
Abstract
In stroke patients, local sampling of pial blood within the occluded vasculature before recanalization by mechanical thrombectomy emerged as powerful tool enabling insights into ultra-early stroke pathophysiology. Thereby, a strong intravascular inflammatory response hallmarked by hyper-acute neutrophil recruitment, altered lymphocyte composition and platelet activation could be observed. These human findings mirror experimental stroke. Here, neutrophil and T-cell activation are driven by platelets involving engagement of platelet glycoprotein receptor (GP)Ib, GPVI and CD84 as well as α-granule release orchestrating infarct progression. Thus, targeting of early intravascular inflammation may evolve as a new therapeutic strategy to augment the effects of recanalization.
Introduction
Large-vessel occlusion (LVO) due to thromboembolism emerging from the heart or extracranial artery stenoses is a major cause of disabling ischemic stroke. 1 Mechanical thrombectomy (MT), the most powerful treatment option for LVO in conjunction with systemic thrombolysis could substantially improve outcomes and the rates of successful recanalization of over 80%.2,3 However, clinical outcome in about half of the recanalized patients is still unfavourable.2,4 Progression of infarction already before this emergency procedure (“penumbral tissue loss”) may lead to futile or even harmful recanalization.4 –6 In addition, even if this procedure has been performed successfully, ischemia/reperfusion (I/R) injury is likely to perpetuate infarct progression not only in the experimental setting but also in human patients.7 –9 The underlying cellular and molecular pathomechanisms of infarct progression before and after recanalization are incompletely understood and alternative/adjunct therapeutic options to recanalization are eagerly awaited.
It is well established that ischemic stroke elicits a strong neuroinflammatory response in men and rodents 10 which has mainly been regarded as a secondary consequence of danger associated molecular patterns (DAMPs) released from dying neurons and glial cells within the ischemic brain parenchyma. 11 This concept has changed. Recent evidence from experimental and human stroke suggests that there is an immediate innate immune reaction commencing already during LVO within the vascular compartment when it is fed by collateral blood flow.12,13 In this review we focus on this hyper-acute inflammatory response phase during occlusive ischemia taking an intravascular perspective. We compare and combine seminal observations in acute stroke patients with descriptive and mechanistic evidence from experimental animal research. With regard to the contribution of immune cells to subacute and chronic stroke evolution the readers are referred to recent comprehensive reviews.14 –16
The local neuroinflammatory response in human hyper-acute stroke
Until recently, observations on stroke-related inflammation were largely restricted to post-mortem brain tissue with an obvious delay from onset, thereby mostly missing the hyper-acute phase,10,17 or to cerebrospinal fluid and venous blood sampling which do not reflect the local cellular and molecular milieu within the ischemic territory. 18 The advent of MT in 2015 has not only revolutionized stroke medicine by improving treatment efficacy, 4 but nowadays allows a glimpse into ultra-early, intravascular stroke pathophysiology. We and few other groups could demonstrate that minute samples of ischemic arterial blood can be aspirated directly from the pial cerebral collateral circulation during occlusive ischemia of hyper-acute human stroke. Briefly, a microcatheter is navigated through the embolic occlusive lesion (typically distal to M1 and/or intracranial ICA occlusions) into the center of the affected vascular field at midinsular M2 position as target sampling location. Then, immediately before recanalization the pial sample can be aspirated in only few seconds without significantly delaying the emergency interventional recanalization procedure.12,19 As the control standard, an additional arterial blood sample is taken at the level of the internal carotid artery under physiologic non-occlusive flow condition and serves as the systemic intraindividual reference to which local cellular and molecular alterations of the occluded ischemic cerebral vascular compartment are compared to. In these local target samples we discovered a substantial increase in absolute leukocyte counts which was strongly driven by neutrophils. 12 This observation could be prospectively replicated in a large independent cohort using the exact same sampling protocol.12,20 Flow cytometric automated detection and counting of leukocyte (sub-)populations again corroborated a strong predominance of neutrophils, and, in addition, disclosed a local shift in the relative distribution of lymphocytes towards CD4+ T-cells.21,22 However, absolute lymphocyte numbers did not differ between local ischemic target and systemic control samples. Taken together these data indicated that LVO elicits a robust intravascular inflammatory response within the ischemic penumbra. Since immune cells do not normally reside within this local vascular compartment 23 they must have infiltrated through the pial collateral channels/anastomoses.24,25
As another principal finding, we could raise evidence of platelet activation within the same vascular compartment. Activated platelets trigger and guide inflammation beyond their fundamental roles in thrombus formation and hemostasis. 26 Platelet activation can be linked to inflammation by engagement of glycoprotein surface receptors and/or release of α-granules. 27 As a strong indicator of local platelet activation we found increased concentrations of the platelet-derived neutrophil-activating chemokine CXC motif ligand (CXCL) 4 (platelet factor 4) and the neutrophil attractant CXCL7 (neutrophil-activating peptide 2) in local ischemic blood samples before recanalization which represent the most abundant platelet releasates from α-granules. 28 Interestingly, CXCL7 is a potent chemoattractant for neutrophils while CXCL4 induces neutrophil degranulation and increases their secretion of matrix metalloproteinase (MMP)-9 and myeloperoxidase (MPO). 29 MMP-9 has been implicated in breakdown of the blood-brain barrier (BBB) in cerebral ischemia 30 , while MPO is indispensable for the formation of neutrophil extracellular traps (NETs). 31 NETs are extracellular DNA lattices able to trap pathogens, but are also instrumental in sterile inflammation and thrombus formation by binding other blood cells and coagulation factors. 32 NET formation has recently been shown to contribute to infarct development in experimental stroke. 33 These findings point to a possible pathophysiological link between very early local platelet activation and neutrophil responses inside the occluded vascular compartment during acute stroke.
Furthermore, platelets are an important source of high-mobility group box 1 protein (HMGB1), 34 one of the most prevalent DAMPs which triggers sterile inflammation by activation of inflammasome pathways. 35 In human pial occlusive intravascular samples local HMGB1 concentrations were increased in addition to another DAMP, calprotectin (S100A8/A9), 36 which is mainly released by leukocytes. 37 These human data suggest a vicious cycle of platelet-leukocyte interactions in acute stroke patients in which local DAMP release by platelets (e.g. HMGB1) and leukocytes (e.g. S100A8/A9) could augment the first wave of neuroinflammation directly within the vascular compartment long before the ischemic brain parenchyma releases large additional amounts of DAMPs due to neuronal and glial damage and thereby fosters secondary intracerebral inflammation. 11
Comparing human observations on hyper-acute neutrophil and platelet responses with experimental stroke
Human observational data derived from cerebral blood sampling in acute stroke must be translated into animal experimentation to establish causal relationships. So far seminal early histopathological studies by Garcia et al. 38 and later Enzmann et al. 17 already described a similar massive leukocyte response to cerebral ischemia in rodents. Intravascular neutrophils were detected as early as 30 min after cerebral LVO with a peak number at 12 h in Wistar rats. 38 This was later refined and confirmed in mice. 17 One neuroanatomical feature was neutrophil accumulation in the arteriolar compartment of the leptomeningeal space, that is, within the vascular compartment that overlies the convex brain surface. Neutrophil numbers peaked at around 18–24 h, but neutrophils did not enter the ischemic brain parenchyma in transient middle cerebral artery occlusion (tMCAO) up to this time point. Similarly, permanent focal cerebral ischemia in mice induced an accumulation of rolling and adhering leukocytes predominantly in venules of the ischemic brain receiving collateral blood flow already 40 min after onset and to a lesser extent also in pial arterioles as shown by intravital fluorescence microscopy. 39 Human local blood sampling as described above is performed not directly inside but in the vicinity of the pial arteriolar network where these experimental observations were made. Based on their predominant intravascular location it was suggested that neutrophils act detrimentally at the neurovascular unit during the acute stroke phase rather than within the brain parenchyma. 17 Accordingly, in-vivo imaging studies employing two photon-microscopy observed microvascular obstruction of 20–30% of capillaries in the infarct core and penumbra by neutrophils adhering to distal capillary segments during reperfusion after tMCAO. 40 Depletion of circulating neutrophils using an anti-Ly6G antibody 24 h before infarct induction restored microvascular perfusion. Likewise in-vivo microscopy revealed leukocytes travelling slowly through the microvasculature or getting stuck after recanalization which led to “traffic jams”. 41 Stall dynamics could be modulated by anti-Ly6G antibody as early as 2 h after application even before settlement of neutropenia. Although these studies clearly demonstrate that neutrophils contribute to plugging, the molecular mechanisms underlying the effects of anti-Ly6G treatment remain elusive. Ly6G is only expressed on mouse, but not human neutrophils, 42 and Ly6G’s endogenous ligand and physiological roles are still unknown. 43 One study suggested that Ly6G modulates neutrophil migration to sites of inflammation via a ß2-integrin dependent mechanism. 43 Upon activation neutrophil expel NETs 32 and recently, Denorme and colleagues could show that platelet derived HMGB1 causes NET formation in a mouse tMCAO model. 33 Moreover, blocking of NET formation by a NET inhibitory peptide ameliorated stroke outcome without affecting neutrophil recrutiment. 33 Previous experimental studies took an alternative approach by targeting neutrophil-endothelial cell interactions thereby blocking adhesion and transmigration into the brain parenchyma. Blockade of intracellular adhesion molecule-1 (ICAM-1), Mac-1 (CD11b/CD18) and selectins showed a variable degree of protection, but primarily in transient MCAO models with controlled reperfusion (reviewed in 44 ). Three clinical stroke trials, however, showed no clinical benefit (reviewed in15,16,44). Importantly, these clinical studies were undertaken before the advent of mechanical thrombectomy in 2015 including patients with permanent vessel occlusion, and under the premise that neutrophils immediately infiltrate the brain parenychma mediated by ICAM-1, Mac-1 or selectin engagement and directly damage neurons. This view has been challenged.17,45 Novel evidence suggests, that neutrophils driven by platelet releasates such as HMGB1, CXCL4 and CXCL7 can act immediately and locally within the intravascular compartment after LVO by release of MMP-9, MPO and NET formation among others and thereby damage the BBB30,33 (Figure 1). This may not require direct cellular neutrophil/endothelial cell interactions, in contrast to migration into the brain parenchyma in the subacute stroke phase.
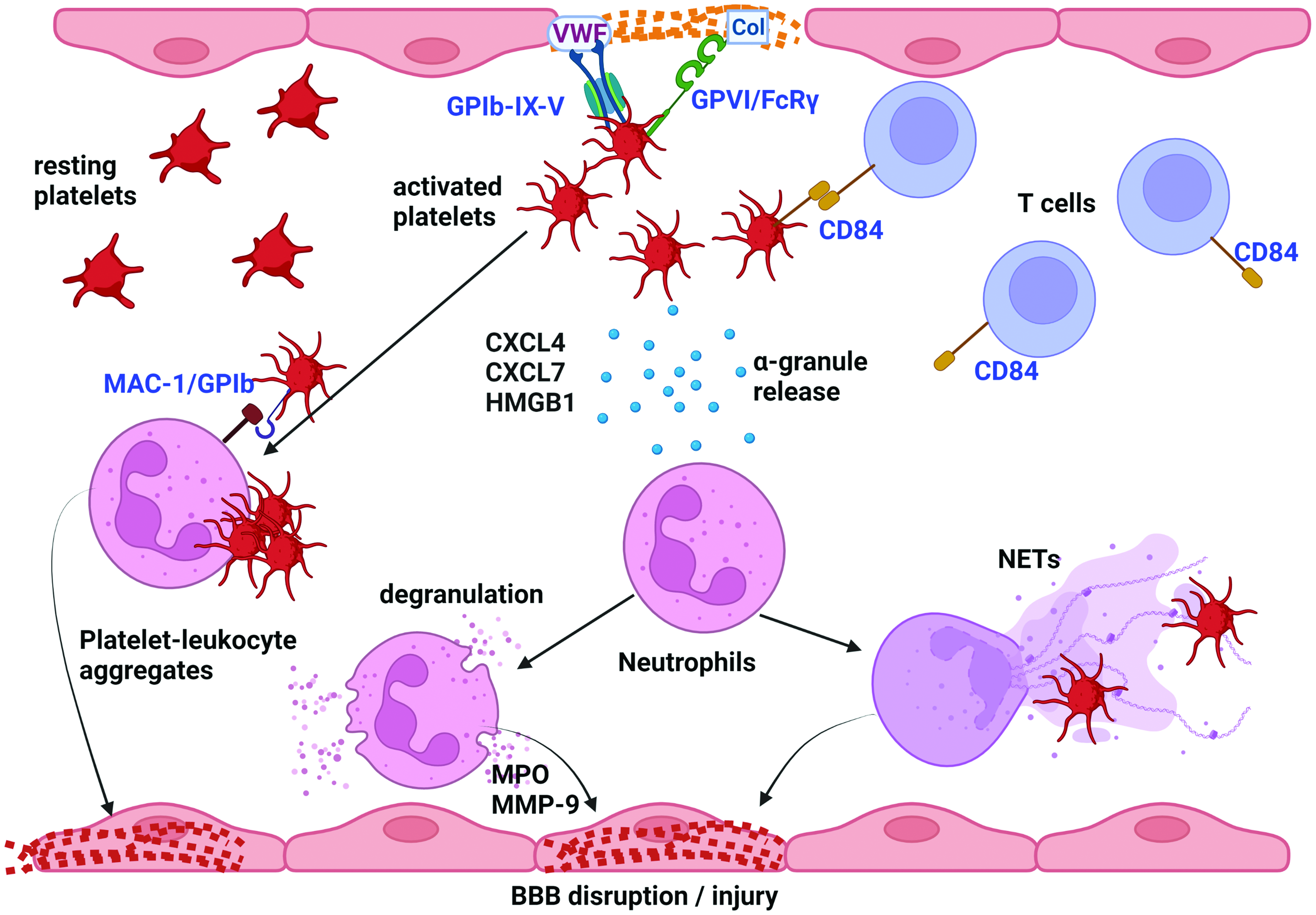
Both in human and experimental stroke, ischemia induces a rapid intravascular inflammatory response dominated by neutrophils, but also T cells, that is guided by platelets. Experimental studies in mice have shown that during middle cerebral artery (MCA) occlusion as well as in the reperfusion phase platelet tethering via GPIb (GPIb-IX-V complex binding to von Willebrand Factor (VWF)) and firm adhesion via GPVI (GPVI/FcRγ binding to collagen (Col)) contribute to infarct progression. This can be partly explained by platelet release of α-granules which contain a plethora of proteins, most abundantly CXCL4, CXCL7 and HMGB1. An increased concentration of α-granule releasates could be measured in pial blood samples drawn from the secluded MCA territory before mechanical recanalization in hyper-acute stroke patients. CXCL7 is a potent chemoattractant for neutrophils and CXCL4 can induce granule release from neutrophils which contain MPO and MMP-9. On the other hand, HMGB1 is a potent inducer of NET formation. MMP-9 and NETosis can cause disruption of the blood-brain-barrier (BBB) and perivascular tissue damage. Collectively these combined clinical and experimental data suggest that platelet activation further drives inflammation and formation of platelet-leukocyte aggregates, which may additionally cause obstruction of brain capillaries. In addition, T cells interact with platelets via CD84, a homophilic cell adhesion molecule, in mouse stroke and by so far unknown effector mechanisms aggravate infarct progression during recanalization. Figure created with BioRender.com.
Now, the striking similarities between human and experimental findings in terms of intravascular neutrophil accumulation under LVO already before recanalization warrant further mechanistic studies focusing on a vascular mode of inflammation not exclusively depending on reperfusion but commencing before it. In ultra-early stroke neutrophils in concert with platelet activation could contribute to progressive infarction by multiple mechanism, e.g. toxic effects damaging the BBB, dynamic stalls at the level of the microcirculation and/or increased viscosity of pial blood which in turn increase vascular resistance and impair collateral blood flow.
Another important observation underpinning our human observations relates to platelets. Garcia and colleagues using a rat stroke model described early and abundant platelet aggregates in arterial rather than the venous cerebral vessels which completely obstructed these vessels only at 2 days of stroke onset. 38 Accordingly, our group could recently show by light sheet microscopy that infarct progression after recanalization in the transient MCAO model in mice did not require thrombus formation. 46 This is in accordance with previous findings showing that platelet aggregation via glycoprotein (GP) IIb/IIIa is dispensible for lesion formation in stroke. 47 It is well established that platelets guide inflammation via GPIb signaling. GPIb is expressed exclusively on the surface of megakaryocytes and platelets and facilitates tethering to the vessel wall by binding to von Willebrand factor. 48 Moreover, GPIb exhibits binding sites for Mac-1 expressed on leukocytes which allows formation of platelet-leukocyte complexes. Mice treated with GPIb fab developed smaller infarcts in models of permanent ischemia up to 4 h occlusion time 13 and largely prevented ischemia/reperfusion injury after tMCAO.47,49 In both settings treatments targeting solely platelets diminished local inflammatory responses in the ischemic cerebral vasculature, including reduced numbers of neutrophil, platelet-neutrophil aggregates and T-cells. These findings laid the foundation of the concept of thrombo-inflammation i.e. the cooperative detrimental action of platelets and immune cells as driving force of acute infarct progression (Figure 1). 7 Firm adhesion and activation of platelets requires further involvement of the glycoprotein receptor VI (GPVI), 50 the principal collagen receptor on platelets. Blocking of GPVI similar to GPIb ameliorated infarct progression under LVO, 51 as well as I/R injury upon recanalization.47,52 GPVI signalling is an important step for platelet granule release. There is strong evidence that granule release is involved in stroke development since Nbeal knock-out mice which lack α-granules are protected against I/R injury in tMCAO.53,54 These experimental data again match with the increased concentrations of CXCL4, CXCL7 and HMGB1 present in human ischemic blood samples before recanalization. 28 These represent the most abundant proteins stored in platelet α-granules. 27 In summary, observations derived from both human pial blood samples under occlusion and experimental stroke in close parallels point to a concerted detrimental action of platelets and leukocytes (i.e. thrombo-inflammation) that emerges from the vascular compartment which awaits further exploration. Deeper mechanistic insight on the functional role of leukocytes in stroke development has already been gained for T-cells.
The peculiar role of T-cells on progressive stroke development
As desribed above by flow cytometry-based analysis of local pial blood samples, a shift in the composition of lymphocytes was seen within the ischemic intracranial vascular compartment. While the relative proportion of CD4+ T-cells increased, the number of B-cells decreased.21,22 Interestingly, immuno-deficient mice that lack any lymphocyte population are protected from progressive infarction under occlusion in the mouse pMCAO model 13 and also from I/R injury after recanalization 55,56 in the tMCAO model. This protection could be reversed in both models by adoptive transfer, particularly, of CD4+ T-cells among others. Of note, lack of B-cells had no influence on stroke development. 57 Immuno-deficient mice displayed no altered hemostasis and responded properly to conventional stimuli of thrombus formation. 56 This further indicated that T-cells contribute to infarct development independent from thrombus formation, an effect that relied on the presence of platelets. 58 Recently, the missing molecular link between T-cell and platelet effects in acute stroke could have been identified providing further mechanistic proof and a novel therapeutic target to tackle the deleterious T-cell-platelet interaction: CD84 is a homophilic cell adhesion molecule expressed on both T-cells and platelets, and CD84-deficiency on either of both cell types conferred stroke protection. 59 Moreover, high levels of CD84 expression on platelets in the circulation were associated with poor outcome in human stroke patients. 59 This serves as another example how experimental findings are in accordance with (albeit limited) human evidence and illustrates that the development of ischemic brain lesions is not simply the consequence of ongoing/recurrent thrombosis after recanalization but partly caused by complex inflammatory processes amenable to treatment.
Conclusion
We are facing an exciting new area of stroke research which allows verification of key experimental findings in human stroke patients through the methodical breakthrough of local sampling. Importantly, local sampling has already allowed for the assessment of concentrations of pharmacological agents (e.g. alteplase) within the collateral circulation. 60 By this approach it could be proven that despite complete LVO systemically applied substances reach concentrations equivalent to systemic levels within the penumbra. Given that platelet-driven neuroinflammation commences early under occlusion 28,39 and selected platelet blockade is able to reduce inflammation and delay infarct progression before recanalization in experimental animals, 13 these data pave the way for the development of alternative treatments that may be given upon stroke onset before MT. 6 Emerging targets are platelet GPIb and GPVI which are critical molecules guiding T-cell and neutrophil inflammation, but appear dispensible for hemostasis in the ischemic brain. 47 After passing uneventful application of a GPVI inhibitor in normal subjects, 61 the potential of further ameliorating stroke outcome in acute stroke patients by blocking GPVI in combination with standard care is currently under investigation in a clinical phase 2 trial (ACTIMS trial NCT03803007). Moreover, pial blood sampling has the potential to evaluate early (inflammatory) biomarkers to predict outcome before massive brain tissue destruction has occurred, and to monitor local drug concentrations applied in addition to MT in clinical trials.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Work cited in this review was supported by the Deutsche Forschungsgemeinschaft (project number 374031971 CRC/TR 240 and project number 413657723 – UNION CVD).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.