostic phase III trial of 180 patients with severe aSAH, SD variables were included in each multiple regression model for early, delayed, and total brain damage, seven-month outcome, and death. This study concluded that SDs are an independent biomarker of progressive brain injury. 9
Hence, our study fills the gaps between human and large animal studies concerning SDs as a pathomechanism of brain injury, and how the size of infarction is related to SDs.
In previous studies dedicated to SDs, a correlation between SDs occurrence and brain temperature was observed. There was an increase of 0.2°C 25 min preceding the onset of SD and 0.4°C when a cluster occurred. A higher probability of developing SDs was observed during episodes of brain temperatures over 38°C. 27 Additionally, the hypothermia effect was examined against ischemic SDs and demonstrated to delay the appearance of such SDs. Nonetheless, the infarct progression was not ceased. 5
Using ECoG and IOS, we demonstrated that hypothermia not only reduces the SD rate, but also their expansion over the cortex. Those results were reflected in how the infarct zone of pig brains was reduced under cold temperatures in comparison to normothermia. We aimed to reveal the vascular dynamics in vivo during infarct progression and the SD development using IOS. IOS is able to depict the brain blood volume and intrinsic changes of brain tissue produced by edema.
Finally, we proved that both IOS and ECoG are capable of documenting the effects exerted by therapeutic hypothermia against brain ischemia. Moreover, our work elucidates the vascular changes during an ischemic insult and subsequent anomalous phenomena (SDs), and how SDs are affected in their frequency and expansion by cold temperatures since the early brain infarct phase. In future studies, a reversible vascular occlusion model should be implemented to fully comprehend the vascular and tissular damages caused in both permanent and temporal occlusion models. Based on our results in combination with prior literature, the right time for cooling, doses, and time window should be personalized based on biomarkers to assure success in clinical trials. 11 It was suggested to establish therapeutic hypothermia at 33–34°C, increase the time window to therapy initiation to obtain promising results, decrease the treatment duration to avoid any adverse effect, to apply more focal brain cooling therapy, and to increment the feasibility to establish therapeutic hypothermia as a standard intervention12,28,29 and avoid peripheric side effects. These recommendations should be tested in humans and animal models to observe potential risks, disadvantages, and opportunity areas. It is necessary to find reliable biomarkers to correctly guide hypothermia based on the underlying pathophysiology of the infarct progression, as well as the action mechanism of therapeutic hypothermia. As supported by our study, SDs could be used as a marker to guide hypothermia therapy.
Criteria for selecting experimental conditions
The experimental temperatures were selected based on the evidence that temperatures of 33–35°C are the most effective. In a systematic study, a temperature of 34°C yielded superior outcomes compared to temperatures ranging from 32–37°C. A temperature of 34°C during a 4-hour period showed better neuroprotective effects on infarct size, edema, and neurologic outcomes and a sustained benefit over the subsequent 5 days. 30 In preclinical studies, the highest efficacy was demonstrated when treatment was started before or at the onset of ischemia, with the efficacious effect seen up to 2.5–3 hours.31,32 Hypothermia lasting at least 24 hours has shown the most consistently positive results and is the standard in current trials.33,34 Humans do not tolerate temperatures much below 32°C without significant cardiac effects. 35 In our setting, we could achieve the target temperature (32–33°C) within 2–3 hours with minimal side effects in the observed duration, and the experiment could be extended up to 18 hours. However, continuous ECoG and IOS data could only reliably be analyzed for 14 hours, due to artifact effects and loss of signal quality.
Effect of hypothermia on SDs
The effect of mild hypothermia (temperature 32–35°C) on SDs is controversial. Previous studies have evaluated volume reduction using mild hypothermia, but without analysis of SDs.22,24 Sasaki et al. 5 showed that SDs in the permanent MCAo lissencephalic animal model were not significantly reduced within 2 hours of hypothermia (30°C) in spontaneously hypertensive rats, although SD propagation was slowed. Similarly, Chen et al. 36 reported a significant correlation between animal body temperature and the number of SDs in a Wistar rat transient MCAo model. Until now, the effect of mild hypothermia on SDs has not been studied in a large gyrencephalic animal model. The porcine permanent MCAo model provides several benefits, including comparable brain anatomy and physiology to those of humans.37,38 In addition, we have refined the methodology used in our ischemic swine model over the years, focusing on techniques that aim to shorten the duration of surgical procedures and reduce the loss of blood, warranting increased reproducibility of our results.17,19,20,39
In our study, we observed a significant decrease in MAP in the hypothermic group. However, MAP remained above 70 mmHg, which is considered a conservative lower limit for MAP maintenance in neurointensive care. 40 Furthermore, the probability of SD remaines similar over an MAP range of 70–100 mmHg.40,41 We applied vasopressors to maintain an MAP in the normal range. The effect of vasopressors on SDs is not well studied. Intravascular endothelin-1(ET-1), one of the most known vasoconstrictors, did not trigger or increase the susceptibility to SDs in mice. 42 In contrast, topically applied norepinephrine has blocked the propagation of SDs induced mechanically in a rat model.43,44 Further, caffeine did not affect SD in a KCl rat model.43,45
Mechanisms underlying neuroprotection against SDs during hypothermia
Neuronal damage during acute ischemic stroke is considered to occur in two phases: ischemic damage and ischemic-reperfusion damage. 1 In the present study, we induced hypothermia during the first phase, in which hypothermia is suggested to be most effective.1,46 The mechanisms underlying the neuroprotective effects of hypothermia are extensive and cannot be listed exhaustively in this discussion. Many of these mechanisms can counteract the induction of SDs. First, an SD is an energy-demanding event that increases metabolism 47 and oxygen consumption. 48 On the contrary, hypothermia reduces neuronal metabolism, minimizes glucose utilization and oxygen consumption, and decreases the formation of acidic end-products of anaerobic metabolism. 49 Second, SDs are time locked to the release of glutamate, 50 while hypothermia inhibits glutamate-mediated activation of voltage-gated calcium channels.51,52 Third, SDs have been linked to an increase in inflammatory response; in contrast, hypothermia inhibits the inflammatory response caused by the exposure of immune cells to damaged and necrotic tissue.53,54 Fourth, SDs are the main phenomena responsible for cytotoxic edema formation through the induction of microvascular constriction and swelling of the neuronal soma and neuronal dendrites (so-called dendritic beading), 55 while hypothermia inhibits edema formation. 56 This occurs, in part, following a reduction in the N-methyl D-aspartate (NMDA) receptor activation, thereby modulating excessive sodium influx and hyperosmolar water pull. It is known that SDs require the NMDA receptor to expand and it has been shown that SD expansion can be blocked via NMDA receptor antagonists.39,57–59 Fifth, SDs are associated with increased ICP, 60 while hypothermia has been shown to prevent the increase in ICP associated with medium and large strokes. 61 Sixth, SD frequency has been correlated with infarct expansion and studies have shown that the incidence of SDs is dramatically increased when the body temperature exceeds 38.4°C. They also indicate up to a 3-fold higher probability of SD occurrence compared to normothermia.27,40 Studies of ischemic stroke have shown that elevated temperatures during the reperfusion period worsen clinical outcomes.62–64 Simply preventing fevers with prophylactic acetaminophen is associated with improvements 3 months after stroke occurrence. 65 Therefore, hypothermia may prevent fever peaks, thereby reducing SD frequency and its deleterious effects.
Study limitations
In this MCAo model, we did not investigate neurological outcomes, which are the main cause for the mismatch between experimental and clinical studies of the effect of therapeutic hypothermia. Instead, we used infarct volume as a prognostic factor of ischemic damage; however, neurologic evaluation is necessary to enhance the credibility and transferability of the results. Reperfusion in stroke therapy takes precedence and hypothermia is more effective after thrombolysis therapy. The current ischemic model has been performed without reperfusion; hence, potential adverse events during the reperfusion phase could not be evaluated. Experimental studies have suggested that longer durations of hypothermia might increase its neuroprotective effects. We induced hypothermia for 18 h with minimal side effects. Moreover, hypothermia undoubtedly causes discomfort in conscious patients, which might be overcome through sedation and mechanical ventilation. However, such an approach will further increase the risk of pneumonia or other side effects, which were not evaluated due to the short duration of hypothermia in the current study. Further, age and age-associated comorbidities can affect results in clinical and preclinical studies. In our model, we used young female animals, which do not properly simulate the complex clinical situations of human stroke patients.
Conclusions
ECoG and IOS are useful techniques for monitoring SDs during infarct progression. Therapeutic hypothermia produces a reduction in SD frequency and expansion in the gyrencephalic swine brain, while reducing infarct volume. Hypothermia can be a neuroprotective tool for targeting SDs. Further studies are needed before establishing a clinically adequate therapeutic model.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dr. Edgar Santos Marcial was funded by the Physician Scientist Program from the University of Heidelberg. Dr. Edgar Santos Marcial and Dr. Lena Maier-Hein were funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; project number: 462569370). Francisco L. Ramírez-Cuapio was supported by the Consejo Nacional de Ciencia y Tecnología of Mexico (CONACyT, National Council of Science and Technology; reference no. 2019-000021-01EXTF-00514). Roberto Díaz-Peregrino was supported with a scholarship granted by the Consejo Mexiquense de Ciencia y Tecnología del Estado de México (COMECYT, Council of Science and Technology from the Estate of Mexico. Reference: 2021BPS2-E0405).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
MK: Performed the experiments and analysis and wrote the manuscript. FLRC: Performed the experiments and analysis and wrote the manuscript. MAGH: Performed the experiments and analysis. RSP: corrected the manuscript and provided scientific support. RDP: corrected the manuscript and provided scientific support. NH, LMH, and JW: Corrected the manuscript and provided scientific support. ES: Designed the project, performed the experiments, and wrote the manuscript.