
Abstract
Tibia fracture (BF) enhances stroke injury and post-stroke memory dysfunction in mouse. Reduction of neuroinflammation by activation of α-7 nicotinic acetylcholine receptor (α-7 nAchR) reduced acute neuronal injury and sensorimotor dysfunction in mice with BF 1-day after stroke. We hypothesize that reduction of neuroinflammation by activation of α-7 nAchR improves long-term memory function of mice with BF 6-h before stroke. The mice were randomly assigned to saline, PHA-568487 (α-7 nAchR agonist) and methyllycaconitine (antagonist) treatment groups. The sensorimotor function was tested by adhesive removal and corner tests at 3 days, the memory function was tested by Y-maze test weekly for 8 weeks and novel objective recognition test at 8 weeks post-injuries. We found PHA-568487 treatment reduced, methyllycaconitine increased the number of CD68+ cells in the peri-infarct and hippocampal regions, neuronal injury in the infarct region, sensorimotor and long-term memory dysfunctions. PHA-568487 treatment also reduced, while methyllycaconitine treatment increased atrophy of hippocampal granule cell layer and white matter damage in the striatum. In addition, PHA-568487 treatment increased neuron proliferation in granule cell layer. Our data indicated that reduction of neuroinflammation through activation of α-7 nAchR decreased neuronal damage, sensorimotor and long-term memory dysfunction of mice with BF shortly before stroke.
Keywords
Introduction
Memory dysfunction is one of the major health problems for elderly, which increases the burdens of individuals and society as the population aging. Stroke and fracture of long-bones or hips are risk factors for memory dysfunction. If these conditions present simultaneously, the individuals will have less optimal outcome.
The risk of stroke increases in the first year after hip fracture, 1 and remains high in the following 10 years. 2 Patients with post-fracture stroke have a poor functional recovery in the first year than patients with either bone fracture or stroke alone. 3 Alternatively, post-stroke fracture of long-bones increases late-onset comorbidities and mortality in elderly. 4 Strategies for preventing or treating stroke may adversely influence bone healing. 5
Our previous studies showed that in mouse, tibia fracture (BF) before or after ischemic stroke augments stroke-related brain injuries, inflammatory cell-infiltration and the levels of pro-inflammatory cytokines in the peri-infarct region and hippocampus analyzed 3 days after the injuries.6–9 In young adult mice, BF causes a short-term (<1 week) memory dysfunction.10,11 We found that young mice developed long-lasting (≥8 weeks) spatial memory dysfunction when BF preceded ischemic stroke by 6 h, which was associated with an accumulation of CX3 chemokine receptor 1+ (Cx3cr1+) and CD68+ cells in the hippocampal stratum lacunosum moleculare (SLM) region. 9
Alpha-7 nicotinic acetylcholine receptors (α7-nAChRs) are widely distributed on the surface of systemic macrophages 12 regulating inflammatory processes. Activation of α7-nAChRs has a neuroprotective effect on ischemic and hemorrhagic stroke. 13 We showed that activation of α7-nAChRs8,14 reduces neuroinflammation and neuronal injury in mice subjected to BF 1-day after ischemic stroke at 3-days after the injuries.
In this study, we demonstrated that activation of α7-nAChRs not only reduced neuroinflammation and the neuronal damage in the infarct region, but also reduced neuroinflammation in the hippocampi and long-term memory dysfunction in mice subjected to BF 6-h before ischemic stroke.
Methods
Animals
Eight-week-old CD1 female and male mice were purchased from Charles River Laboratories (South San Francisco, CA) and kept in the animal facility at Zuckerberg San Francisco General Hospital. Mice were fed standard rodent food and water ad libitum and were housed in sawdust-lined cages in an air-conditioned environment with 12-h light/dark cycles.
All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California, San Francisco, and conformed to National Institutes of Health guidelines and ARRIVE guidelines.
Mice were randomly assigned to short-term (51 male mice) and long-term groups (75 mice, 47 male and 28 female), and three treatment groups using a randomization tool (http://www.graphpad.com/quickcals/randomize1.cfm): (1) Saline, (2) α7-nAChR selective agonist PHA 5,68,487 (PHA, Tocris Bioscience, Bristol, UK), (3) α7-nAChR antagonist methyllycaconitine (MLA, sigma-Aldrich Chemicals Company, St Louis, Mo, USA). The experimental design is illustrated in Figure 1. Saline, PHA (0.8 mg/kg) or MLA (6 mg/kg) were injected intra-peritoneally (i.p) right before BF and one day after pMCAO. These doses were selected based on the results on our previous study. 14
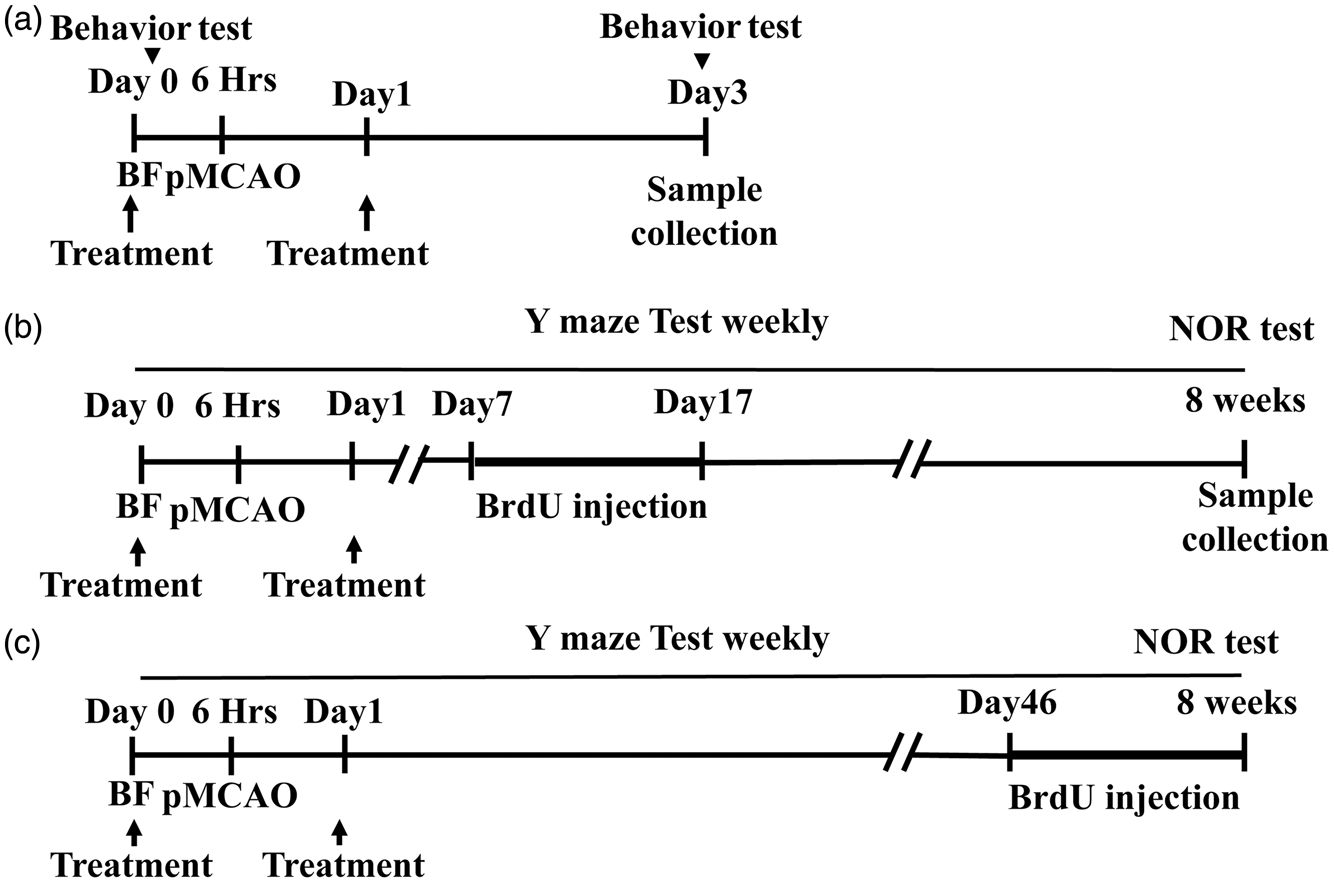
Experimental design. BF was performed 6 h (hrs) before pMCAO. Drugs were injected (treatment) intra-peritoneally (i.p) right before BF and one day after pMCAO. (a) Short-term group. The brain samples were collected 3 days after surgeries and sensorimotor function analysis (behavior test). (b and c) Long-term group. Mice were subjected to Y-maze test weekly and NOR test at 8 weeks after the surgeries. The brain samples were collected 8 weeks after surgeries and behavior tests. BrdU were injected daily from 7 to 17 days (b) and from 46 to 56 days (8 weeks, c) after the surgeries.
All surgeries were performed under anesthesia with 2% isoflurane inhalation and in aseptic conditions. Buprenorphine (0.1 mg/kg of body weight) was given at the beginning of and 6 h after each surgery and as needed afterwards. Rectal temperature was monitored and maintained at 37 ± 0.5°C with a thermal blanket during the surgeries. Blood pressure was measured using CODA Non-Invasive Blood Pressure System (Kent Scientific Corporation, CT, USA), at baseline, first anesthesia, 5 min and recovery after BF, second anesthesia, 10 min and recovery after permanent distal middle cerebral artery occlusion (pMCAO). No difference was found among mice in different treatment groups (Supplementary Figure I).
BF
After the mice were anesthetized with isoflurane inhalation, a longitudinal incision was made from the knee to the midshaft of the tibia. The subcutaneous tissues were dissected, and muscles isolated until the patellar tendon and the tibia periosteum were revealed. A 0.5 mm hole in the proximal tibia was drilled with a 25-gauge needle just beneath and medial to the patellar tendon to enter the intramedullary canal. A 0.38 mm stainless steel rod was inserted in the canal until resistance was felt. Subsequently, the fibula and the muscles surrounding the tibia were isolated, the periosteum stripped over a distance of 10 mm circumferentially and an osteotomy was performed with scissors at the junction of the middle and distal third of the tibia. The wound was closed. The mice were allowed to recover spontaneously from anesthesia in warmed cages.
pMCAO
Six hours after BF, the mice were re-anesthetized. Under a surgical microscope, a 1 cm incision was made between the left orbit and tragus. A piece of 2 mm2 skull was removed. The arachnoid was then opened, and the middle cerebral artery (MCA) was occluded with an electrocoagulation. The surface cerebral blood flow was monitored by a laser Doppler flow-meter (Vasamedics, Little Canada, MN, USA). Mice were excluded if the surface cerebral blood in the ischemic core were 15% above the baseline or massive bleeding occurred. Six mice were excluded in this study. A total of 9 mice died (3 in saline group, 2 in PHA group and 4 in MLA group) during and after surgeries and were replaced with additional mice.
Behavior tests
The sensorimotor dysfunction was analyzed by adhesive removal test 15 and corner test 16 1 day before (baseline) and 3 days after the surgery.
For adhesive removal test, two pieces of different color adhesive tapes (0.3x0.3 cm) were placed on each of the forepaws. The time the mouse took to remove the tapes were recorded. The maximum testing time was 120 s. Mice were trained twice daily for 4 days before BF procedure to obtain an optimal level of performance. The mice took longer time to remove the tape from the paw on the opposite side of the stroke injury 3 days (right paw in this study) after pMCAO.
For corner test, mice were placed between two 30 × 20 cm boards. Both sides of their vibrissae were stimulated as they approached the corner. The mice would then move up and turn to face the open end. Normal mice will make equal numbers of right and left turns. The stroke mice could not sense the stimulation on the stroke injury side, and hence, made more turns to the stroke side (left turns in this study). Three sets of 10 trials were conducted. Turning not incorporated in a rearing movement was excluded. The operator had maneuvered the mice for 5 min daily one week before the actual test to prevent mouse being agitated by the operator.
Memory function were evaluated by Y maze test weekly after surgeries for eight weeks and by novel objective recognition (NOR) test at 8 weeks after surgeries. 9
For Y maze test, mice were placed in the center of a Y-shaped maze (Stoelting, Chicago, IL, USA) that have three white, opaque plastic arms set at a 120-degree angle from each other and allowed to freely explore the three arms for 10 min until 20 entries have been achieved. Mice entered fewer than 20 times during the 10 min-trial were excluded. Four mice were excluded in this study. Normal mice are likely to visit a new arm in the maze rather than the recently visited arm. The number of arm entries and the number of alternative entries were recorded to determine the percentage of spontaneous alternations.
The NOR test was performed in four days. In the first and second days, each mouse was allowed to explore freely a rectangular arena for 10 min. In the third day, two identical items were placed in the arena. Each mouse was allowed to explore the arena for 5 min. In the last day, one of the objectives was replaced with a different one. The explorations were recorded by a night vision camera (ELP 1 Megapixel Day Night Vision, China) and the exploration processes were analyzed with SMART 3.0 video tracking software (Panlab, Spain). Normal mice will spend more time exploring the novel object than the familiar one. If exploration time of all objects are same, it can be interpreted as a memory dysfunction. 17 The preference was determined by dividing the exploration time on the novel object by the whole exploration time. Nine mice were excluded due to standing on the objective for more than 1 min or failure to reach a 20-s minimum exploration time on both objects. 18
Bromodeoxyuridine labeling
To observe neurogenesis, we randomly assigned the long-term group mice into two groups: (A) received BrdU (100 mg/kg, Sigma-Aldrich, St Louis, MO) daily through i.p. injection from 7–17 days after the surgeries; and (B) received BrdU (100 mg/kg) i.p. daily between 46–56 days post-surgeries. All mice were euthanized at 8 weeks (56 days) after the surgeries.
Measurement of infarct and atrophy volumes
The brain samples were collected 3 days or 8 weeks after the surgeries. Infarct volume/atrophy volumes were evaluated using a protocol adapted from previously published studies.7,19 A series of 20 µm-thick coronal sections starting 2 mm caudal to the frontal pole were made using a Leica CM1950 Cryostat (Leica Microsystems, Wetzlar, Germany). One in 10 (200 µm apart) sections were stained with cresyl violet and imaged. The infarct areas were outlined and quantified using IMAGE J (National Institutes of Health, Bethesda, MD). The infarct volumes were calculated by multiplying the sum of infarct areas from all sections by 200 µm. The atrophic areas were calculated by subtraction the area of ipsilateral hemisphere from the area of contralateral hemisphere. The atrophy volumes were reconstructed by multiplying the sum of atrophic areas from all sections by 200 µm.
Histological and immunohistochemical staining
We used the slides collected from bregma −1.2 to −1.4 mm for analyzing cells in the peri-infarct and striatum regions; and slides collected from bregma −1.6 to −2.3 mm for analyzing cells in the hippocampi. Sections were incubated overnight at 4°C with primary antibodies specific to: CD68 (1:500, Abd Serotec, Kidlington, USA), NeuN (1:500, Millipore, MA, USA), BrdU (1:500, Invitrogen, CA, USA), myelin basic protein (MBP, 1:500, Sigma-Aldrich) and then incubated with Alexa Fluor 488 or 594 fluorescent-dye conjugated secondary antibodies (1:500, Invitrogen) at room temperature for 2 h. All slides were covered with Vectashield Mounting medium with DAPI (Vector Laboratories Inc., Burlingam, CA).
Fluoro-jade C (Millipore) was used to detect damaged neurons. The staining was performed according to the manufacturer's instruction. For detection of BrdU+ cells, brain sections were pre-incubated with 2 N HCl at 37°C for 18 min and then rinsed in 0.1 M boric acid (pH 8.5) at 25°C for 8 min to depurinate the DNA before incubated with primary antibody. Sections of intestine were used as positive controls for BrdU staining. Negative controls were performed by omitting the primary or secondary antibodies. The pictures were taken using a fluorescence microscope (Boprevo BZ-9000, Keyence, Osaka, Japan).
Quantification of cells in peri-infarct and hippocampus regions
For quantification of CD68+ and Fluoro-Jade C+ neurons in the peri-infarct region, six images were taken from two coronal sections (200 µm apart) per brain in the areas indicated in our previous publications.6,7,14,20 CD68+ and Fluoro-Jade C+ neurons were quantified using Adobe Photoshop CS6 as previously described. 20
For qualification of cells in hippocampal SLM region, the number of CD68+ in the contralateral or ipsilateral sides of stroke were quantified in two coronal sections per brain (200 µm apart), three areas per section on each side. BrdU+ neurons in dentate gyrus (DG) were counted in two sections 200 µm apart of each brain.
Quantification of white matter bundle in the striatum and the volume of granular cell layer in hippocampus
Two sections were selected and stained with an antibody specific to myelin basic protein (MBP). The MBP positive area in the striatum was measured as previously reported. 21 The GCL volumes were measured following the Cavalieri principle 22 using 2 DAPI stained sections 200 µm apart. The sum of GCL volumes were calculated using the equation described previously 22 and the ratios of ipsilateral and contralateral GCL were compared among the different treatment groups.
Protein extraction and enzyme-linked immunosorbent assay
After being anesthetized through isoflurane inhalation, mice were perfused with 1X PBSto remove blood. Brains were collected. The hippocampi were dissected out and homogenized in 1X cell lysis buffer (R&D Systems, Minneapolis, MN), supplemented with 1 mM PMSF (Cell signaling). The lysates were centrifuged at 13,000 rpm for 25 min, and the supernatants, collected. Protein concentrations were measured using the Bradford Protein Assay (Bio-Rad Laboratories, Hercules, CA, USA). IL-1ß was quantified using Quantitkine ELISA Kits (R&D Systems).
Statistical analyses
All quantifications were performed by at least two researchers who were blinded to the group assignment. Data are presented as mean ± standard deviation (SD). Histogram was used to test if the data were normally distributed. Data were log-transformed before analysis if they were not normally distributed. Differences among multiple groups were analyzed by one-way analysis of variance (ANOVA) followed by multiple comparisons with Tukey's correction using GraphPad Prism 6. Y-maze and IL-1β data were analyzed by two-way ANOVA followed by multiple comparisons and Tukey's correction. A p value < 0.05 was considered significant. The sample size was estimated according to our previously published data 9 and indicated in figure legends.
Results
Activation of α7-nAChRs reduces the CD68+ cells in the peri-infarct and hippocampal SLM regions and levels of IL-1β in hippocampi ipsilateral to stroke injury
To analyze neuroinflammation, the CD68+ microglia/macrophages were quantified in the peri-infarct and hippocampal regions on brains collected 3 days after the surgeries. Compared to saline-treated mice, PHA-treated mice had fewer (p < 0.001) and MLA-treated mice had more (p = 0.024) CD68+ microglia/macrophages in the peri-infarct regions. PHA treated mice also had fewer CD68+ microglia/macrophages than MLA treated mice (p = 0.034, Figure 2(a) and (b)).
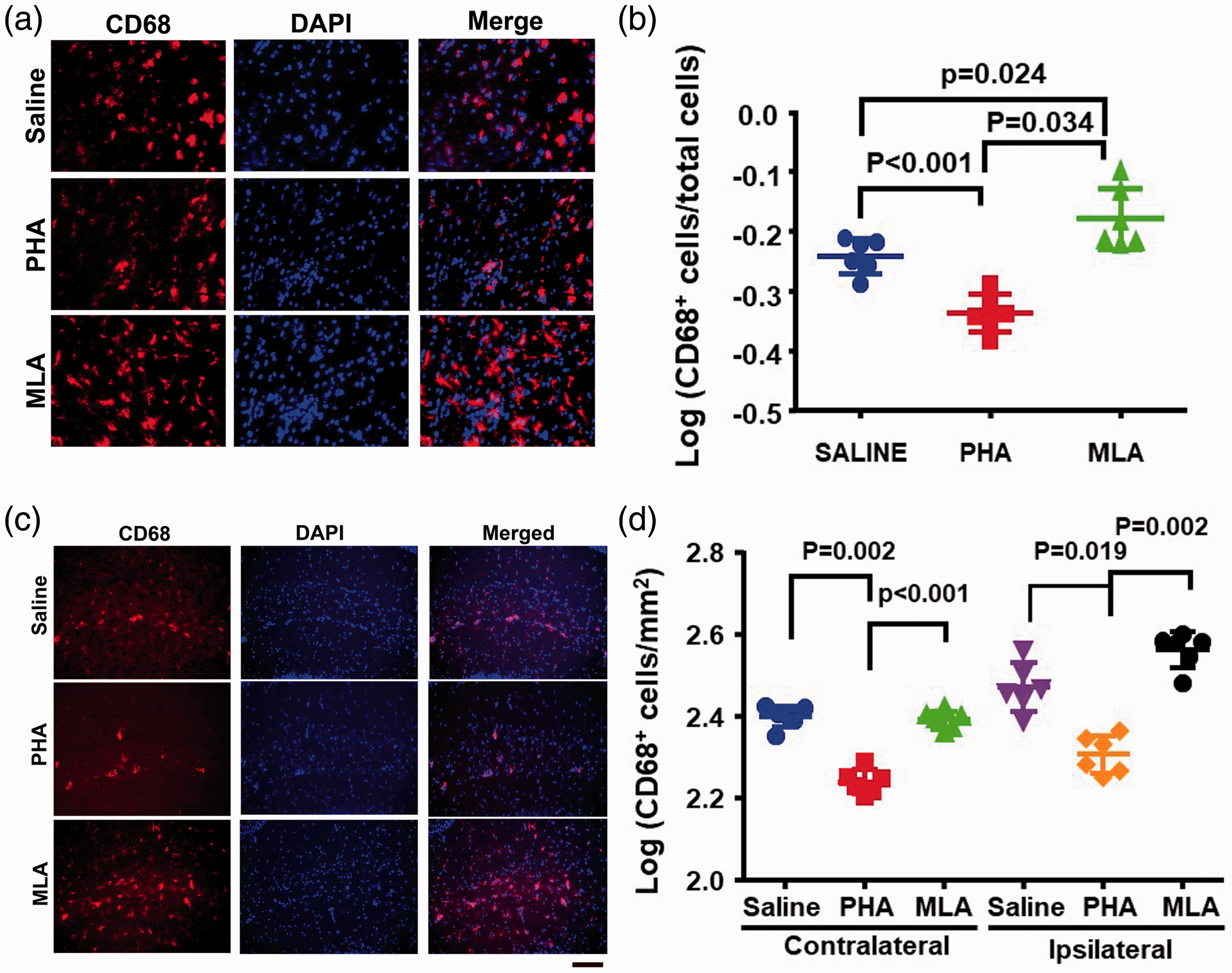
Activation of α7-nAChR reduced CD68+ cells in the peri-infarct region and the hippocampi. (a and c) Representative images of CD68+ cells (red) in the peri-infarct region (a) and hippocampi ipsilateral to the stroke injury side (c). The nuclei were stained with DAPI (blue). Scale bar = 50 µm. B & D. Quantification of CD68+ in the per-infarct regions (b) and SLM regions (d). n = 6.
In the hippocampi, more CD68+ cells were accumulated in the ipsilateral side than contralateral side of stroke injury in the saline group (p = 0.03, Figure 2(c) and (d)). Compared to saline group, PHA group had few CD68+ cells in both contralateral (p = 0.002) and ipsilateral side (p < 0.001) of hippocampal SLM regions of stroke (Figure 2(c) and (d)). PHA group also had fewer CD68+ cells than MLA group in both contralateral (p<0.001) and ipsilateral side (p = 0.002) of hippocampal SLM regions. These data suggest that activation of α7-nAChRs reduces neuro-inflammation in the peri-infarct and hippocampal regions.
To test if activation of α7-nAChRs at the onset of the injuries can reduce neuroinflammation in the hippocampi in the later stage of the injuries, we analyzed IL-1β level in the hippocampi collected 8-weeks after the injuries. We found that compared to saline treated mice, the PHA treated mice have lower levels of IL-1β (p = 0.012) in the hippocampi ipsilateral to the stroke lesion (Supplementary Figure II). These data indicate that activation of α7-nAchRs at the onset of the injuries reduced neuroinflammation in in the lateral stage of the injuries.
Reduction of neuroinflammation alleviated sensorimotor and long-term memory dysfunction in mice with BF and stroke injuries
The adhesive removal test and corner test were performed 3 days after surgeries to evaluate the sensorimotor function. In adhesive test, mice took longer time to remove adhesives from their right paws (baseline vs. saline: p < 0.001) after the surgeries. PHA treated mice used shorter time than saline treated mice to remove the adhesives from their right paws (p < 0.001), while the MLA treated mice used longer time to remove the adhesives (p < 0.001, Figure 3(a)). Mice in all groups took similar time to remove adhesives from their left paws (p = 0.74, Supplementary Figure III). In corner test, mice took more left turns after surgeries (baseline vs. saline: p < 0.001). PHA group made fewer left turns (p = 0.015), while the MLA group made more left turns (p = 0.041) than saline group (Figure 3(b)). These data are similar to those obtained from our previous study on mice that had subjected to BF one day after pMCAO. 14 Since we showed previously that the performances of mice subjected to BF one day after pMCAO have recovered to normal level in these tests 14 days after the surgeries, 20 we did not perform these tests on mice 8 weeks after the surgeries in this study.
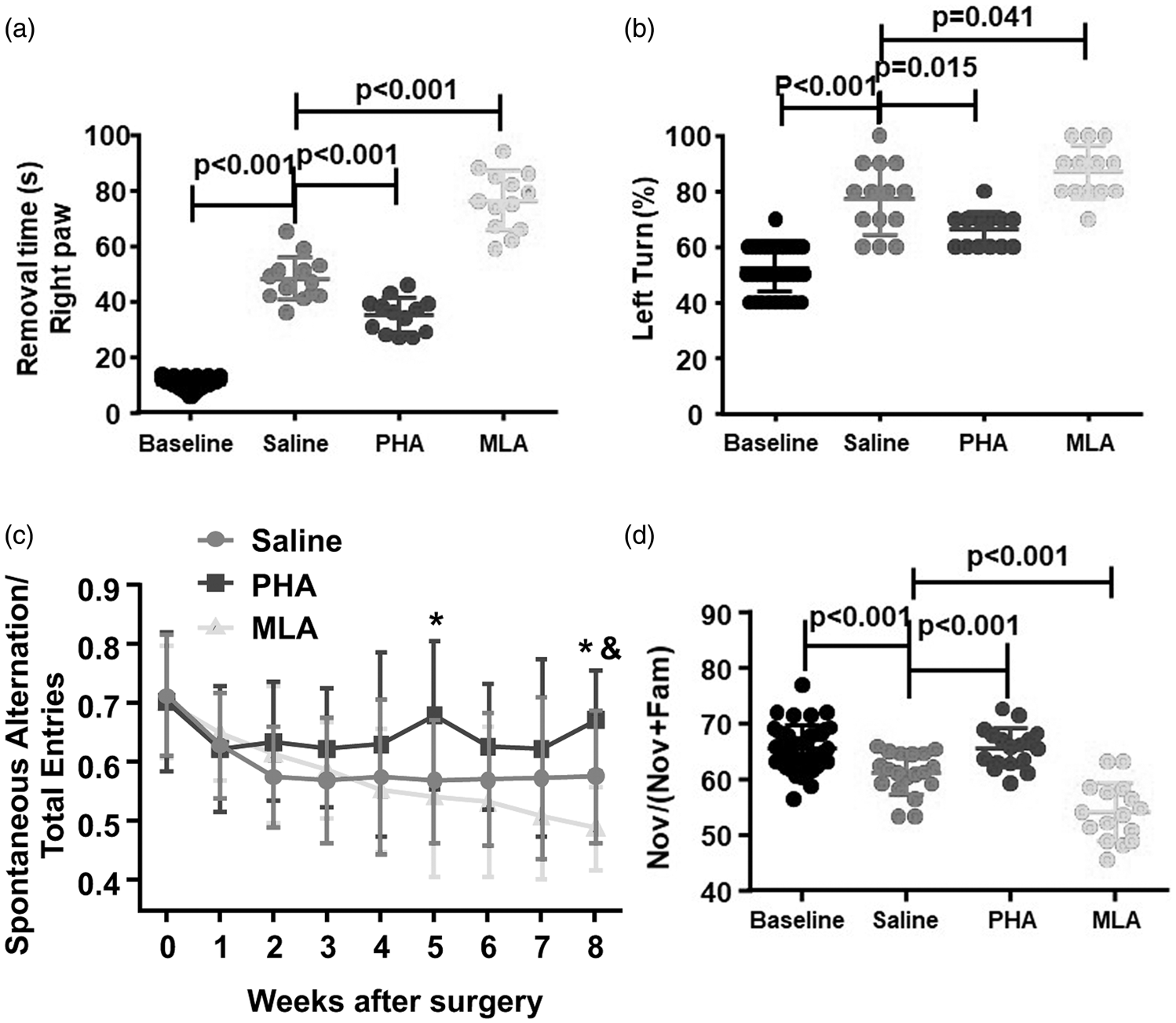
Reduction of inflammation improved sensorimotor and long-term memory function. (a) Quantification of the time mice used to remove adhesives from their right paws (contralateral to stroke side). s: Second. (b) Quantification of percentage of the left turns in the corner test. For (a and b), n = 40 (base line), n = 14 (saline treated group), n = 13 (PHA group), n = 13 (MLA group). (c) Quantification of spontaneous alternations in Y-maze test. * indicate that the differences between PHA and saline group are significant. & indicates the difference between MLA and Saline groups is significant. n = 24 (saline group), n = 22 (PHA group), n = 20 (MLA group). (d) Quantification of time that mice spent on the novel object in NOR tests. (NOV/(NOV+Fam): time spent on novel object (NOV)/time spent on novel and familiar object (Fam). n = 42 (baseline), n = 19 (Saline group), n = 17 (PHA group), n = 16 (MLA group).
To test the cognitive/memory function, Y-maze test was performed weekly and novel objective recognition (NOR) test was performed at 8 weeks after the surgeries. In Y-maze test, the saline group made fewer spontaneous alternations in the first two weeks after the surgeries than baseline level and the deficit lasted to the end of the experiment (8 weeks after the surgeries, Figure 3(c)). PHA treated mice made fewer spontaneous alternations than baseline level in the first week and maintained the level in the next seven weeks. MLA treated mice made fewer spontaneous alternations compared to the baseline in the first week and their performance decline continuously throughout the rest of weeks (Figure 3(c)). The differences between saline group and PHA group were significant in the fifth week (p = 0.014) and eighth week (p = 0.019) post-injuries. MLA treated group made few spontaneous alterations than saline group in the eighth week (p = 0.05). The differences between PHA treated and MLA treated groups were detected from the 5th–8th weeks. There were no differences among different treatment groups on the total entries at all time-points (Supplementary Figure IV(a)), indicating that motion abilities of mice in all groups were comparable. Male and female mice in all group performed similarly (Supplementary IVB). In NOR test, compared to baseline, saline group spent shorter time exploring the novel object (p < 0.001) after the surgeries. PHA group spent longer time exploring the novel object (p < 0.001), while the MLA group spent shorter time on the novel object (p < 0.001) than saline group (Figure 3(d)). There were no differences on running velocities and distances among different groups (Supplementary Figure V(a) and (b)). No gender difference (p = 0.55, Supplementary Figure V(c)) had been observed.
These data suggest that reduction of neuroinflammation through activation of α7-nAChR alleviates short-term sensorimotor deficits and long-lasting memory dysfunction of mice subjected to BF 6 h before pMCAO.
Reduction of neuroinflammation reduced infarct volume and the number of damaged neurons in the peri-infarct regions
To test if reduction of neuroinflammation by activation of α7-nAChR reduces neuronal damage, the infarct and atrophic volume were quantified on cresyl violet-stained brain sections collected 3 days and 8 weeks after the injuries. Compared to saline group, PHA group had smaller (p = 0.025), while MLA group had larger (p = 0.003) infarct volumes (Figure 4(a) and (b)). However, the atrophic volumes were similar among all groups (P = 0.1, Figure 4(c) and (d)). The possible explanation is that the atrophy volumes were very small at 8 weeks after the injuries, which is difficult to measure accurately.
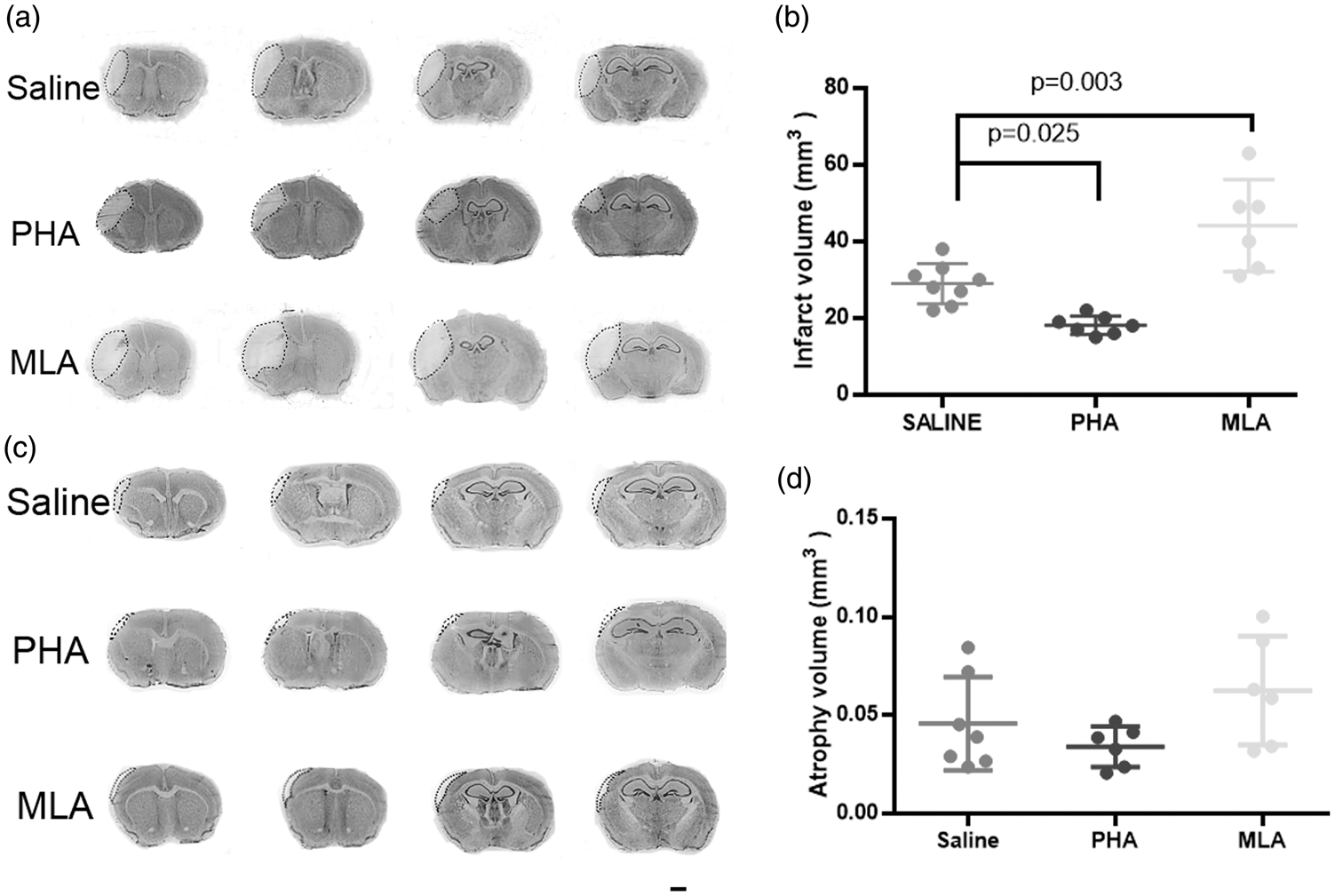
Reduction of neuroinflammation reduced infarct volumes. (a and c) Representative images of cresyl violet-stained brain sections collected three days (a) and eight weeks after BF and pMCAO. Scale bar = 1 mm. (b) Quantification of infarct volume. N = 8 (saline), n = 7 (PHA), and n = 6 (MLA) groups. (d) Quantification of atrophic volume. N = 7 (saline), n = 6 (PHA), n = 6 (MLA) groups. The atrophic volumes were similar among groups (p > 0.05).
The damaged neurons were also quantified on Fluoro-Jade C (damaged neurons) and anti-NeuN (stains the nuclei of neurons) antibody stained brain sections collected three days after the injures. Compared to saline group, PHA group had fewer Fluoro-Jade C+ neurons (p = 0.024), while MLA group had more Fluoro-Jade C+ neurons (p < 0.001). PHA group also had fewer Fluoro-Jade C+ neurons than MLA group (p < 0.001, Figure 5). Together, these data indicate that reduction of neuroinflammation through activation of α7-nAChRs reduces neuronal damage.
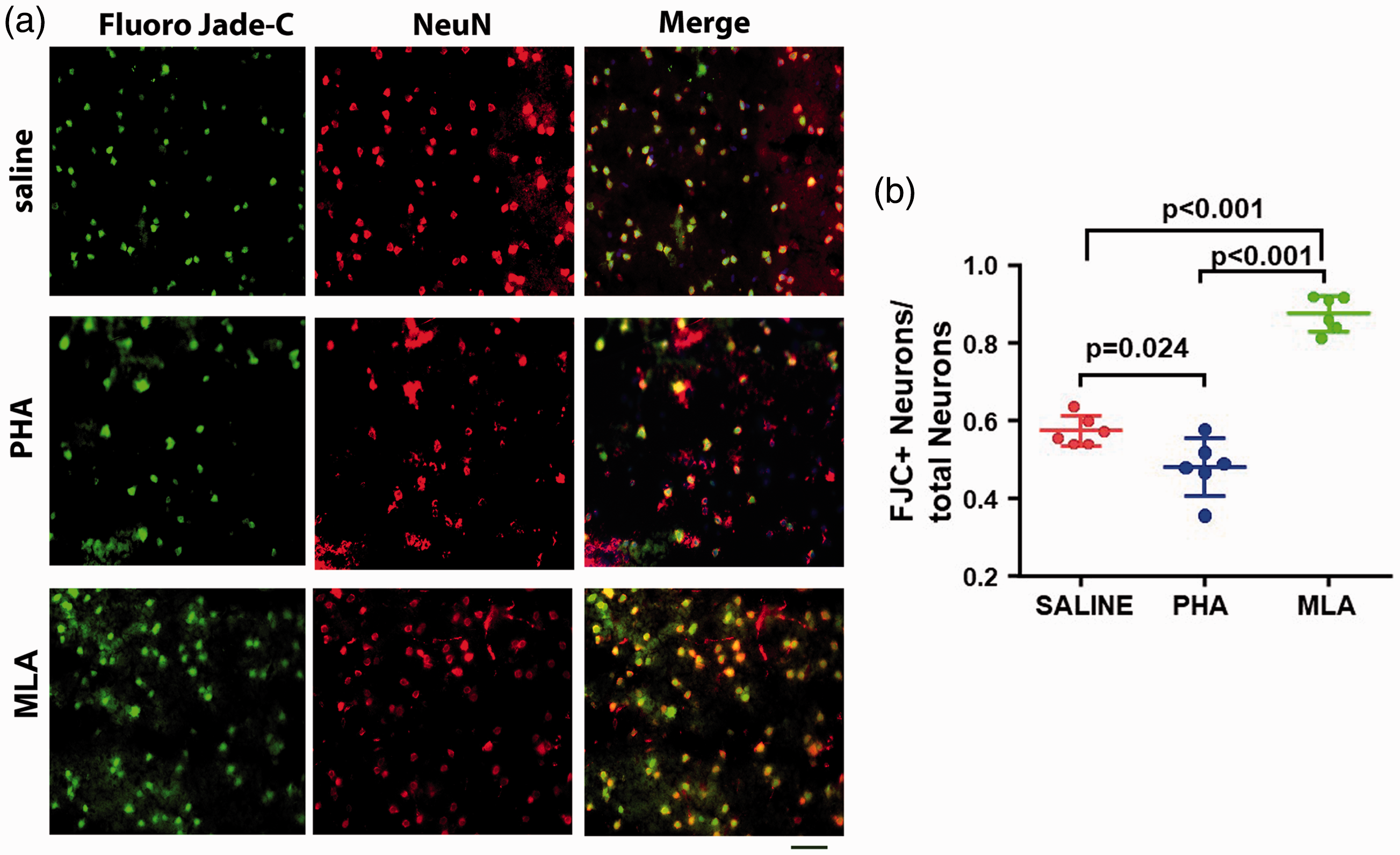
Reduction of neuroinflammation reduced Fluoro-Jade C positive neurons in the peri-infarct region. (a) Representative images of Fluoro-Jade C (green) and anti-NeuN antibody (red) stained sections. Scale bar= 50 µm. (b) Quantification of Fluoro-Jade C positive neurons. N = 6.
Reduction of neuroinflammation reduced white matter damage in the striatum
To assess white matter damage, we quantified the percentage of white matter bundle area in total of the striatum areas (MBP positive areas/total areas of striatum X100) 8 weeks after the injuries. On the ipsilateral side, PHA group had larger MBP positive area (p = 0.032) while MLA group had smaller area (p = 0.011) than saline group (Figure 6(a) and (b)). PHA group also had larger MBP positive area (p = 0.002) then MLA group on the ipsilateral side of stroke. These data suggest that reduction of neuroinflammation reduced white matter damage in the striatum ipsilateral of stroke injury.
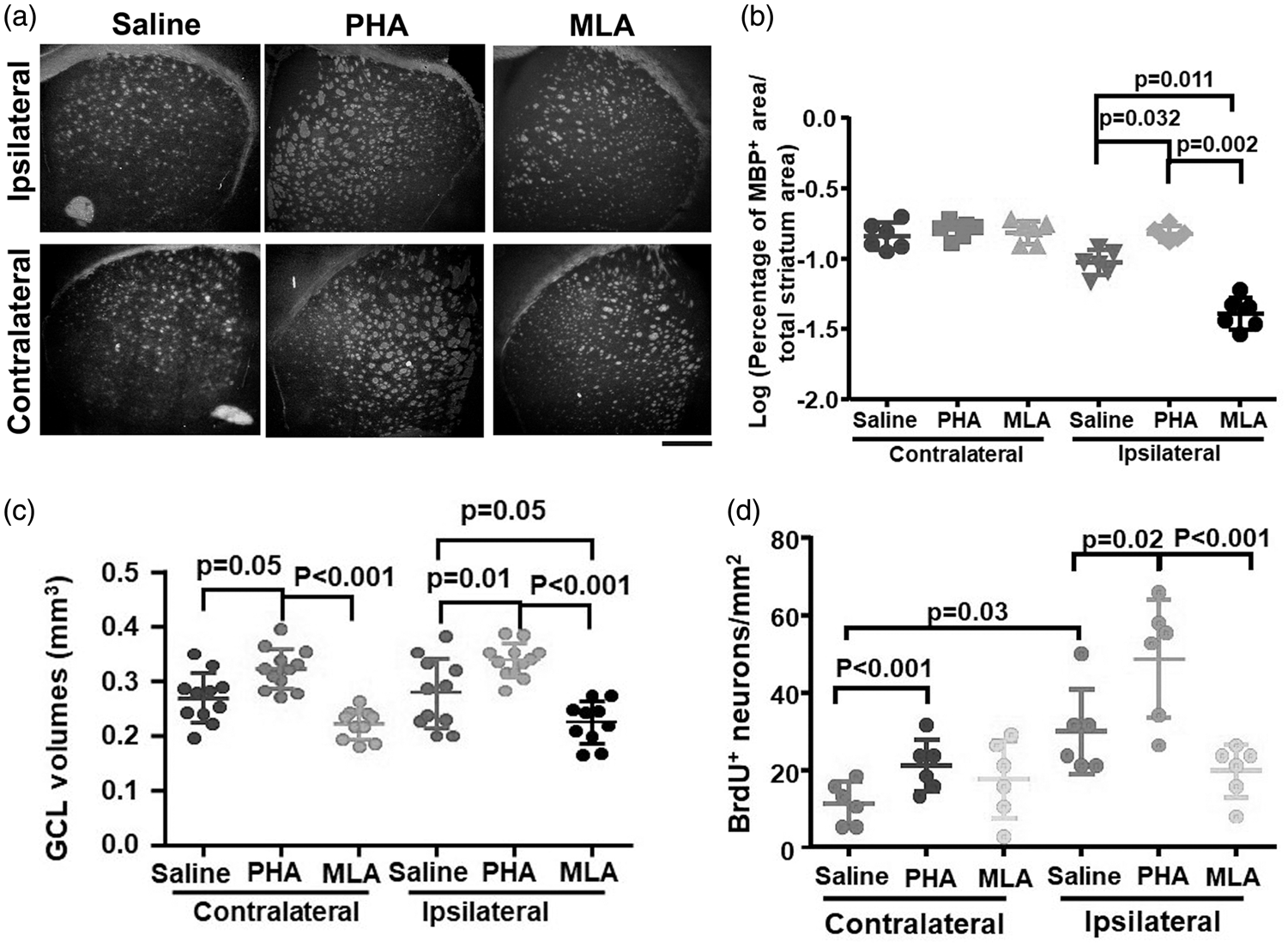
Reduction of neuroinflammation reduced white matter damage in the stratums, GCL atrophy and increased neuron proliferation in DGs. (a) Representative images of anti-MBP antibody stained brain sections. Scale bar: 500 µm. (b) Quantification of MBP+ areas in the stratums. N = 6. (c) Quantification of GCL volumes 8 weeks after the injuries. N = 11 for saline treated group, n = 12 for PHA treated group, and n = 10 for MLA treated group. (d) Quantification of BrdU+ neurons in DGs of mice received BrdU between 7–17 days after the injury. N = 6 for each treatment group.
Reduction of neuroinflammation reduced atrophy of hippocampal granule cell layer
We found previously that GCL volumes were reduced 8 weeks after BF and pMCAO. 9 To test if reduction of neuroinflammation by PHA treatment reduces GCL atrophy, we measured GCL volume 8 weeks after the injuries. We found that PHA group had larger GCL volumes on both contralateral (p = 0.05) and ipsilateral (p = 0.01) sides than saline group (Figure 6(c)). Conversely, MLA group had smaller GCL volumes on the ipsilateral side than saline group (p = 0.05) and smaller GCLs on both contralateral and ipsilateral sides than PHA group (p < 0.001, Figure 6(c)). The was no difference of GCL volumes between male and female mice in same groups (Supplementary Figure VI). These results suggest that reduction of neuroinflammation prevents the GCL atrophy after BF and stroke, which might be one of the mechanisms explaining why PHA treatment reduces long-term memory dysfunction in mice subjected to these injuries.
Reduction of neuroinflammation promotes neuron proliferation on the ipsilateral dentate gyrus between 7–17 days after BF and pMCAO
To investigate whether reduction of neuroinflammation increases neuron proliferation in DG, we treated mice with BrdU daily between 7–17 days and 46–56 days (last 10 days of the experiment) after the surgeries to label proliferating cells. The proliferating neurons were mostly detected in mice received BrdU between 7–17 days after the injuries. The saline group had more proliferating neurons in the ipsilateral side than the contralateral side of DGs (p = 0.03, Figure 6(d)). Compared to saline group, PHA group had more proliferating neurons in both contralateral (p < 0.001) and ipsilateral DGs (p = 0.02). MLA group and saline group had similar numbers of BrdU+ neurons on both sides of DGs (Figure 6(d)). PHA treated mice also had more BrdU+ neurons on the ipsilateral side than MLA treated mice (p < 0.001). There were few BrdU+ neurons in DGs in all mice injected with BrdU between 47–56 days. No difference was detected among groups (p = 0.09). These data suggest that reduction of neuroinflammation increased neuron proliferation in DGs in the subacute stage of BF and stroke injuries, which could be another underlying mechanism for its effect on improvement of memory/cognitive function of mice subjected to these injures.
Discussion
In this study, we demonstrated that reduction of neuroinflammation through activation of α7-nAChRs reduced the ischemic brain injury in the peri-infarct region and improved long-term memory function of mice subjected to BF 6 h before ischemic stroke. Reduction of neuroinflammation by PHA, an α7-nAChRs agonist, treatment reduced white matter injury in the striatum, and GCL atrophy, and increased neurogenesis in the DGs in the subacute stage of the injuries. All of these could contribute to its effect on the improvement of the post-stroke memory/cognitive function in mice.
Peripheral trauma, including bone fracture, can cause neuroinflammation and memory impairment.10,23 For the majority of patients, post-trauma or post-surgery neuroinflammation and memory dysfunction resolve promptly with no sequelae. However, for patients with some risk factors, such as metabolic syndrome, Alzheimer’s disease and aging, neuroinflammation can persist and cause persistent and even permanent memory dysfunction. 24 Memory dysfunction increases the risk of mortality. 25 Animal studies show that BF increases inflammatory cytokines in the blood, and inflammatory cytokines and macrophages in the hippocampus that is associated with a short-term (<7 days) memory dysfunction in young mice.10,26 Post-BF neuroinflammation and memory dysfunction are more severe in rats that have metabolic syndrome which can persist up to 3 months. 27 We noticed that unlike BF or stoke only mice, the memory dysfunction in young mice subjected to BF shortly before ischemic stroke lasted beyond 8-weeks. 9 Therefore, the augmented systemic or neuronal inflammation may cause long-term memory dysfunction in patients with bone fracture and stroke.
It has been found that the cholinergic anti-inflammatory pathway inhibits cytokine release through a mechanism that requires the α7 subunit-containing nAChRs. 28 Activation of the vagal cholinergic anti-inflammatory pathway attenuates neuroinflammation, protects against ischemic stroke-related cerebral damage,29,30 and has therapeutic benefits in patients with Alzheimer's 31 and Parkinson's diseases. 32 We showed previously that activation of α7-nAChR through PHA treatment reduced acute neuroinflammation, stroke injury, and sensorimotor dysfunction in mice subjected to ischemic stroke alone and mice subject to BF one day after ischemic stroke. 14 In this study, we found that activation α7-nAChR decreased acute neuroinflammation in the peri-infarct region and hippocampal region, reduced neuronal injury, and sensorimotor dysfunction in mice subjected to BF 6 h before ischemic stroke. In addition, we showed in this study that reduction of CD68+ positive cells in the hippocampal SLM regions reduced GCL atrophy, white matter injury in the striatum, and increased neurogenesis in DG in the subacute stage of BF and stroke. More importantly, reduction of neuroinflammation through activation of α7-nAChR alleviated the long-term memory dysfunction.
The damage tissue at the bone fracture site releases high mobility group box 1 (HMGB1). 33 By interaction with pattern recognition receptors on circulating bone marrow (BM)-derived monocytes, 33 HMGB1 induces the synthesis and release of pro-inflammatory cytokines into blood through upregulation of NF-κB 34 leading to systemic inflammation. Inflammatory cytokines in the blood disrupts blood brain barrier (BBB), allowing BM derived cells entering into the hippocampus. BBB breakdown can also cause leakage of blood contents, such as plasminogen and albumin into brain parenchyma, which activates microglia and astrocytes exacerbating neuroinflammation.35–39 The neuroinflammation can abrogate memory-enhancing synaptic plasticity. 40 Administration of an antibody to HMGB1 prevented the development of the post-BF memory dysfunction, 33 and reduced the negative impact of BF on ischemic stroke injury. 7 A recent study showed that microglia play an important role in forgetting memory through eliminating synapses. 41 Future studies are needed to analyze if increased number of active microglia/macrophages in the hippocampus leads to excessive synapses elimination, which in turn causes memory dysfunction. Nonetheless, our data indicate that neuroinflammation triggered by peripheral inflammation is a mechanism of memory dysfunction of mice subjected to BF and stroke. Inhibition of inflammation could alleviate long-term post-stroke memory dysfunction caused by BF.
It has been reported that WM damage 42 and ipsilateral hippocampal atrophy is associated with long-term memory dysfunction after ischemic stroke. 43 Our previous studies showed WM 44 and GCL atrophy 9 in mice 8 weeks after BF and stroke. In this study, we showed that inhibition of inflammation through PHA treatment reduced WM and GCL atrophy and increased neurogenesis in DG at the subacute stage of the injuries. Further studies are needed to unveil the mechanism of cholinergic anti-inflammatory pathway in preventing WM and GCL atrophy.
It is well known that inflammation has bi-physiological roles in determining the outcome of stroke.45–47 Simply inhibiting inflammation may not improve long-term outcomes of stroke patients. 48 Here, we showed that activation of α7-nAChR at the time of and soon after the injuries reduced BF and stroke induced neuroinflammation, brain injuries and improved long-term memory function. Therefore, the timing and dose of the treatment may influence the outcomes of anti-inflammation therapies in stroke patients.
Limitations of these study include, the HMGB1 levels in the blood were not measured. It is not clear if PHA treatment reduced HMGB1 levels in mouse blood. Our recent paper showed that BF enhanced BBB breakdown in the hippocampus. 49 We did not analyze if PHA treatment improved BBB integrity. B-lymphocytes have been shown to be involved in delayed cognitive dysfunction in stroke victims and in mice that have subjected to pMCAO followed with 1 h hypoxia treatment. 50 T cells invasion has also been found in the ischemic region in an ischemic reperfusion model for an extended period.51,52 Future study should analyze if T and B lymphocytes also contribute to the long-term memory dysfunction post BF and stroke. We should also analyze if BF exacerbates the damage of neural fiber and synapses and if PHA treatment reduces the damage in future.
Summary
Our research showed that reduction of neuroinflammation through activation of α7-nAChR reduced neuronal injury; improved sensorimotor function and long-term memory/cognitive function of mice subjected to BF shortly before stroke. Therefore, activation of α7-nAChR could be one of the potential therapies for improving the overall outcomes and long-term cognitive function of patients with BF and stroke.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X21996177 - Supplemental material for Reduction of neuroinflammation alleviated mouse post bone fracture and stroke memory dysfunction
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X21996177 for Reduction of neuroinflammation alleviated mouse post bone fracture and stroke memory dysfunction by Kang Huo, Meng Wei, Meng Zhang, Zhanqiang Wang, Peipei Pan, Sonali S Shaligram, Jinhao Huang, Leandro B Do Prado, Julia Wong and Hua Su in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Acknowledgements
We thank Tuanpeng Sun and Kaige Ma for helping on data analysis.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
KH and HS: participated in conception and design of the study; KH, MW, SS, MZ, ZW, PP, JH, LP and JW participated in acquisition and analysis of data, KH and HS drafted the manuscript.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.