
Abstract
Hemorrhagic stroke is a leading cause of death. The causes of intracerebral hemorrhage (ICH)-induced brain damage are thought to include lysis of red blood cells, hemin release and iron overload. These mechanisms, however, have not proven very amenable to therapeutic intervention, and so other mechanistic targets are being sought. Here we report that accumulation of endogenously formed zinc protoporphyrin (ZnPP) also critically contributes to ICH-induced brain damage. ICH caused a significant accumulation of ZnPP in brain tissue surrounding hematoma, as evidenced by fluorescence microscopy of ZnPP, and further confirmed by fluorescence spectroscopy and supercritical fluid chromatography-mass spectrometry. ZnPP formation was dependent upon both ICH-induced hypoxia and an increase in free zinc accumulation. Notably, inhibiting ferrochelatase, which catalyzes insertion of zinc into protoporphyrin, greatly decreased ICH-induced endogenous ZnPP generation. Moreover, a significant decrease in brain damage was observed upon ferrochelatase inhibition, suggesting that endogenous ZnPP contributes to the damage in ICH. Our findings reveal a novel mechanism of ICH-induced brain damage through ferrochelatase-mediated formation of ZnPP in ICH tissue. Since ferrochelatase can be readily inhibited by small molecules, such as protein kinase inhibitors, this may provide a promising new and druggable target for ICH therapy.
Introduction
Stroke is a leading cause of death in the world, and intracerebral hemorrhage (ICH) is the type of stroke with the greatest mortality rate. 1 However, so far, no effective therapy is available for ICH. 2 ICH leads to brain damage including blood brain barrier (BBB) disruption and infiltration of blood components into the brain parenchyma. 3 The lysis of red blood cells, which causes brain edema and neurological deficit, has been recognized as the primary cause of ICH-induced brain damage.4–6 Hemin has also been shown as a cause of brain damage by releasing redox-active iron, depleting NADPH and glutathione, producing free radicals and membrane lipid oxidation.6,7 In addition, hypoxia has been reported following ICH, 8 , 9 and following ischemia or hypoxia, zinc, one of the most abundant transition metals in brain tissue, is released from glutamatergic terminals into the brain’s extracellular space, causing brain damage.10,11
Zinc, essential for normal cellular development and survival, 12 is concentrated in synaptic vesicles in the central nervous system, and released from glutamatergic terminals after synaptic activation. 13 However, a pathological release of excess zinc is toxic. 14 Labile zinc accumulation in mitochondria is particularly important in ischemic stroke, and is directly linked to brain injury. 15 In conditions of low iron and high zinc levels, such as in anemia or lead poisoning, zinc can be mistakenly inserted into protoporphyrin by ferrochelatase instead of iron, forming zinc protoporphyrin (ZnPP) and not heme. 16 Since ferrochelatase is located in the mitochondria, 17 an excess of free zinc could lead to ZnPP formation, especially in hypoxic tissue where the heme biosynthetic pathway will be induced resulting in increased protoporphyrin and ferrochelatase.
Not only is endogenous ZnPP formation theoretically possible in ICH, but its administration has negative effects on several inflammatory conditions in stroke. 18 ZnPP is well-known as a potent inhibitor of heme oxygenase (HO), responsible for heme degradation,19,20 while HO is also known to be involved in neuroprotection. 21 Thus, ZnPP may be neurotoxic through the inhibition of HO. This study aimed to investigate whether ZnPP is endogenously formed following ICH and whether its generation contributes to ICH-induced brain damage.
Material and methods
Rat model of collagenase-induced intracerebral hemorrhage
The Laboratory Animal Care and Use Committee of UNM approved all experimental protocols. The animals were used in compliance with the NIH Guide for Care and Use of Laboratory Animals. The animal data reporting of the current study has followed the Animal Research: Reporting in Vivo Experiments (ARRIVE) guidelines. 22 Male Sprague Dawley rats (Charles River Laboratories) weighing 290–320 g (age 10–12 weeks) were anesthetized with isoflurane (5% for induction, 2% for maintenance) in N2/O2 (70:30%) during surgical procedures, and the body temperature was maintained at 37.5 ± 0.5 °C using a heating pad. To induce an ICH, a small burr hole was made at 3.5 mm right of the bregma. 0.2 U collagenase (Sigma, USA) in 1 µl sterile saline was injected to 6.0 mm deep through the burr hole. 23 The needle was kept in its position for 5 min after collagenase injection to prevent backflow. 44 rats were used in this study. The success of the ICH models was verified by magnetic resonance imaging (MRI). All animal ICH model was successful; thus, no animal was excluded from analysis.
Mouse model of collagenase-induced intracerebral hemorrhage
Male C57BL/6 (Charles River Laboratories) weighing 16–20 g were anesthetized with isoflurane (5% for induction, 1.5% for maintenance) in N2/O2 (70:30%) during surgical procedures, and the body temperature was maintained at 37.5 ± 0.5 °C. 0.075 U collagenase (Sigma, USA) in 0.5 µl sterile saline was injected to 3.7 mm deep through the burr hole at 2 mm right of the bregma. 24 10 mice were used in this study.
Mouse model of blood injection intracerebral hemorrhage
Male C57BL/6 (Charles River Laboratories) weighing 16-20 g were anesthetized as described in collagenase ICH model. 30 µl tail blood without anticoagulant was injected at a speed of 2 µl/min to 3.7 mm deep through the burr hole at 2 mm right of the bregma. The needle was kept for 10 min after injection to prevent backflow.24,25 25 mice were used in this study.
Magnetic resonance imaging
The success of the ICH model and brain damage (including hematoma-caused hypointense and perihematomal brain damage-caused hyperintense 26 ) was estimated by T2-weighted images using a 4.7-Tesla MRI scanner (Bruker BioSpec), which was equipped with a 40-cm bore, a 660 mT/m (rise time within 120 µm) gradient and shim systems (Bruker Biospin MRI, USA). Animals were anaesthetized with 2% isoflurane during MRI. The body temperature was maintained at 37.0 ± 0.5 °C. T2-weighted images were acquired with a fast spin-echo sequence (rapid acquisition with relaxation enhancement (RARE)) (Repetition Time (TR)/Echo Time (TE) = 5,000 ms/56 ms, Field of View (FOV) = 40 mm × 40 mm, slice thickness = 1 mm, inter-slice distance =1.1 mm, number of slices =12, matrix = 256 × 256, number of average = 3). T2* weighted images for detecting hematoma were acquired using FLASH sequence with the following parameters: TR/TE = 250/5 ms, filp angle = 30 degree, matrix: 200×200, FOV: 20 mm×20 mm, slice thickness = 1 mm, imaging averages = 9, to increase signal to noise ratio. The operator was blinded to the experimental conditions.
N,N,Nʹ,Nʹ-tetrakis-(2-pyridylmethyl)ethylenediamine administration
N,N,N’,N’-tetrakis-(2-pyridylmethyl) ethylenediamine (TPEN) (Sigma, USA), a specific zinc chelator, was dissolved in dimethyl sulfoxide (DMSO) to 25 mg/ml and then further diluted in physiological saline to a final concentration of 2.5 mg/ml. 10 mg/kg TPEN was intraperitoneally injected at 1 h before collagenase injection or at 30 min after blood injection. Saline with 10% DMSO was used as control.
N-methyl protoporphyrin IX administration
N-methyl protoporphyrin IX (NMPP) (Santa Cruz, USA), a ferrochelatase inhibitor was dissolved in DMSO to 200 mg/ml and then further diluted in physiological saline to a final concentration of 20 mg/ml. 100 mg/kg NMPP was intraperitoneally injected at 1 h before collagenase injection or at 30 m after blood injection. Saline with 10% DMSO was used as control.
Behavioral tests
Three types of behavioral functional tests were performed. The observer was blinded to the experimental conditions. (1) Corner test was performed as described.24,27 The number of right turns in 10 total turns were calculated. (2) Foot-fault test was performed as described. 27 The number of contralateral forelimb foot faults made by traversing the grid surface was calculated. (3) 14 points Neurological Severity Scores (mNSS) was performed as described. 28
Rat model of focal cerebral ischemia and zinc protoporphyrin treatment
Middle cerebral artery occlusion (MCAO) surgery was used to induce focal cerebral ischemia through intraluminal filament methods as we described previously. 29 The animals underwent right MCAO for 24 h. ZnPP (10 µg/kg) in 5 µl saline or 5 µl saline was injected to 6.0 mm deep at 3.5 mm right of the bregma through a burr hole on normal or MCAO rat, 10 m after MCAO onset. 30 rats were used in this study.
Tissue processing
Animals were transcardially perfused under anesthesia with cold PBS and then 4% paraformaldehyde 24 h after collagenase injection. The brain was removed and fixed in 4% paraformaldehyde, and was placed in 20% sucrose solution. After incubation in sucrose, the brains were embedded in optimum cutting temperature (O.C.T.) compound for cryosectioning. Brain cryosections (16-µm-thick) were prepared for ZnPP, neuron, astrocytes and cell death measurement.
Staining for labile zinc
Brain cryosections were stained with the zinc-specific membrane-permeable fluorescent dye Fluozin-3 (Life Technologies, USA). The cryosections were washed in PBS and incubated with Fluozin-3 (5 µmol/L) for 15 m at room temperature. After a thorough washing in PBS, images were acquired by a fluorescence microscope (Olympus IX71) with a GFP dichroic mirror through the Olympus Cellsens software.
Zinc protoporphyrin measurement by fluorescence microscopy and spectroscopy
To detect ZnPP, 24 h after collagenase injection, the auto-fluorescence of ZnPP (excitation = 420 nm, emission = 588 nm) in brain cryosections were visualized using a fluorescence microscope (Olympus IX83) with customized ZnPP dichroic mirror. Additionally, to examine the brain by fluorescence spectroscopy, brains were harvested and cut into 2-mm thick coronal slices with a rat brain coronal matrice (Braintree Scientific, USA). The tissue slices 4–10 mm away from the tip of the frontal lobe, which contained visible blood in the hemorrhagic hemisphere, was collected. Then hemorrhagic and contralateral hemisphere was homogenized on ice in ethanol, separately. After centrifugation at 13,000×g, the fluorescence intensity (excitation = 420 nm, emission = 588 nm) was determined using a SpectraMax M2 Multi-Mode Microplate Readers (Molecular Devices, USA). 30 Furthermore, blood was also drawn 24 h after collagenase injection. Following treatment with heparin, 50 µl blood was diluted in 1 ml ethanol, centrifuged, and the fluorescence intensity of ZnPP in the ethanol extract supernatant was measured.
Hemorrhage measurement by fluorescence microscopy
To visualize the hemorrhage in brain tissue, erythrocytes were stained by incubating cryosections (16-µm-thick) with 0.2% (W/V) NaBH4 in PBS for 15 min. 31 After a 5-min rinsing in PBS, the section was mounted in ProLong™ Gold Antifade Mountant with DAPI (ThermoFisher Scientific, USA). Images were acquired by a fluorescence microscope (Olympus IX71) with TRICT (Hemorrhage, NaBH4) and DAPI (nucleus) dichroic mirrors through the Olympus Cellsens software.
Detection of zinc protoporphyrin in brain tissue by supercritical fluid chromatography-mass spectrometry
To detect ZnPP by supercritical fluid chromatography-mass spectrometry (SFC-MS), brains were harvested 24 hrs after collagenase injection. The hemorrhagic hemisphere brain tissue within 4–10 mm from the tip of the frontal lobe was homogenized on ice in ethanol, then centrifuged at 13,000 g for supernatant. The brain with saline injection was used as control. SFC-MS analyses were performed with Acquity UPC2 Ultra-Performance Liquid Chromatograph coupled with a single quadrupole-mass detector (SQD) from Waters Corporation, USA. The LC-MS system was controlled by MassLynx Version 4.1 software. SFC condition: Acquity UPC2 HSS C18 column (2.1 × 100 mm, 1.8 µm) at 40 °C. Mobile phase A: CO2 in liquid state. Mobile phase B: methanol. Gradient: 0.00 min: A/B (100/0); 2.00 min: A/B (75/25); 5.90 min: A/B (75/25); 5.91 min: A/B (100/0). Flow rate was at 2.00 mL/min of the mobile phase were set at 1.75 mL/min. The right hemisphere of a saline injected rat brain was used as control. ZnPP were detected in positive electrospray ionization mode (ESI+). The ion source temperature was 150 °C. N2 was used as the desolvation gas at a flow rate of 500 L/h at 350 °C. Voltages of the capillary and the cone were 3 kV and 45 V, respectively. MS detection was in the m/z range of 300 to 700. The ZnPP protonated ion was observed at m/z of 626.0. Identification of ZnPP was made on the basis of its matching peak with standard ZnPP. Three rats in each group were used in the experiments.
Measurement of cerebral pO2 by electron paramagnetic resonance
For measurement of local cerebral pO2 in the anesthetized rats before and 24 h after injection of collagenase, electron paramagnetic resonance (EPR) oximetry was conducted according to our published methods.32–35 Under anesthesia, a pin hole on the parietal skull was made at 4.5 mm right of the bregma. A lithium phthalocyanine (LiPc) crystal (approximate diameter 0.2 mm) was placed at a depth of 6 mm using a microdialysis guide cannula (CMA microdialysis, Stockholm, Sweden). The location of LiPc implanting was chosen based on our experience, at the edge of ICH-enveloping rim. The rats were allowed to recover from implantation 72 h before EPR measurement using a Bruker EleXsys E540 EPR spectrometer equipped with an L-band bridge (Bruker Instruments, USA). Three 36 rats were measured in the experiments and all of them were successfully implanted with LiPc within 1 mm from the edge of ICH-enveloping rim.
Double staining of dead cell and neuron or astrocytes
A standard terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling (TUNEL) procedure for cryosection was performed to stain the dead cell by Click-iT TUNEL Alexa Fluor 594 Imaging Assay kit (Thermo Fisher Scientific, USA). To visualize neuron or astrocytes, NeuroTrace 500/525 Green Fluorescent Nissl stain (Thermo Fisher Scientific, USA) or GFAP Monoclonal antibody-Alex 488 (Thermo Fisher Scientific, USA) was used. Histological images were captured with FITC or TRITC dichroic mirror by a fluorescence microscope (Olympus IX71) via the Olympus Cellsens software.
Hypoxia treatment of blood
Blood was drawn from normal Sprague Dawley rats. 50 U/ml heparin was used to treat the blood for anticoagulation. Then the blood was subjected to hypoxia by incubating in a humidified airtight chamber (Billups-Rothberg Inc., USA) equipped with an air lock and flushed with 95% N2/5% CO2 for 15 min. The chamber was sealed and kept at 37 °C for 3 h.
Primary culture of rat astrocytes
Primary astrocytes were isolated from the cortices of day 1 Sprague-Dawley rats as previously described. 37 Briefly, the brains of postnatal day 1 rats were excised, and meninges and blood vessels were removed. The forebrains were placed in DMEM (Life Technologies, USA). The tissue was minced and placed in 0.05% trypsin (Life Technologies, USA) at 37°C for 30 min. Trypsinization was completed by adding 10% (v/v) FBS (Life Technologies, USA) in DMEM. After dissociating tissue and passing through a 40 µmol/L strainer, the cells were seeded in the flasks at a density of 1.5 × 103 cells/mm2 in growth medium (90% Dulbecco’s Modified Eagle medium containing 4.5 g/L glucose, and 10% Fetal Bovine Serum) and placed in the cell culture incubator at 37°C with 95% air/5% CO2.
Primary culture of rat neuron
Primary neuron were isolated from the cortices of embryonic day 17 Sprague-Dawley rat’s brain as described previously. 38 Briefly, the brains were collected and placed in Hank’s balanced salt solution (HBSS). Following the removal of meninges and blood vessels, forebrains were transferred to HBSS with 0.6% (w/v) trypsin for 30 min. After rinsed in HBSS for twice, the dissociated cells were passed through a 40 µmol/L strainer and then placed into 96-well microtiter plates with glutamine free Dulbecco's Modified Eagle Medium (DMEM)-Eagle's salts (Life Technologies, USA), supplemented with Ham's F12 (Life Technologies, USA), 10% heat-inactivated fetal bovine serum (FBS) (Life Technologies, USA), and 200 IU/ml penicillin/streptomycin (Sigma, USA), at a density of 5 × 103 cells/well.
Cytotoxicity assay
Neuron or astrocytes (5 × 103 cells/well) were seeded into 96-well microtiter plates. Following 3 hrs normoxia or hypoxia treatment, the death rate were measured using Cytotox 96 nonradioactive cytotoxicity assay kit (Promega).
Experimental design and statistical analysis
Collagenase and blood-induced ICH in rats and mice have predominately been used as model systems for ICH.39–46 In particular, studies have shown that the gender does not affect hemorrhage evolution. 47 Therefore, male rats and mice were used in this study to test our hypothesis and make direct comparisons to literatures. We first investigated zinc accumulation following ICH. Then, zinc chelator TPEN was used to investigate the role of zinc in ICH-induced brain damage. Autofluorescence, fluorescence spectrophotometric and SFC-MS analysis were used to demonstrate the generation of endogenous ZnPP. Ferrochelatase inhibitor NMPP was used to investigate the generation mechanism and the toxicity of endogenous ZnPP under ICH. The Shapiro-Wilk test was used to test the normality of all data sets. The Student’s t-test or ANOVA was used to analyze the differences in means from groups (listed in each figure legend). A value of P < 0.05 was considered statistically significant.
Results
Zinc accumulated around the hemorrhagic area at 24 h following intracerebral hemorrhage induction
We recently reported that ischemic stroke causes excess zinc release, leading to brain damage.29,48 Hypoxia, the cardinal feature of ischemic stroke, also appears during hemorrhage. 9 Thus, we hypothesized that zinc would likely be released in ICH. To verify this, an ICH rat model using collagenase intracerebral injections was utilized. Since the size of the blood clot impacts survival and the degree of neurological disability and enlargement of the blood clot occurs in the first 24 hours, 49 we selected 24 hours after ICH onset as the time point in our experiments. At this time, the brain was collected and a zinc-specific fluorescence probe, Fluozin-3, was used to visualize the labile zinc accumulation in brain tissue. As expected, there was a minimal fluorescence signal in the non-hemorrhagic hemisphere while a dramatically increased signal was observed around ICH-enveloping rim (Figure 1(a)). To confirm that the Fluozin-3 signal was caused by zinc accumulation, the zinc specific chelator TPEN was i.p. injected 1 hr before ICH induction. Chelating zinc significantly reduced the Fluozin-3 fluorescence signal (within 200 µm from the edge of ICH-caused signal dephasing area was analyzed, Figure 1(a)). These results demonstrate that labile zinc accumulates at the edge of the hemorrhagic area after ICH.
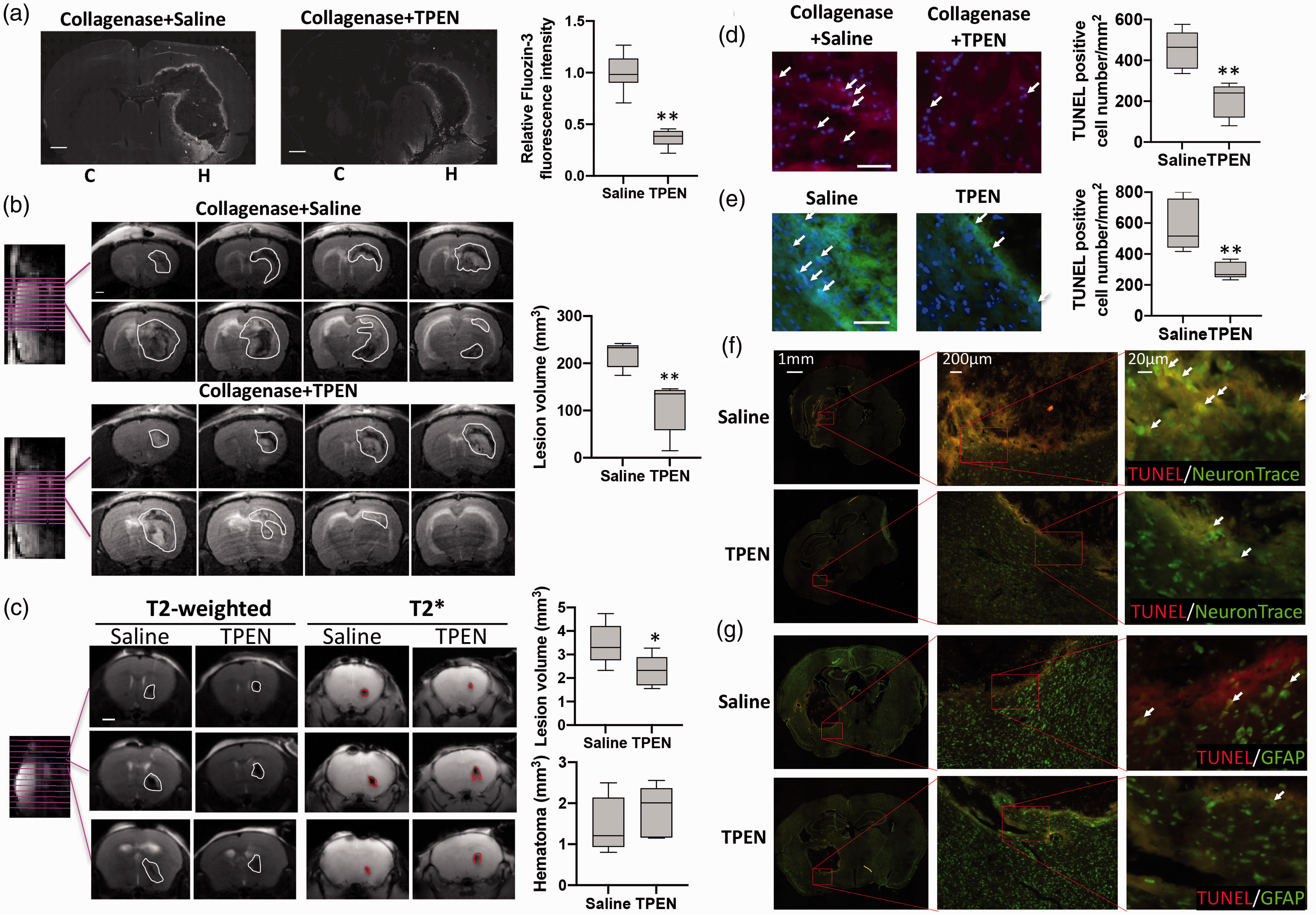
Hemorrhage resulted in zinc accumulation, which was associated with brain damage. (a) Labile zinc in brain slices of collagenase-induced ICH rat was detected by fluorescence probe Fluozin-3. “Relative Fluozin-3 fluorescence intensity” indicates the fluorescence intensity at the area within 200 µm from the edge of ICH in each brain slice normalized to the average value of that at the non-ICH hemisphere of saline group. 5 animals in each group, one slice from each animal, and four points on each slice were included in the intensity analysis. C: contralateral hemisphere; H: hemorrhagic hemisphere. (b) Collagenase induced brain injury was assessed by T2-weighted MRI at 24 h after ICH onset. White line circles the brain damage (including hematoma caused hypointense and perihematomal brain damage caused hyperintense). Scale bar: 1 mm. Box plot shows the total lesion volume calculated from T2-weighted MRI image. n = 5. (c) blood injection-caused brain injury and hematoma were assessed by t2-weighted and t2* mri at 24 h after ich onset. white line circles the brain damage and red line circles hematoma. scale bar: 1 mm. box plot shows the total lesion volume and hematoma volume calculated from t2-weighted and t2* mri image. n = 5. cell death in the collagenase- (d, red) or blood injection- (e, green) induced hemorrhagic brain was measured by TUNEL assay kit. Arrow indicates TUNEL positive cells. Scale bar: 50 µm. Box plot shows the density of TUNEL positive cell at the area within 200 µm from the edge of collagenase-induced ICH in rat brain slices
Zinc accumulation is associated with brain damage after hemorrhagic stroke
To evaluate the effect of zinc accumulation on brain damage, T2-weighted MRI was utilized. MRI showed the total brain damage area as hypointense (hematoma) and hyperintense (perihematomal brain damage) 26 lesion (Figure 1(b), circled area) in the hemorrhagic hemisphere 24 hrs after ICH. Notably, the brain damage was significantly reduced by chelation of zinc using TPEN, confirming that elevated free zinc is involved in ICH-induced brain damage. To further verify the detrimental role of zinc accumulation on the brain cells, we measured brain cell death in the brain cryosections using TUNEL assay. As shown in Figure 1(d), the number of TUNEL positive cells within 200 µm from the edge of ICH-caused signal dephasing area in rats pre-treated with TPEN was about half of those in saline control group, indicating that zinc is involved in ICH-induced brain damage. Moreover, to eliminate the possibility that the reduction of brain damage in TPEN pre-treated ICH brain was caused by the interruption of collagenase-induced hematoma through zinc chelation, autologous blood injection ICH mice model was also used. TPEN was i.p. injected at 30 min after blood injection. MRI results shown that post-treatment of TPEN hardly changed the hematoma volume (Figure 1(c), T2*), while it significantly decreased total brain damage at 24 h after ICH onset (Figure 1(c), T2-weighted). By staining the dead cell on brain sections (collected at 24 h after ICH onset), a noticeable decrease of cell death number (within 100 µm from the edge of ICH-caused signal dephasing area) was found in TPEN post-treated ICH brain (Figure 1(e)), indicating that zinc is involved in cell death and brain damage in ICH. Furthermore, NeuronTrace (for neuron) and GFAP (for astrocytes) were co-stained with TUNEL, respectively. The co-staining experiments showed that TUNEL-positive cells were largely co-localized with NeuroTrace-positive cells (Figure 1(f)), while only a few co-localized with GFAP-positive cells (Figure 1(g)). These results indicate that neuron is the major cell type damaged by zinc in ICH.
Zinc protoporphyrin is generated following hemorrhagic stroke
To substantiate our hypothesis on ZnPP formation in ICH, we imaged ZnPP levels in the hemorrhagic rat brain at 24 hrs after collagenase injection using fluorescence microscopy. 50 We found minimal ZnPP fluorescence in the non-hemorrhagic hemisphere, while there was intense signal of ZnPP observed around the ICH-enveloping rim (Figure 2(a)). Noticeably, the ZnPP signal around the hemorrhagic area (Figure 2(a), the slices from the same animal’s brain in Figure 1(a)) was strongly co-localized with the increase in free zinc (Figure 1(a)). Pretreatment of the animal with zinc chelator TPEN significantly decreased ZnPP fluorescence (Figure 2(a)), demonstrating that ZnPP generation co-localizes with increased free zinc. Similar results were also observed in blood injection ICH mice with TPEN injection at 30 min after ICH onset (Figure 2(b)). These results with different ICH models confirm that zinc takes an important part in the formation of ZnPP.
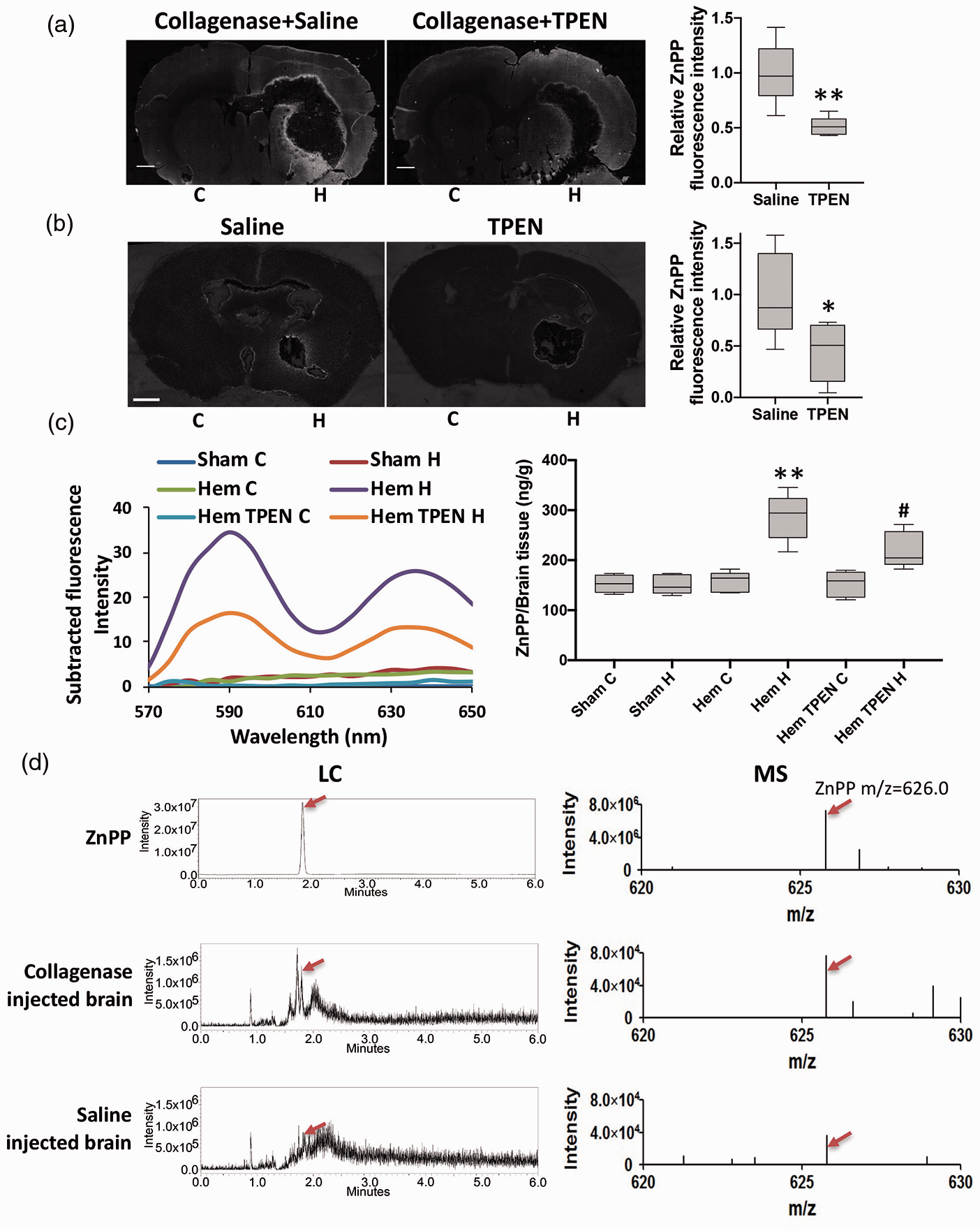
Endogenous ZnPP was generated following hemorrhagic stroke. ZnPP in the collagenase- (a) and blood injection- (b) induced ICH brain slices was imaged by recording its autofluorescence. Scale bar: 1 mm. Relative ZnPP fluorescence intensity is the fluorescence intensity at the area within 200 µm from the edge of collagenase- or within 100 µm from the edge of blood injection-caused signal dephasing area in each brain slice normalized to the average value of that at the non-ICH hemisphere of saline group. The plot's horizontal line represents the median; the box encompasses the 25th to 75th percentiles; and the error bars encompass the 10th to 90th percentiles. n = 5 (4 point/slice; 1 slice/rat; 5 rats/group). **P < 0.01, student’s t-test. (c) ZnPP fluorescence spectrum from each hemisphere of rats was measured by a fluorescence spectrophotometer (Sham: hemisphere tissue from saline injected rats; Hem: hemisphere tissue from ICH rats; Hem TPEN: TPEN pretreated ICH rats; C: contralateral hemisphere; H: hemorrhagic hemisphere). Subtracted fluorescence intensity was calculated by the fluorescence intensity of each group subtracting that of the contralateral hemisphere in sham rats. Quantification of ZnPP was calculated according to ZnPP standard curve using fluorescence intensity at 588 nm. n = 5. **P < 0.01, versus Sham H, #P < 0.05, versus Hem H, student’s t-test. (d) SFC-MS scanning showed the supercritical fluid chromatograph (LC) and mass spectroscopic (MS) profile of commercial pure ZnPP, collagenase or saline injected brain (n = 3). Peak identity (red arrow) was assigned by retention time and chromatographic pattern of authentic standard.
To validate chemical identity of the fluorescent species imaged as ZnPP (and not another fluorescent product), we extracted each hemorrhagic hemisphere tissue and recorded the fluorescence emission spectrum. The intensity of the spectrum with characteristic 588 and 636 nm maxima of ZnPP were increased in the hemisphere of hemorrhagic brain and TPEN treatment significantly lessened the increase of ZnPP (Figure 2(c)). Final confirmation of ZnPP formation in ICH was obtained by SFC-MS spectroscopy of brain extracts. Authentic ZnPP standard gave a peak at m/z 626.0 in MS and retention time 1.85 min on LC, and the same signal was observed in extracts of hemorrhagic and control brains (Figure 2(d)). ZnPP signals in hemorrhagic brains had higher intensities in both LC and MS against control brains. These results provide the direct evidence for the formation of ZnPP in the brain parenchyma following ICH.
Cerebral tissue hypoxia following ICH
In addition to excess free zinc, we speculated that hypoxia might be another important condition in driving ZnPP formation in ICH, since hypoxia could initiate an increase in the heme biosynthetic pathway. However, there is limited experimental evidence for decreased brain tissue oxygenation after ICH, 9 and none in our ICH model. To investigate whether hypoxia is present at the ICH-enveloping rim following ICH, brain tissue partial pressure of oxygen (pO2) levels in the hemorrhagic rats were measured by in vivo EPR oximetry. 35 We found that brain tissue pO2 level at the edge of hemorrhagic area, where the EPR oximetry LiPc probe was implanted (Figure 3(a), arrow) and where high concentrations of ZnPP were detected (Figure 2(a)), was decreased from 33.4 ± 3.1 mmHg before collagenase injection to 22.7 ± 1.7 mmHg at 24 hrs after collagenase injection (Figure 3(a)). Since the brain is known to upregulate erythropoietin expression (and thus heme biosynthesis) by hypoxia, 51 lowered pO2 could drive heme synthesis, and thus increasing protoporphyrin and ferrochelatase levels.
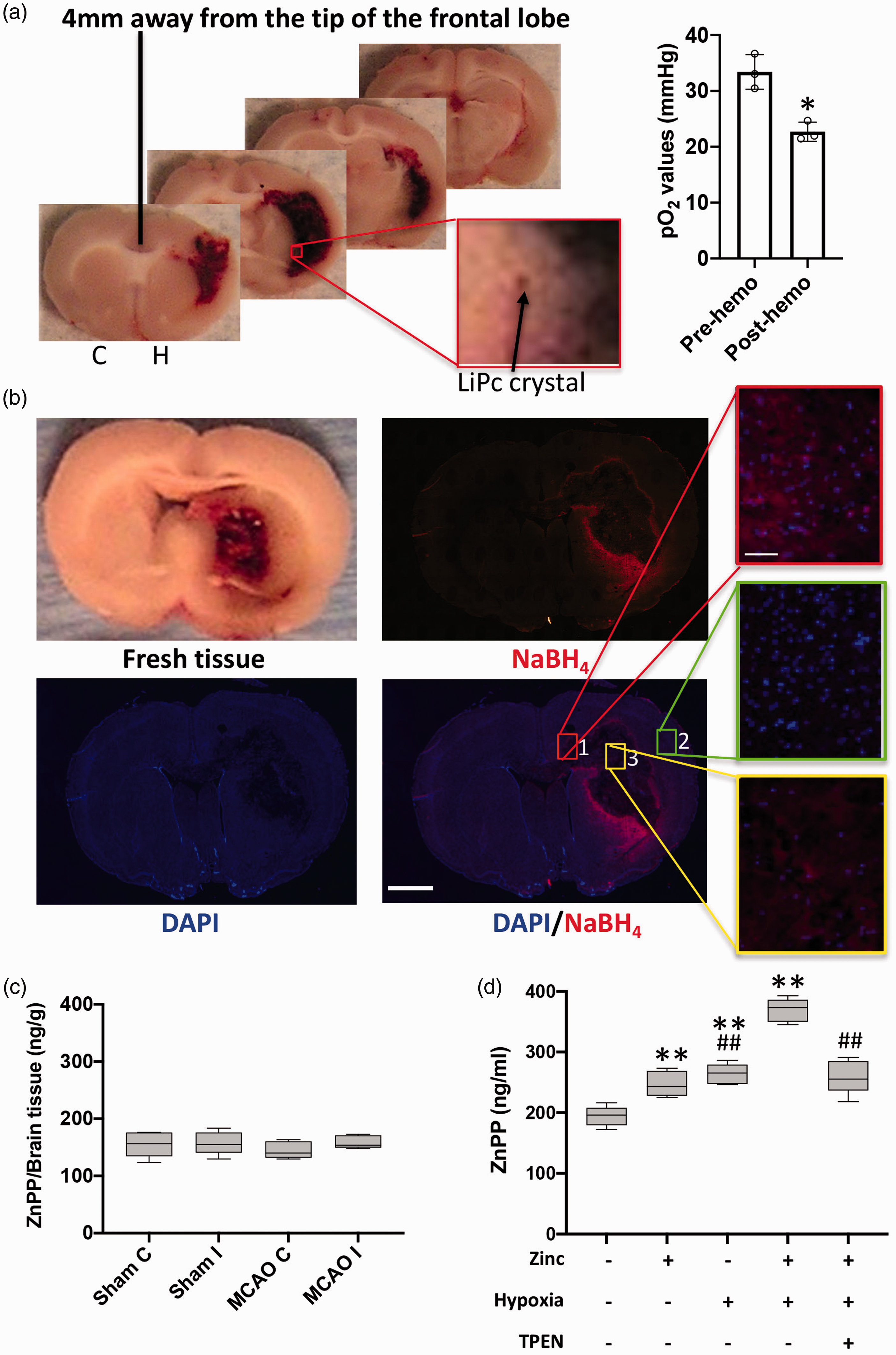
Hypoxia, zinc and blood are required for ZnPP generation following ICH. (a) EPR oximetry probe LiPc crystal (black arrow) was implanted in brain before collagenase injection (c: contralateral hemisphere; H: hemorrhagic hemisphere). Tissue oxygen level before (Pre-hemo) and 24 hrs after (Post-hemo) collagenase injection was measured by in vivo electron paramagnetic resonance oximetry (n = 3). Dot plot shows the tissue oxygen level of the site where the LiPc crystal was implanted in each brain. Data are presented as mean±SD. *P < 0.05, student’s t-test. (b) Rat brain was collected 24 hour after collagenase injection. A 2-mm-thick fresh tissue slice (6 mm away from tip of frontal lobe) shows hemorrhage. Using 16-µm-thick cryosections, hemorrhage was visualized by NaBH4 treatment (red), while brain cells was stain by DAPI (blue). Scale bar for whole brain is 2 mm. Three insets represent selected areas (1, 2, and 3) from the DAPI and NaBH4 merged image. Scale bar is 30 µm. (c) ZnPP fluorescence in each hemisphere was measured by fluorescence spectrophotometer (Sham: sham rat tissue; MCAO: MCAO rat tissue; C: contralateral hemisphere; I: Ischemic hemisphere). n = 5. (d) ZnPP fluorescence intensity in whole blood was measured by fluorescence spectrophotometer. Quantification of ZnPP was calculated according to ZnPP standard curve of fluorescence intensity at 588 nm (n = 5). The plot's horizontal line represents the median; the box encompasses the 25th to 75th percentiles; and the error bars encompass the 10th to 90th percentiles. **P < 0.01, versus normoxia only, ##P < 0.01, versus hypoxia plus zinc, student’s t-test.
Blood, zinc and hypoxia are required for zinc protoporphyrin formation
We observed in Figure 2(a) that ZnPP was highly concentrated around the ICH-enveloped rim area, whereas the areas in the center of hemorrhage and outside of the ICH-enveloped rim had much lower ZnPP level. On a fresh tissue slice (Figure 3(b)), the center of hemorrhagic area was fully filled with blood clot, while blood infiltration to the tissue decreased rapidly with increasing distance from the blood clot. These observations suggest that ZnPP generation may also be related to blood distribution in the tissue. To test this postulation, we utilized NaBH4 to visualize the erythrocytes
31
and DAPI to stain the cell nucleus. Three areas were selected for detailed examination:
To provide additional support for the hypothesis that blood (hemorrhage), hypoxia and zinc are required for ZnPP formation, whole blood was used as an in vitro model system. Although most cells in blood are enucleated mature erythrocytes, there are a small portion of nucleated cells (such as white cells) that are capable of synthesizing heme that is needed for zinc to react with to form ZnPP. 100 µmol/L zinc (to mimic the ICH-mediated zinc release) was added to whole rat blood under normoxia and hypoxia. Greater ZnPP generation under hypoxia conditions was observed, compared to normoxia (Figure 3(d)). Addition of zinc further increased ZnPP generation. Furthermore, treatment with zinc chelator TPEN decreased the zinc-induced ZnPP generation in hypoxic blood. Together, these results demonstrate that blood, free zinc and hypoxia are all required for ZnPP formation.
Zinc protoporphyrin toxicity is dependent upon hypoxia
To investigate ZnPP toxicity under ICH relevant conditions, we exposed primary neurons to increasing concentrations of ZnPP under hypoxia or normoxia, and compared the effects on cell death with exposure to hemin, a recognized toxic factor in ICH, under identical conditions. Neither ZnPP nor hemin induced significant cell death in normoxia at any concentration tested. However, under hypoxia, both ZnPP and high dose hemin showed significant dose-dependent increases in toxicity to neurons (Figure 4(a)). Surprisingly, ZnPP toxicity under hypoxia was much greater than that of hemin, which has long been recognized as a major cause of brain damage following ICH. Analogous experiments with primary astrocytes (Figure 4(a)) exhibited similar results, but only ZnPP showed a dose dependent increase in toxicity in hypoxia while hemin did not. These results demonstrate that ZnPP is highly toxic to both neurons and astrocytes under hypoxia but not normoxia.
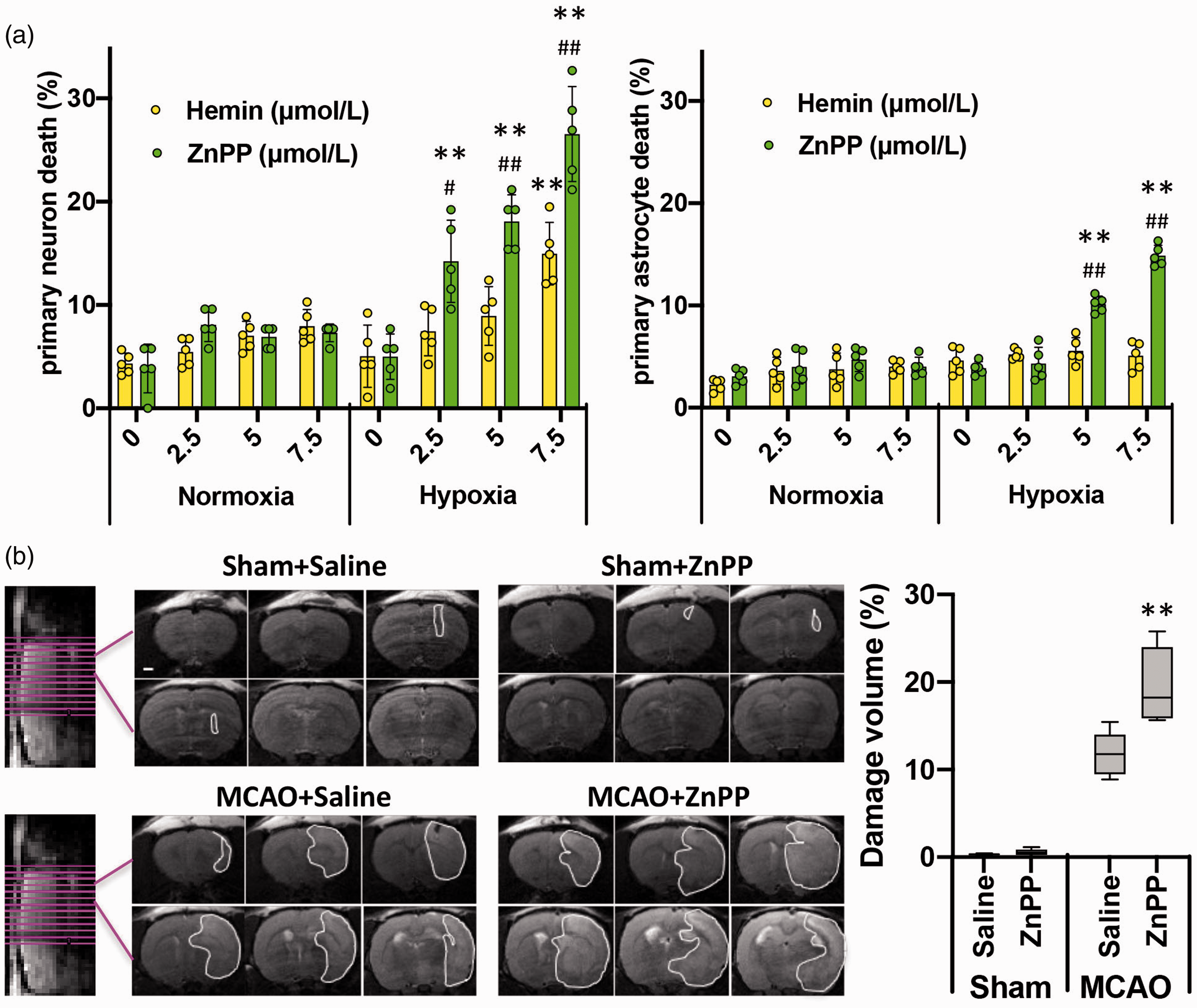
ZnPP is toxic only to hypoxic brain cells and tissue. (a) Primary neuron or astrocyte was incubated with indicated concentrations of ZnPP or Hemin. After exposed to 3 h normoxia or hypoxia, cell death was measured. The percentage of dead cells are presented as mean±SD, n = 5. **P < 0.01, versus hypoxia only group, #P < 0.05; ##P < 0.01, versus same concentration Hemin in hypoxia group, student’s t-test. (b) Brain injury by T2-weighted MRI. White line circles the brain damage. Box plot shows the total lesion volume calculated from the MRI image. Scale bar: 1 mm. The plot's horizontal line represents the median; the box encompasses the 25th to 75th percentiles; and the error bars encompass the 10th to 90th percentiles. **P < 0.01, versus MCAO saline group, student’s t-test.
To investigate ZnPP toxicity in vivo, the MCAO rat model was used, which provides hypoxic conditions without significant endogenous ZnPP formation in brain (Figure 3(b)). Exogenous ZnPP was intracerebrally injected to the ischemic side of MCAO and control rats. The T2-weighted MRI images (Figure 4(b)) showed that injection of ZnPP to normal, non-ischemic rats caused little brain damage. The small lesion in both saline control and ZnPP injected rats was likely due to mechanical damage caused by the needle or unabsorbed solution. Most importantly, the brain infarct area in MCAO rats was further exacerbated by ZnPP injection. These findings demonstrate that, as in the cellular models, ZnPP-induced brain damage occurred only under hypoxia, and recapitulated the exacerbated damage seen upon endogenous ZnPP biosynthesis.
Ferrochelatase inhibition decreased zinc protoporphyrin generation and brain damage after intracerebral hemorrhage
The final reaction in heme biosynthesis is iron insertion into protoporphyrin IX by mitochondrial ferrochelatase. Since ZnPP was generated, we hypothesized that under the hypoxic conditions with excess free zinc in ICH, ferrochelatase may insert zinc to protoporphyrin IX to generate ZnPP. Because ferrochelatase can be potently inhibited by NMPP, 52 we predicted that if our hypothesis was valid, NMPP treatment should decrease ZnPP formation and reduce brain tissue damage in ICH. As shown in Figure 5, NMPP treatment, 1 hr before ICH induction by collagenase (a) or 30 min after ICH induction by blood injection (b), reduced the ZnPP fluorescence by about half of that without NMPP treatment, indicating that the ICH-induced ZnPP generation is indeed caused by ferrochelatase-catalyzed insertion of zinc into protoporphyrin.
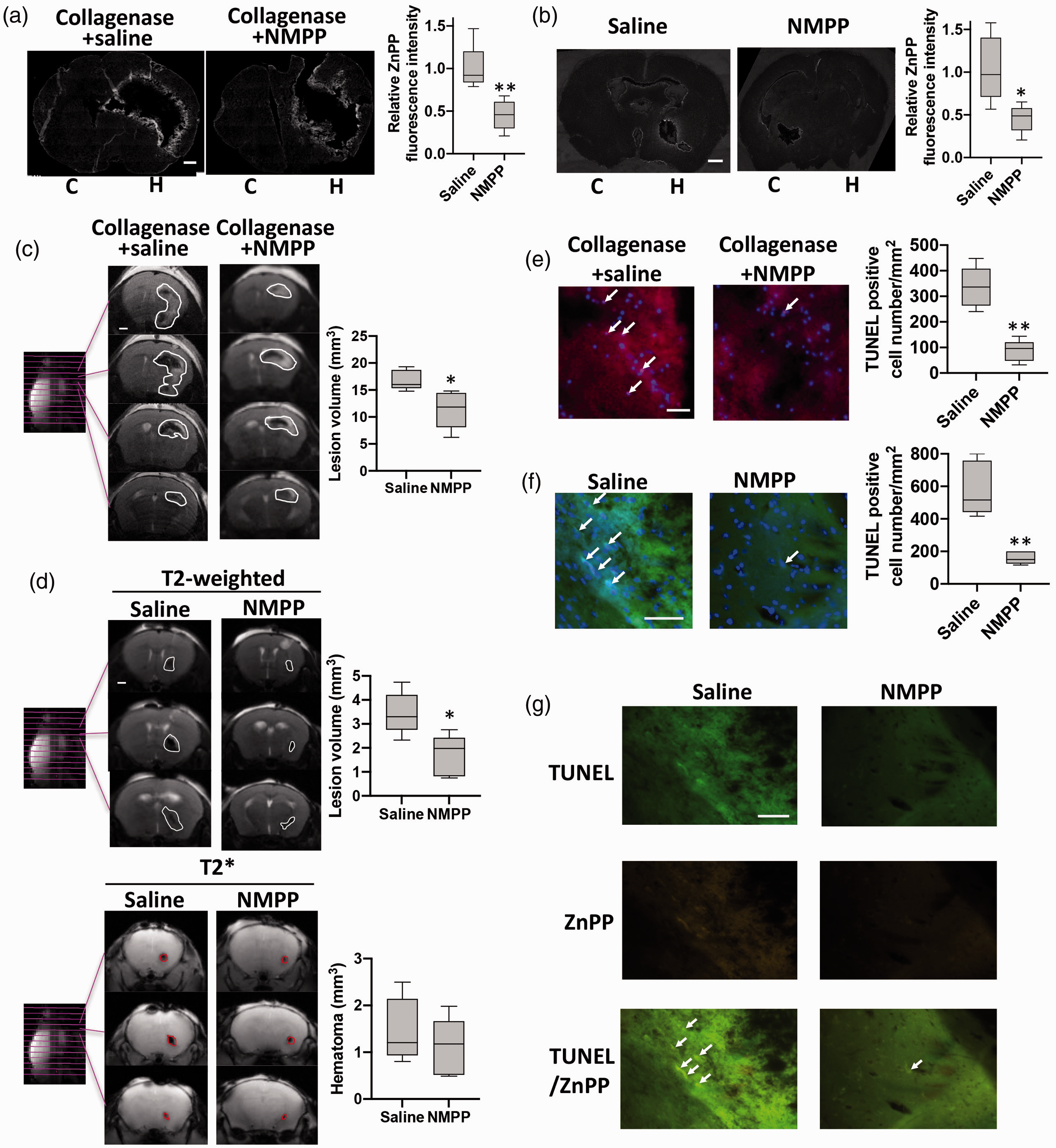
ZnPP is generated through ferrochelatase catalysis, and inhibition of ferrochelatase decreases ZnPP generation and toxicity in ICH animal models. (a) ZnPP in the collagenase- (a) and blood injection- (b) induced hemorrhagic brain was detected by imaging its autofluorescence on brain slices. Scale bar is 1 mm. Relative ZnPP fluorescence intensity is the intensity at the area within 200 µm from the edge of collagenase- or within 100 µm from the edge of blood injection-caused signal dephasing area in each brain slice normalized to the average value of that at the non-ICH hemisphere of saline group. n = 5 (4point/slice; 1slice/rat; 5 rats/group). **P < 0.01, student’s t-test. (c) Collagenase induced brain injury by T2-weighted MRI at 24 h after ICH onset. White line circles the brain damage. Scale bar: 1 mm. Box plot shows the total lesion volume calculated from MRI image. n = 5. (d) Blood injection-caused brain injury and hematoma were assessed by T2-weighted and T2* MRI at 24 h after ICH onset. White line circles the brain damage and red line circles hematoma. Scale bar: 1 mm. Box plot shows the total lesion volume and hematoma volume calculated from T2-weighted and T2* MRI image. n = 5. Cell death in the collagenase- (e, red) or blood injection- (f, green) caused hemorrhagic brain measured by TUNEL assay kit. Arrow indicates TUNEL positive cells. Scale bar: 50 µm. Box plot shows the density of TUNEL positive cell at the area within 200 µm from the edge of collagenase-induced ICH in rat brain slices (e) or within 100 µm from the edge of blood injection-induced ICH in mouse brain slices (f), n = 5 (4point/slice; 1slice/rat; 5rats/group). The plot's horizontal line represents the median; the box encompasses the 25th to 75th percentiles; and the error bars encompass the 10th to 90th percentiles. **P < 0.01, student’s t-test. (g) Arrows indicate the co-localized TUNEL (green) and autofluorescence of ZnPP (yellow) signals on ICH brain section, which was collected at 24 h after blood injection. Scale bar: 50 µm.
To evaluate the effect of reducing ZnPP formation through inhibition of ferrochelatase on brain damage in ICH, rats were pretreated with NMPP before collagenase-induced ICH, and brain damage was assessed by T2-weighted MRI. Our results showed that NMPP pre-treatment significantly reduced the extent of brain damage (Figure 5(c)), indicating endogenous-formed ZnPP promotes brain damage following ICH. To confirm the brain damage reduction by NMPP is not model dependent and post-ICH onset NMPP administration could still reduce brain damage, blood injection ICH with NMPP administration at 30 min after blood injection was performed in mice. MRI results shown that post-treatment of NMPP hardly changed the hematoma volume (Figure 5(d), T2*), while it significantly decreased total brain damage at 24 h after ICH onset (Figure 5(d), T2-weighted).
We also measured brain cell death to investigate the toxicity of ZnPP in ICH. As shown in Figure 5(e), the number of TUNEL positive cells with NMPP pretreatment decreased to just 14% of those in saline treated group with collagenase-induced ICH. This drastic reduction of cell death from NMPP treatment indicates that ZnPP plays a critically important role in ICH-induced brain damage. Similarly, TUNEL positive cells number with NMPP post-treatment decreased to 27% of those in saline treated group in blood-injected ICH mice (Figure 5(f)). Moreover, TUNEL and ZnPP signals were well co-localized, and NMPP treatment reduced both ZnPP and TUNEL (Figure 5(g)). These results furtherly confirmed ZnPP is involved in cell death and brain damage following ICH.
Blocking endogenous zinc protoporphyrin formation improves neurological outcome
Corner turn was used to assess the sensorimotor dysfunction (Figure 6(a)). Compared with saline-treated ICH mice, treatment with NMPP at 30 min after blood injection reduced the right turns at 7 days after ICH onset. However, little difference was observed at 14 days after ICH, since the mice in both groups turned randomly. The functional deficits of the left forelimb were assessed by foot-fault test. Post-treatment with NMPP slightly, but not significantly, improved the functional deficits at 14 days after ICH onset when compared with saline-treated group (Figure 6(b)). NMPP post-treated mice had much lower neurological severity scores at 7 and 14 days after ICH onset than the saline post-treated mice (Figure 6(c)). The improve of neurological outcome by NMPP suggests that endogenous ZnPP plays a major role in ICH-induced behavioral dysfunction.
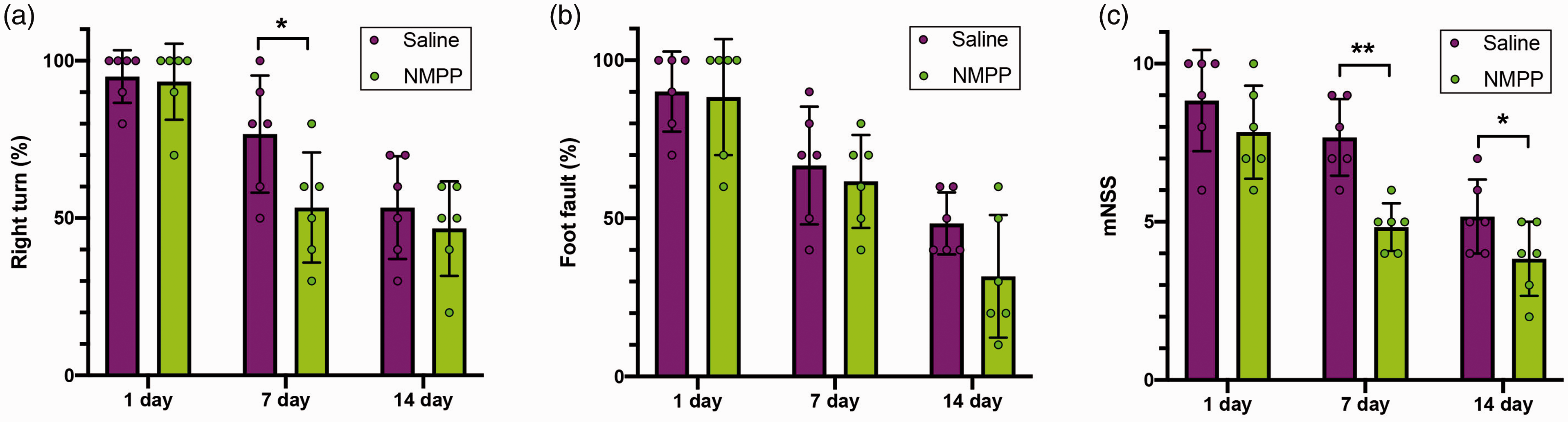
N-methyl protoporphyrin IX (NMPP) treatment improved functional outcomes in blood injection-induced ICH mice. NMPP or saline was injected intraperitoneally at 30 min after blood injected into the brain. The corner turning test (a), foot fault test (b) and the mNSS score (c) were measured at indicated time following ICH onset. Data are presented as mean±SD. *P < 0.05; **P < 0.01, one-factor ANOVA test.
Discussion
Hemorrhagic stroke is associated with high mortality rates and long-term disability. However, the mechanisms of ICH-induced brain damage are not well understood, nor are there effective treatments for ICH. Currently, brain damage in ICH is thought to be mediated through red cell lysis and toxicity of released hemoglobin and particularly its degradation products, heme, free iron and inflammation.3,4,53–55 Base on this, iron chelation,
56
and the combination of iron chelation and other treatments, such as minimally invasive surgery with hematoma lysis,
57
lactoferrin,
58
interleukin-4 transducer and transcription 6 axis activator,
59
have been investigated as promising treatment of ICH. In this study, we showed that endogenous ZnPP formation in ICH is also an important mediator of brain damage. Thus far, ZnPP has only been used as an exogenous inhibitor of heme oxygenase (HO)-1 to investigate the role of HO-1 in ICH-induced brain damage. However, nothing has been reported regarding
It was known that formation of ZnPP increases under certain conditions, such as exposure to lead or iron deficiency.60,61 However, by utilizing multiple approaches, we demonstrate that ZnPP was generated in ICH brain, via the reaction of zinc with protoporphyrin. Our results show that ICH-induced zinc and ZnPP were similarly distributed in the hemorrhagic tissue, and chelation of zinc by TPEN significantly decreased the generation of ZnPP, indicating the zinc dependence of ZnPP formation. Moreover, ZnPP generation required the presence of both brain tissue and blood, as hypoxic neurons are essential for zinc release, 13 while blood provides protoporphyrin. Zinc has been shown to be inserted in protoporphyrin IX by the catalysis of ferrochelatase, forming ZnPP in heme biosynthesis. 62 By demonstrating that inhibiting ferrochelatase with a common inhibitor (NMPP) significantly decreased the generation of ZnPP, our results have elucidated the molecular mechanism of ZnPP formation via ferrochelatase-catalyzed binding of zinc with protoporphyrin.
As to how ZnPP might exert its effects in ICH, ZnPP is a known potent inhibitor of HO-1 with levels as low as 0.15 µmol/L significantly decreasing HO-1 activity and with 50% inhibition at about 2.5 µmol/L. 63 From our measured ZnPP levels in the homogenized entire hemisphere of ICH brain (Figure 2(b)), the overall concentration was about 0.3 µmol/L, and local ZnPP concentrations in the ICH-enveloping rim are likely much higher when considering its special distribution (Figure 2(a)). Therefore, ZnPP is present at levels that could realistically inhibit HO-1 in brain tissue, especially in the ICH-enveloping rim. On one hand, HO-1 is well-known to be neuroprotective, 64 particularly in ICH. 65 , 66 In ICH, HO-1 catalyzes the degradation of heme to produce carbon monoxide, bilirubin, and ferrous iron (Fe2+). While the first two products are neuroprotective,67,68 it is well studied that formation of excess reactive oxygen species (ROS) is critically involved in ICH brain damage.69,70 ROS removal is an important function of HO-1. 71 Thus, endogenous formation of ZnPP in ICH may lead to brain damage through the inhibition of HO-1. Recently, we reported that a synergistic interaction between Zn and ROS forms a feedback loop, resulting in much increased level of each, leading to severe brain damage in ischemic stroke. 72 Since ZnPP could increase ROS in ICH through inhibiting HO-1 activity, it may also increase Zn level and trigger the damaging feedback loop of ROS and Zn in ICH. Thus, ICH-generated ZnPP may cause brain damage through inhibiting HO-1, triggering the fatal feedback loop of ROS and Zn. Nevertheless, blocking of ZnPP generation may lead to Zn accumulation, which may increase MMP synthetization. 73 Evidences show that MMP contributes to brain damage and perihematomal edema following both stroke.73,74 Thus, inhibition of ferrochelatase may increase brain damage through MMP activation. One the other hand, Fe2+ is released during the HO-1 catalyzes the degradation of heme, which can cause oxidative damage.75–77 The inhibition of HO-1 by ZnPP may also decrease brain injury. However, the oxidation of Fe2+ to Fe3+ following ICH may prevent Fe from being inserted into protoporphyrin, since Fe3+ is not a substrate for ferrochelatase, 78 which will result in accumulation of elevated level of labile Fe. Thus, although HO-1 inhibition by ZnPP may reduce Fe release from HO-1 catalyzed degradation of heme, formation of ZnPP may cause free Fe accumulation. Additionally, in some conditions, ZnPP also can induce HO-1, 79 , 80 which may cause excessive Fe release, leading to brain damage. Furthermore, while overload of Fe in ICH has been considered as a major cause of brain damage3,4 and oxidation of Fe2+ to Fe 3 may promote ZnPP generation in ICH, the oxidation of Fe has been well known to trigger Fenton reaction and generate highly toxic ROS, causing brain damage.81,82 Thus, ICH-generated ZnPP and iron may cooperate with each other, leading to brain damage. Apparently, the exact mechanism of how ZnPP causes brain damage following ICH, and the interplay between HO-1, Zn, Fe, and ferrochelatase, will need further investigation.
Hemorrhagic stroke complications seriously threaten patient’s life. Unfortunately, currently available treatments do not effectively reduce the complication. Although red blood cell lysis and iron overload are recognized as major causes of ICH-induced brain damage,3,4 intervention by iron chelation has only yielded limited clinical efficacy. 56 The mechanism of ICH brain injury may be much more complex than we have known before, probably involving interaction and cooperation among Zn, ZnPP, Fe and ROS. Thus, chelation of Fe alone would have limited clinical efficacy. Our study suggests ferrochelatase inhibition significantly blocked ZnPP generation and decreased ICH-induced brain damage, suggesting inhibition of ferrochelatase as a promising treatment strategy for ICH. Moreover, it has recently become clear that a major off-target activity of several protein kinase drugs is the potent inhibition of ferrochelatase, 83 thus, the FDA approved protein kinase inhibitors may have the potential for ICH treatment.
In summary, it is currently known that brain damage in ICH is caused by red cell lysis, hemoglobin and its degradation products, heme and free iron. However, our results shown that ZnPP, generated endogenously following ICH, critically contributes to brain damage. Our findings presented here reveal a novel mechanism by which endogenous formation of ZnPP contributes to the brain tissue damage seen in ICH. Further elucidation of this pathway might lay basic science foundations for later translational studies to provide promising treatments for this condition.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from National Institutes of Health to KJ Liu (P30GM103400) and University of New Mexico BBHI Mini Grant Award (BBHI 2017-1006). The chromatographic/MS data was obtained at the Facility for Metabolic Chromatography at the University of New Mexico, which was established and funded by the National Science Foundation (NSF) award #IIA-1301346.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Rong Pan contributed to the concept, design the experiments, acquisition and analysis data, draft the article; Song Yu and Haikun Zhang contributed to acquisition data; Graham S. Timmins contributed to the concept, revise the article; John Weaver contributed to acquisition the EPR data; Yirong Yang contributed to acquisition the MRI data; Xixi Zhou contributed to acquisition the MS data; Ke Jian Liu contributed to the concept, design the experiments, revised the article and approved the version to be published.