
Abstract
Mitochondria may be transferred from cell to cell in the central nervous system and this process may help defend neurons against injury and disease. But how mitochondria maintain their functionality during the process of release into extracellular space remains unknown. Here, we report that mitochondrial protein O-GlcNAcylation is a critical process to support extracellular mitochondrial functionality. Activation of CD38-cADPR signaling in astrocytes robustly induced protein O-GlcNAcylation in mitochondria, while oxygen-glucose deprivation and reoxygenation showed transient and mild protein modification. Blocking the endoplasmic reticulum – Golgi trafficking with Brefeldin A or slc35B4 siRNA reduced O-GlcNAcylation, and resulted in the secretion of mitochondria with decreased membrane potential and mtDNA. Finally, loss-of-function studies verified that O-GlcNAc-modified mitochondria demonstrated higher levels of neuroprotection after astrocyte-to-neuron mitochondrial transfer. Collectively, these findings suggest that post-translational modification by O-GlcNAc may be required for supporting the functionality and neuroprotective properties of mitochondria released from astrocytes.
Keywords
Introduction
Mitochondrial function is essential for maintaining homeostasis in the high energy-consuming central nervous system (CNS),1,2 and mitochondrial dysfunction plays a key role in almost all types of CNS injury and disease.3–5 Recently, mitochondrial transfer between cells has been proposed as a non-cell-autonomous help-me mechanism for protecting against injury and disease in mammalian organs.6–11 Mesenchymal stem cells can transfer mitochondria to rescue metabolism in target cells and ameliorate tissue injury. 12 In the CNS, intercellular mitochondrial transfer between astrocytes and neurons or microglia may regulate transcellular mitophagy, 13 neuroprotection,14,15 and immunomodulation. 16 Additionally, neuroglial interaction via fragmented dysfunctional mitochondrial transfer may cause neurodegeneration. 17 Furthermore, given the presence of extracellular mitochondria in cerebrospinal fluid, their function and integrity may reflect brain metabolic state and recovery after CNS injury.18,19 What remains unknown is how mitochondria maintain their functionality during the process of release into extracellular space.
O-GlcNAcylation is the process that attaches O-linked β-N-acetylglucosamine to serine and/or threonine side chains of the polypeptide backbone in nuclear and cytoplasmic proteins. 20 The addition and removal of O-GlcNAc moieties cycles regulated by O-GlcNAc transferase and O-GlcNAcase rapidly occur in response to cellular metabolic changes. 21 It has been suggested that O-GlcNAcylation is also ubiquitously distributed in mitochondrial proteins and this process is involved in the regulation of mitochondrial homeostasis, oxidative stress response, energy production, mitochondrial membrane potential and motility.22,23 Moreover, dysfunction in this process may play a role in neurodegenerative disorders and aging pathology.24–27 Our previous study showed that a CD38-mediated metabolic pathway may coordinate the release of functional extracellular mitochondria from astrocytes. 14 The gap in knowledge is how extracellular mitochondria can retain their functionality outside the cell. Since post-translational O-GlcNAcylation is known as a process that maintains and boosts mitochondrial function,22,24 we performed this proof-of-concept study to test the hypothesis that O-GlcNAcylation in mitochondrial protein is one of the reasons why CD38-stimulated extracellular mitochondria are able to retain functionality.
Methods and materials
Reagents
β-Nicotinamide adenine dinucleotide sodium salt (Sigma Aldrich, N0632-1G), cyclic ADP ribose (Sigma Aldrich, C7344-5MG), 8-Bromo-cyclic adenosine diphosphate ribose (B5416-250UG), Dantrolene sodium salt (Tocris, 0507), Brefeldin A (Abcam, ab193369), Protein transport inhibitor (Brefeldin A plus monensin cocktail, Thermo Fisher Scientific, 00-4980-93), Mitotracker Deep Red FM (Thermo Fisher Scientific, M22426), CellLight Mitochondria-GFP, BacMam 2.0 (Thermo Fisher Scientific, C10600).
Animal study
All in vitro experiments using primary cell cultures were approved by the Massachusetts General Hospital (MGH) Institutional Animal Use and Care Committee in accordance with National Institutes of Health guidelines and with the United States Public Health Service's Policy on Human Care and Use of Laboratory Animals and following Animals in Research: Reporting In vivo Experiments (ARRIVE) guidelines.
Primary neuron cultures
Primary neuron cultures were prepared from cerebral cortices of E17-day-old Sprague-Dawley rat embryos. Briefly, cortices were dissected and dissociated using papain dissociation system (Worthington Biochemical Corporation, LK003150). Cells were spread on plates coated with poly-D-lysine (Sigma, P7886) and cultured in Dulbecco's modified Eagle medium (NBM, Life Technology, 11965-084) containing 25 mM glucose, 4 mM glutamine, 1 mM sodium pyruvate, and 5% fetal bovine serum at a density of 2 × 105 cells/mL (1 mL for 12 well format, 0.5 mL for 24 well format). At 24 hrs after seeding, the medium was changed to Neurobasal medium (Invitrogen, 21103-049) supplemented with B-27 (Invitrogen, 17504044) and 0.5 mM glutamine. Cells were cultured at 37 °C in a humidified chamber of 95% air and 5% CO2. Over 95% of purity of neuron cultures was determined by MAP2 staining in our previous study. 14
Primary astrocyte cultures
Primary astrocyte cultures were prepared from cerebral cortices of 2-day-old neonatal Sprague-Dawley rats or E17 C57Bl6 mice. Briefly, dissociated cortical cells were suspended in Dulbecco's modified Eagle medium (DMEM, Life Technology, 11965-084) containing 25 mM glucose, 4 mM glutamine, 1 mM sodium pyruvate, and 10% fetal bovine serum and plated on uncoated 25 cm2 flasks at a density of 6 × 105 cells/cm2. Monolayers of type 1 astrocytes were obtained 12–14 days after plating. Non-astrocytic cells such as microglia and neurons were detached from the flasks by shaking and removed by changing the medium.
Oxygen–glucose deprivation (OGD) and reoxygenation
OGD experiments were performed using a specialized, humidified chamber (Heidolph, incubator 1000, Brinkmann Instruments, Westbury, NY) kept at 37 °C, which contained an anaerobic gas mixture (90% N2, 5% H2, and 5% CO2). To initiate OGD, culture medium was replaced with deoxygenated, glucose-free Dulbecco's modified Eagle medium (Life Technology, 11966-025). After 2 h challenge, cultures were removed from the anaerobic chamber, and the OGD solution in the cultures was replaced with maintenance medium. Cells were then allowed to recover for 24 h for O-GlcNAcylation in astrocytes or neurotoxicity assay.
Cell viability assays
Neuronal injury was measured by Cell Counting Kit 8 cytotoxicity assay (DOJINDO, CK04-13). The relative assessments of neuronal injury were normalized by comparison with control cell as 100% cell survival.
Mitochondria membrane potential measurement
To monitor mitochondrial health, JC-1 dye (invitrogen, T-3168) was used to assess mitochondrial membrane potential. Astrocyte conditioned media were incubated with JC1 (0.8 µM) for 30 min at 37 °C. JC1 dye exhibits potential-dependent accumulation in mitochondria, indicated by fluorescence emission shift from green (Ex 485 nm/Em 516 nm) to red (Ex 579 nm/Em 599 nm). Mitochondria membrane potential was determined by the fluorescent ratio with a fluorescent microplate reader.
FACS analysis
Standard FACS analysis was performed by BD Fortessa. Astrocyte-conditioned medium (ACM) was collected from rat cortical astrocytes. Briefly, ACM were centrifuged by 2,000 rpm for 5 minutes in order to exclude cellular debris. When ACM were collected from astrocytes labeled by mitochondria GFP, samples were directly analyzed by BD Fortessa following the centrifugation. In order to detect mitochondrial DNA and mitochondrial O-GlcNAc, ACM were incubated with Mitotracker Deep Red (50 nM) and O-GlcNAc FITC antibody (2 µg/mL) for 30 min at 37 °C. FACS analysis was performed using an unstained or phenotype control for determining appropriate gates, voltages, and compensations required in multivariate flow cytometry.
Western blot analysis
Each sample was loaded onto 4–20% Tris-glycine gels. After electrophoresis and transferring to nitrocellulose membranes, the membranes were blocked in Tris-buffered saline containing 0.1% Tween 20 and 0.2% I-block (Tropix, T2015) for 90 min at room temperature. Membranes were then incubated overnight at 4 °C with following primary antibodies, anti-β-actin (1:1,000, Sigma-aldrich A5441), anti-TOM40 (1:200, Santacruz, sc-11414), anti-GAP43 (1:500, Santacruz, sc-17790), anti-O-GlcNAc (1:1000, Abcam, ab2739). After incubation with peroxidase-conjugated secondary antibodies, visualization was enhanced by chemiluminescence (GE Healthcare, NA931- anti-mouse, or NA934- anti-rabbit, or NA935- anti-rat). Optical density was assessed using the NIH Image analysis software.
Immunocytochemistry
After staining with primary antibody, fluorescent-tagged secondary antibody, nuclei were counterstained with or without 4,6-diamidino-2-phenylindole (DAPI), and coverslips were placed. Immunostaining images or time lapse images were obtained with a fluorescence microscope (Nikon ECLIPSE Ti-S) interfaced with a digital charge-coupled device camera and an image analysis system.
Electron microscopy analysis
Astrocytic extracellular particles were collected from 3xT75 flasks (∼90% confluence). Following excluding cellular debris (2,000 rpm for 10 min), media were concentrated by Vivaspin 20 according to manufacture’s instruction. Pellets were obtained after a centrifugation at 12,000 rpm for 15 min and fixed in 2.0% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4 (Electron Microscopy Sciences, Hatfield, PA) for one hour at room temperature on a rocker. They were rinsed in cacodylate buffer, gently scraped and pelleted and post-fixed in 1.0% osmium tetroxide in cacodylate buffer for one hour on ice. They were rinsed in buffer and stabilized with a small amount of 2% agarose in PBS to hold them together. They were then dehydrated through a graded series of ethanol to 100%, followed by propylene oxide, 100%. They were infiltrated with Epon resin (Ted Pella, Redding, CA) in a 1:1 solution of Epon:propylene oxide overnight on a rocker at room temperature. The following day they were placed in fresh Epon for several hours and then embedded in Epon overnight at 60 C. Thin sections were cut on a Leica EM UC7 ultramicrotome, collected on formvar-coated grids, stained with uranyl acetate and lead citrate and examined in a JEOL JEM 1011 transmission electron microscope at 80 kV. Images were collected using an AMT digital imaging system (Advanced Microscopy Techniques, Danvers, MA).
mtDNA detection
Genomic and mtDNA were extracted by alkaline lysis/neutralization followed by isopropanol precipitation from cells or conditioned media exosomes (total exosome isolation reagent, Thermo Fisher Scientific, 4478359). Relative levels of mtDNA to genomic DNA were determined by amplifying MT-ND1 gene (Applied Biosystems, Rn03296764) and HBB (beta-globin) gene (Applied Biosystems, Rn04223896).
siRNA experiment (siRNA and plasmid transfection)
The siRNA targeting Slc35b4 (Thermo Fisher Scientific, 4390771) was transfected into rat astrocytes with Lipofectamine 3000 reagent (Thermo Fisher Scientific, L3000015) according to the manufacturer’s instructions. mRNA level of each Slc35 was quantified by RT-PCR using each primer of Slc35a3 (Applied Biosystems, Rn06170878), Slc35b4 (Applied Biosystems, Rn01500306), Slc35d2 (Applied Biosystems, Rn01759558). One day after transfection, cells were treated with NAD for 24 h and subjected to each assay.
Statistical analysis
Results were expressed as mean ± SD. All of experiments were performed with full blinding, allocation concealment and randomization. D’Agostino & Pearson omnibus normality test was used to assess data distribution. When only two groups were compared, unpaired t-test (two-tailed) was used. Multiple comparisons were evaluated by one-way ANOVA followed by Tukey's test. P < 0.05 was considered to be statistically significant.
Results
Astrocytic CD38 signaling increases O–GlcNAc post-translational modification in extracellular mitochondria
Rat cortical astrocytes were grown in primary culture and nicotinamide adenine dinucleotide (NAD) was used to stimulate the CD38-cADPR signaling mechanism that induces the release of extracellular mitochondria. 14 At 0, 3, 12, and 24 hrs after NAD treatment, O-GlcNAc modification of intracellular mitochondrial protein was assessed (Figure 1(a)). Under baseline condition, western blots showed that intracellular mitochondrial protein O-GlcNAcylation was present at very low levels (Figure 1(b)). Stimulation with NAD progressively increased mitochondrial protein O-GlcNAcylation, resulting in a 5-fold change compared to the baseline condition (Figure 1(b)). To further confirm mitochondrial protein O-GlcNAcylation, E1 alpha pyruvate dehydrogenase was labeled by GFP. Immunocytochemistry revealed that CD38 stimulation with NAD for 24 hrs increased O-GlcNAc signals in astrocytic mitochondria (Figure 1(c)). Next, astrocytes were subjected to oxygen-glucose deprivation (OGD) for 2 hrs followed by reoxygenation for 0, 2, or 24 hrs. Western blot analysis demonstrated that OGD and reoxygenation also increased O-GlcNAc post-translational modification, but this response was mild and transient; O-GlcNAc levels peaked around 2-fold at 2 hrs and decreased towards baseline by 24 hrs (Figure 1(d)).
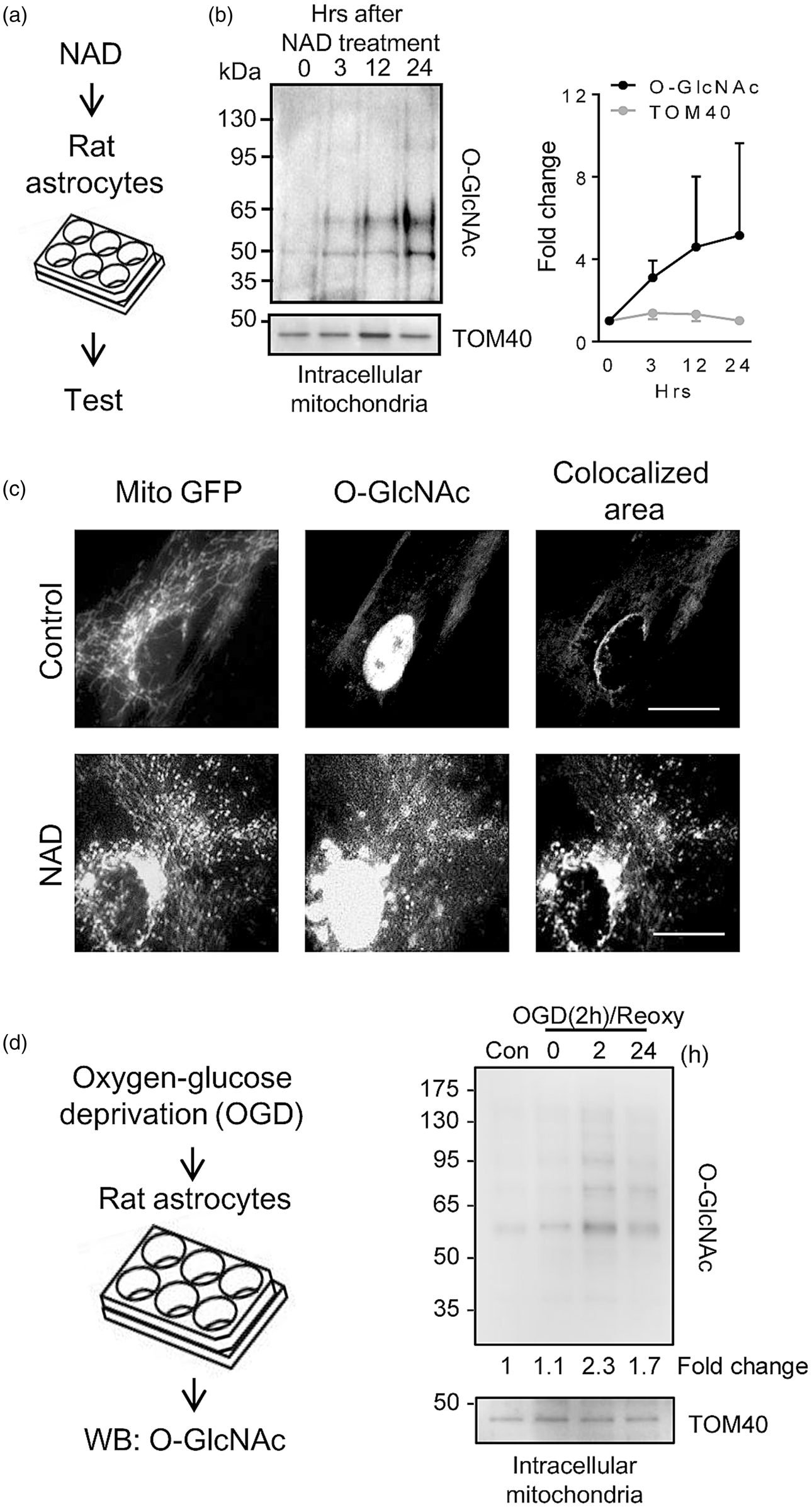
CD38 stimulation increases mitochondrial O-GlcNAcylation in astrocytes: (a) Mitochondrial fraction was obtained from cultured rat cortical astrocytes over time after NAD (5 mM) treatment. (b) Western blot analysis demonstrated that O-GlcNAcylated proteins were steadily increased in isolated mitochondria (n = 4). (c) Fluorescent staining showed that some mitochondria were colocalized with O-GlcNAc. Scale: 20 µm. (d) Rat cortical astrocytes were subjected to oxygen-glucose deprivation for 2 hrs and reoxygenation for up to 24 hrs. Western blot analysis showed that O-GlcNAc modification in mitochondrial proteins was transiently increased during reoxygenation. All values are mean +/– SD.
Furthermore, astrocyte-conditioned media (ACM) were collected at 24 hrs after NAD treatment. TEM analysis confirmed that mitochondria were present in ACM (Figure 2(a)). Flow cytometry of ACM demonstrated that extracellular mitochondria were significantly increased by approximately 2 times in NAD treatment compared to control and this effect was blocked by the cADPR inhibitor 8-Br-cADPR (Figure 2(b) and (c)). There were no apparent changes in cell viability as measured by the standard WST assay, suggesting that these were potentially functional responses and not side effects of cell damage and lysis (Figure 2(d)).
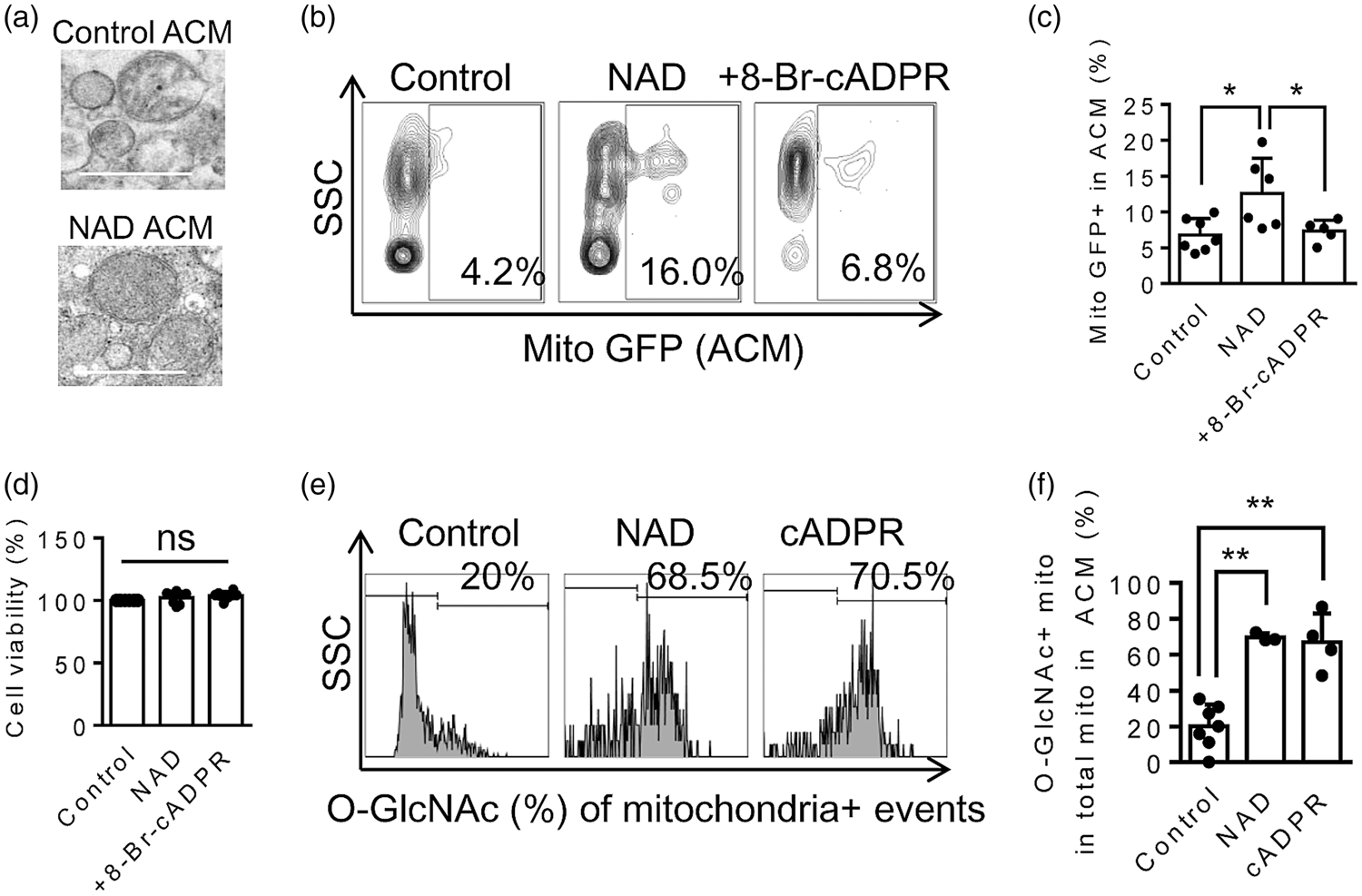
O-GlcNAcylation in extracellular mitochondria: (a) TEM analysis confirmed the presence of extracellular mitochondria in astrocyte-conditioned media (ACM). (b–c) Flow cytometry analysis showed that NAD (5 mM) treatment increased extracellular mitochondria (GFP-labeled mitochondrial pyruvate dehydrogenase E1 Alpha 1), and the effect was blocked by cADPR inhibitor (8-Br-cADPR, 10 µM) (Control; n = 7, NAD; n = 6, NAD + 8-Br-cADPR; n = 5). (d) A standard cell viability assay (WST) showed no cell death after treatment (n = 8). (e–f) Extracellular mitochondria in ACM were labeled by Mitotracker Deep Red (Mito DR, 50 nM) and O-GlcNAc-FITC antibody (2 µg/mL) for 30 min at 37 °C. Flow cytometry analysis showed that under normal condition, around 20% of extracellular mitochondria had O-GlcNAc modification. However, the number was increased up to 70–80% when astrocytes were stimulated by NAD (5 mM) or cADPR (100 µM) (Control; n = 7, NAD; n = 3, cADPR; n = 4). All values are mean +/– SD. *P < 0.05, **P < 0.01. Multiple comparisons were evaluated by one-way ANOVA followed by Tukey's test.
To determine the extent of O-GlcNAcylation, extracellular mitochondria in ACM were labeled by Mitotracker Deep Red and the O-GlcNAc-FITC antibody, then flow cytometry analysis was performed again. Under baseline condition, approximately 20% of extracellular mitochondria had O-GlcNAc modification. After stimulation with NAD or cADPR (as a positive control), extracellular mitochondrial O-GlcNAcylation was increased by up to 75% (Figure 2(e) and (f)).
Mitochondrial protein O-GlcNAcylation in astrocytes is regulated by endoplasmic reticulum (ER) – Golgi trafficking
To track this process of O-GlcNAcylation, we asked whether regulation at the level of endoplasmic reticulum (ER) and Golgi trafficking may be involved. To inhibit ER-Golgi traffic, we used Brefeldin A that disturbs the functions and organization of the Golgi apparatus.28,29 Rat cortical astrocytes were co-incubated with NAD or NAD and Brefeldin A for 24 hrs. Western blot analysis showed that blocking ER-Golgi trafficking with Brefeldin A exclusively reduced mitochondrial O-GlcNAcylation (Figure 3(a)). To quantify O-GlcNAcylation in extracellular mitochondria in ACM, we turned to flow cytometry (Figure 3(b)). Treatment with Brefeldin A did not markedly change NAD-induced mitochondrial secretion, but the proportion of O-GlcNAc-modified extracellular mitochondria was back to baseline levels (Figure 3(c) and (d)).
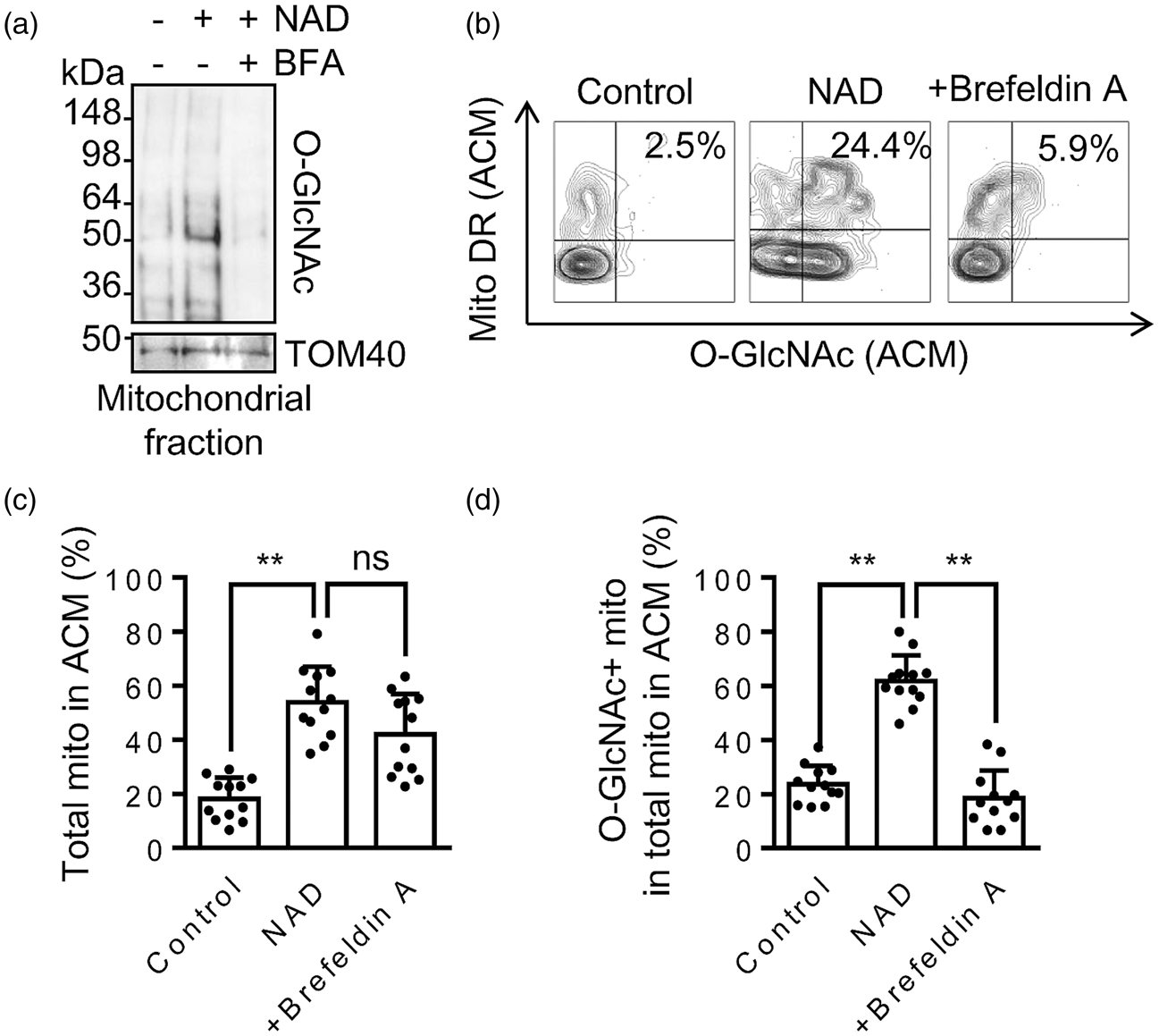
The ER - Golgi traffic regulates mitochondrial protein O-GlcNAc modification: (a) Western blot demonstrated that Brefeldin A (BFA, 5 µg/mL, an inhibitor for the ER-Golgi traffic) inhibited protein O-GlcNAcylation in astrocytic mitochondria. (b–d) FACS demonstrated that NAD (5 mM) increased O-GlcNAc-positive extracellular mitochondria in ACM. Co-treatment with Brefeldin A (BFA, 5 µg/mL) significantly decreased O-GlcNAc-positive mitochondria, but mitochondrial secretion was not affected by BFA (n = 12). All values are mean +/– SD. *P < 0.05, **P < 0.01. One-way ANOVA followed by Tukey's test.
To further probe these trafficking mechanisms, we assessed UDP-GlcNAc antiporters including SLC35A3, SLC35B4, and SLC35D2, all of which are known to transport cytoplasmic UDP-GlcNAc to the Golgi apparatus. 30 Of these 3 genes, astrocytes highly expressed SLC35B4, and this gene was successfully suppressed by 40% with siRNA without affecting expressions of other antiporters (Figure 4(a)). Then, we stimulated astrocytic CD38 with NAD treatment for 24 hrs. Flow cytometry was then used again in order to quantify protein O-GlcNAcylation in extracellular mitochondria in ACM (Figure 4(b)). Gene silencing of SLC35B4 normalized extracellular mitochondrial O-GlcNAc modification without substantially impacting the extent of mitochondrial secretion (Figure 4(c) and (d)).
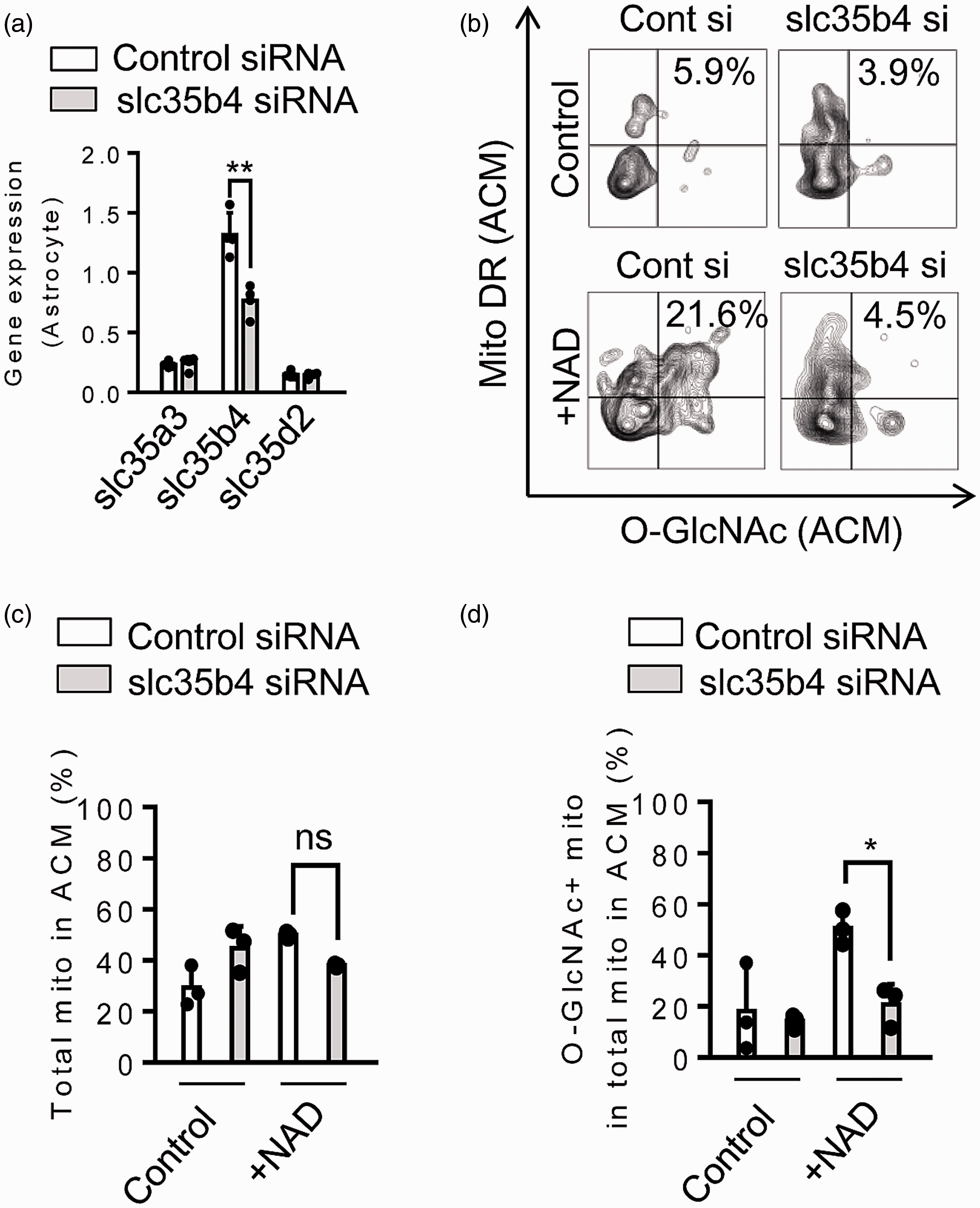
The Golgi-resident slc35b4 may transport O-GlcNAc for mitochondrial protein modification: (a) UDP-GlcNAc antiporter slc35b4 was highly expressed in astrocytes and siRNA treatment selectively inhibited slc35b4 (n = 3). (b) Representative plots from FACS analysis. (c–d) FACS revealed that suppression of slc35b4 decreased O-GlcNAc positive extracellular mitochondria (c) without affecting mitochondrial secretion (d) (n = 3). All values are mean +/– SD. *P < 0.05, **P < 0.01, unpaired t-test (two-tailed).
Mitochondrial protein O–GlcNAcylation in astrocytes is decreased by oxidative stress and aging process
If ER-Golgi trafficking is important for the regulation of protein O-GlcNAcylation in extracellular mitochondria, then this phenomenon should be not only affected by exogenous blockers, but should also be dependent on the overall metabolic health of the astrocytes. Two separate classes of metabolic stress were tested (Figure 5(a)). First, a combination of inflammatory cytokines (IL-1α: 3 ng/mL, TNF-α: 30 ng/mL, C1q: 400 ng/mL) was used to evoke a reactive glial phenotype (A1 astrocyte). Second, astrocytes were cultured for prolonged periods (up to 12 wks) to mimic an aging phenotype. In A1 astrocyte induction and aging, the SLC35B4 antiporter was significantly downregulated by 30% and 50%, respectively (Figure 5(b) and (c)). Next, these two subsets of astrocytes were stimulated by NAD for 24 hrs and mitochondrial protein O-GlcNAcylation was evaluated by western blot. Consistent with SLC35B4 responses, O-GlcNAcylation of mitochondrial proteins was also decreased in both groups of astrocytes (Figure 5(d) and (e)). Taken together, these data suggest that whereas extracellular mitochondrial release per se is not mediated by the conventional ER-Golgi-dependent secretion pathway, the healthy status of ER-Golgi trafficking is essential for the process of mitochondrial protein O-GlcNAcylation.
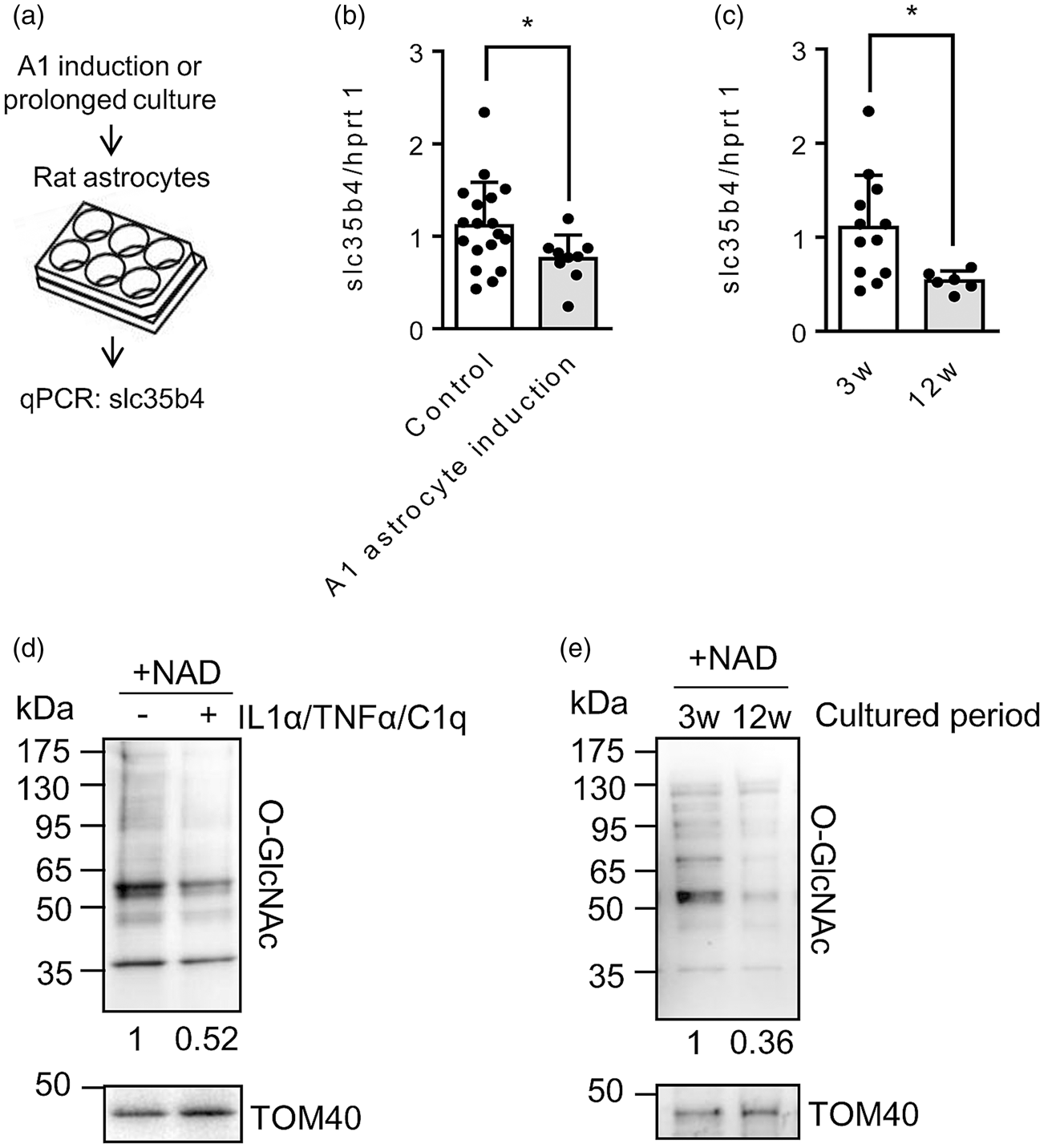
Inflammation and longer period of cell culture may interfere ER-Golgi traffic in astrocytes: (a) Rat cortical astrocytes were pre-exposed to combination of cytokines (IL-1α: 3 ng/mL, TNF-α: 30 ng/mL, C1q: 400 ng/mL) for 24 hrs or cultured for longer period of time (3 weeks or 12 weeks). (b) qPCR analysis demonstrated that slc35b4 was downregulated (Control: n = 18, A1 astrocyte induction: n = 9). (c) Culturing for 12 weeks downregulated slc35b4 (3w: n = 12, 12w: n = 6). (d) NAD (5 mM) was treated to A1 astrocytes for 24 hrs. Western blot analysis confirmed that mitochondrial protein O-GlcNAcylation was decreased in astrocytic mitochondria. (e) Concomitantly, mitochondrial protein O-GlcNAcylation was reduced by 12 week-cell culture compared to 3 week-cell culture. All values are mean +/– SD. *P < 0.05, unpaired t-test (two-tailed).
O-GlcNAcylation supports extracellular mitochondrial functionality and neuroprotective capacity
Ultimately, the importance of mitochondrial O-GlcNAcylation in astrocytes should to be assessed by measurements of extracellular functionality and neuroprotection. NAD was used to stimulate mitochondrial secretion from cortical astrocytes, and membrane potential and DNA content were quantified as surrogate markers of extracellular mitochondrial function and integrity in ACM. When O-GlcNAcylation was blocked by Brefeldin A, both mitochondrial membrane potential and DNA content were significantly decreased by almost 50% in the secreted extracellular mitochondria (Figure 6(a) and (b)). To evaluate neuroprotection, we performed a transfer experiment where astrocytes were stimulated with NAD for 24 hrs, and then mitochondria were collected and transferred onto primary neurons that were damaged by 2 h oxygen-glucose deprivation (Figure 6(c)). The purpose of this experiment was to compare the efficacy of O-GlcNAcylated mitochondria (NAD stimulation alone) versus non-O-GlcNAcylated mitochondria (NAD plus Brefeldin A to block ER-Golgi trafficking and suppress O-GlcNAcylation). At 24 hrs post-transfer, levels of O-GlcNAcylated mitochondria appeared to be higher in recipient neurons (Figure 6(d)). Furthermore, and O-GlcNAcylated mitochondria significantly improved neuroprotection against oxygen-glucose deprivation (Figure 6(e)). Finally, the status of the protected neurons was assessed by examining the synaptic marker GAP43. Transfer of mitochondria collected from astrocytes significantly increased GAP43 in surviving neurons while blocking O-GlcNAcylation with Brefeldin A diminished these effects (Figure 6(f) and (g)).
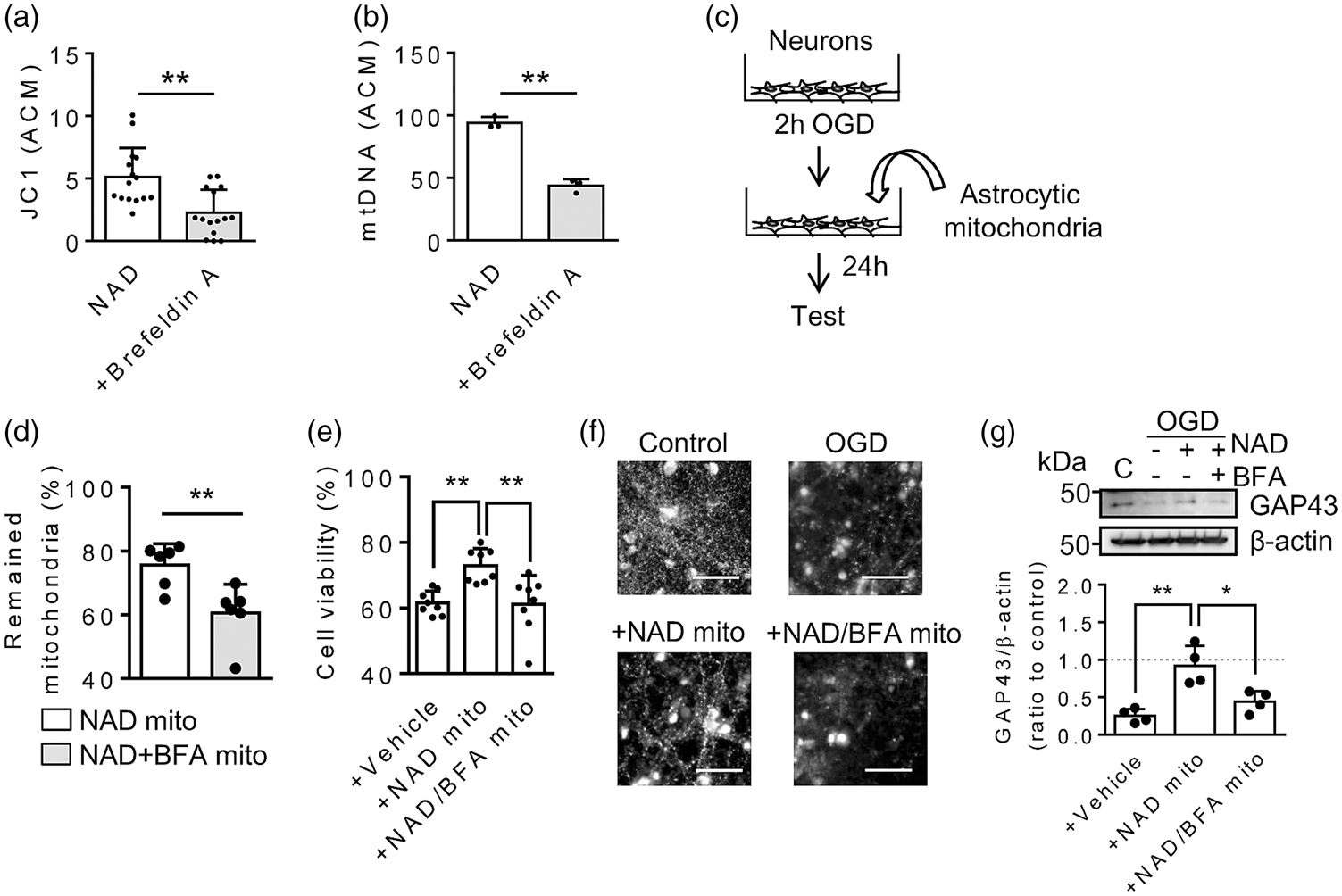
O-GlcNAcylated mitochondria show higher neuroprotection in vitro. (a) JC1 mitochondrial membrane potential in extracellular mitochondria was decreased when O-GlcNAcylation was suppressed by BFA (5 µg/mL) (n = 15). (b) Extracellular vesicles were collected from ACM, then mtDNA content (ND-1) was assessed. BFA significantly decreased mtDNA in ACM (n = 3). (c) We assessed neuroprotective effect of pure mitochondrial subset isolated from astrocytes instead of using astrocyte-conditioned media that potentially contain multi-beneficial soluble factors besides extracellular mitochondria. O-GlcNAc-mitochondria (NAD mito, 2 µg/well) or glycosylation-reduced mitochondria (NAD+BFA mito, 2 µg/well) were treated in rat cortical neurons following oxygen-glucose deprivation (OGD) for 2 hrs. (d) After 24 hrs, NAD mito were more remained in the treated neurons than NAD+BFA mito. (e) Neuronal viability following 2-h OGD was about 60%. Treatment with NAD mito increased neuronal viability, but Brefeldin A treatment canceled mitochondria-mediated neuroprotection (n = 8). (f–g) Immunostaining (f) and western blot (g) demonstrated that a neuroplasticity marker GAP43 was recovered by treatment with O-GlcNAcylated mitochondria, while blocking O-GlcNAcylation diminished the effect (n = 4). All values are mean +/– SD. *P < 0.05, **P < 0.01.
Discussion
Astrocytes play complex and far-ranging roles in the CNS physiology and pathophysiology.31–35 More recently, it has been proposed that astrocytes may also provide a non-cell autonomous source of help-me signals during brain injury and neurodegeneration.36,37 In the context of cerebral ischemia, we previously reported that CD38 may regulate the release of functional extracellular mitochondria from astrocytes. 14 But, the molecular details of how mitochondria sustain their functionality outside cells remain to be fully investigated. In the present study, we provided biochemical and cellular results to support the hypothesis that O-GlcNAc modification in mitochondrial proteins may promote the secretion and maintenance of functionally beneficial extracellular mitochondria from astrocytes following CD38 stimulation or ischemia/reoxygenation (Figure 7).
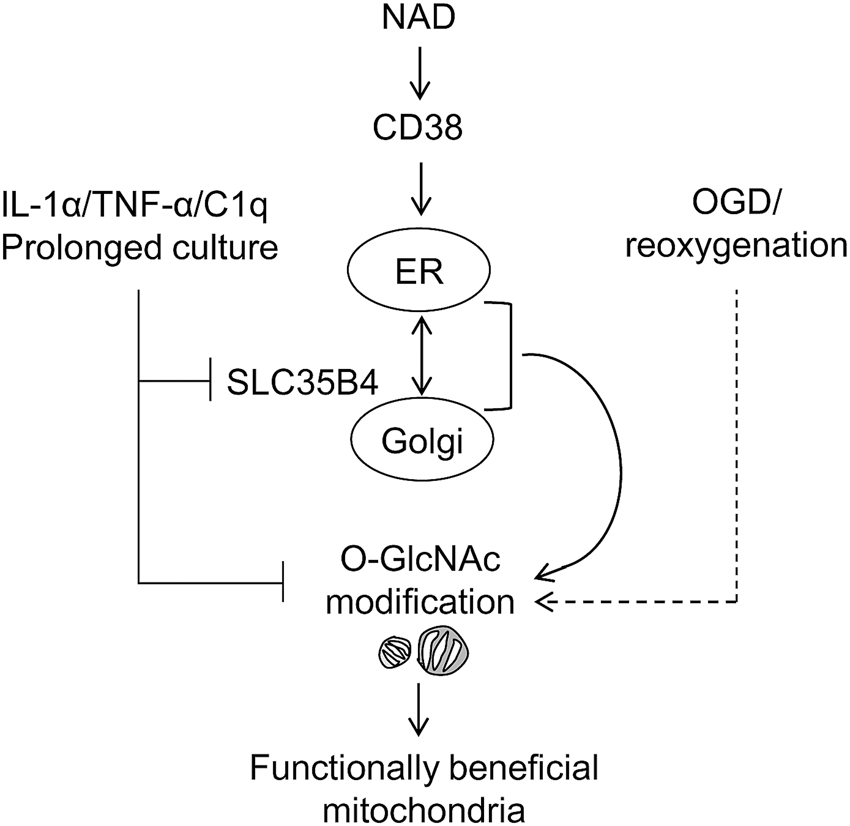
Schematic of O-GlcNAc modification-mediated secretion of functionally beneficial mitochondria. NAD-CD38 pathway activates the ER-Golgi traffic-mediated O-GlcNAcylation in mitochondrial protein. Oxygen-glucose deprivation (OGD) and reoxygenation may be partly involved in the regulation of mitochondrial protein O-GlcNAcylation in astrocytes, while excessive inflammation or aging pathology may retard mitochondrial protein modification with O-GlcNAc through downregulating SLC35B4. Dotted line from the OGD stimulus indicates that multiple wide-ranging and complex signaling pathways in astrocytes so any change in O-GlcNAc response may include CD38-dependent and CD38-independent pathways.
Mitochondria comprise the intracellular cores for cell viability and participate in fundamental mechanisms in the regulation of neurodegeneration as well as neurorecovery in CNS injury.38–43 Recent findings suggest that mitochondria might also be actively released into extracellular space and transferred between cells. Under pathological condition in the CNS, neurons and glia may secrete mitochondria and regulate transcellular mitophagy, 13 neuroprotection14,15 and microglial phenotype, 16 while dysfunctional extracellular mitochondria may act as DAMP signals and expand inflammatory response.17,44 The collective literature suggest that extracellular mitochondria may influence both injury and repair in the neurovascular unit, depending on context, including mitochondrial quality after secretion.
In the brain, CD38 is mainly produced by glial cells45,46 and known to be activated by glutamate, NAD and CD31 antigen.47–49 CD38 catalyzes the synthesis of a calcium messenger, cyclic ADP ribose (cADPR) from nicotinamide adenine dinucleotide (NAD+). 50 cADPR then activates ryanodine receptors (RyRs) in the ER 51 and this activation potentially amplifies intracellular signals within organelle network composed of mitochondria, 52 the Golgi apparatus, 53 vacuole 54 and lysosome. 55 Our initial data suggested that astrocytic CD38 may regulate the secretion of functional mitochondria through calcium-dependent signaling. 14 However, what still remains unclear is the mechanism of how extracellularly secreted mitochondria maintain their function outside cells.
O-GlcNAcylation is the process of a sugar molecule attachment to the oxygen atom of serine (Ser) or threonine (Thr) resides and occurs in nucleus, cytoplasm, ER and Golgi.56,57 The modification affects processes including cellular stress, cell cycle, protein stability and protein turn over. Therefore, dysfunction in this process may lead to the pathology of Alzheimer’s disease including neuronal cell death, neuroinflammation, increased production of hyperphosphorylated tau and Abeta-peptides and memory deficits.24–26 In cerebral ischemia, O-GlcNAcylation is markedly increased in brains of young mice, whereas the activation is diminished in aged brains. 58 Importantly, pharmacologic amplification of O-GlcNAcylation may improve neuroprotection, inhibit pro-inflammatory signaling in microglia and promote stroke recovery.59,60 In this study, we applied the idea of O-GlcNAcylation to extracellular mitochondria released from astrocytes and found that O-GlcNAc post-translational modification regulated by ER-Golgi trafficking plays a key role to maintain extracellular mitochondrial functionality and neuroprotective property. OGD itself also triggered O-GlcNAcylation but these responses were lower and transient compared to exogenous CD38-stimulation. Therefore, it is tempting to speculate that this potentially protective pathway may be induced endogenously after ischemia, but these levels are low and not sustained so that there is still “room for improvement” via further exogenous stimulation of this process to boost extracellular mitochondrial functionality.
Nevertheless, there are a few caveats that warrant further investigation. First, our study demonstrates that stimulated cortical astrocytes may produce functional extracellular mitochondria that are modified by O-GlcNAc. However, astrocytes are highly heterogeneous and pleomorphic within different brain regions. 61 Moreover, activated astrocytes under pathophysiological conditions may be classified into neurotoxic or neuroprotective phenotypes. 17 In addition to astrocytes, microglia also express CD38, 62 and inflammatory microglia may expand neurodegeneration via fragmented dysfunctional extracellular mitochondria. 17 Future studies are required to rigorously investigate how O-GlcNAc responses in extracellular mitochondria may be regulated in various activated astrocytes and microglia from different brain regions under normal versus diseased conditions. Second, our study show that ER-Golgi trafficking may regulate and promote mitochondrial protein O-GlcNAcylation. However, mitochondria have their own O-GlcNAc transferase (mOGT) and UDP-GlcNAc transporter SLC25A33.63,64 How these local pathways interact with the presently described Golgi-ER mechanisms should be explored. Third, how O-GlcNAcylated mitochondria are incorporated into neurons remains to be fully defined. It has reported that extracellular mitochondria can enter cells via endocytosis, 65 integrin-dependent mechanisms 14 or macro-pinocytosis. 66 Whether O-GlcNAc modification affects these entry mechanisms should be addressed in future studies. Fourth, we showed that O-GlcNAc-modified extracellular mitochondria retain higher membrane potentials and are neuroprotective. But how these positive effects operate once mitochondria are absorbed into recipient neurons remains unclear. For example, O-GlcNAcylated proteins activate internal protective pathways including the unfolded protein response (UPR). 67 More recently, studies show that UPR is also activated in mitochondria when mitochondrial OXPHOS is dysfunctional or unfolded proteins accumulate in mitochondria, and UPRmt promotes cell survival and mitochondria recovery.68,69 Ultimately, how O-GlcNAcylated mitochondria induces endogenous intracellular stress response should be dissected, in order to maximize mitochondria-mediated neuroprotection. Fifth, it is important to note that how astrocyte-originated mitochondria integrate with a new mitochondrial network of new physiological environment within recipient neurons remains to be fully understood. Our data showed that at 24 hrs post-transfer, O-GlcNAcylated signatures were retained in the majority of internalized mitochondria within neurons. It has been reported that reduced levels of O-GlcNAcylation may activate LAMP1-mediated autophagic degradation of mitochondria. 70 Is it possible that O-GlcNAc modification may extend mitochondrial viability by allowing them to escape autophagic degradation in neurons? Future studies are warranted to rigorously define mitochondrial survival mechanisms in neurons after mitochondrial transfer. Finally, how O-GlcNAc modification of mitochondria in astrocytes is affected in disease remains to be fully elucidated. Pro-inflammatory factors can induce neurotoxic astrocytes 71 and aging impairs astrocytic function. 72 Our data may be consistent with these ideas since O-GlcNAcylation in extracellular mitochondria was decreased when astrocytes were subjected to insults that mimic inflammation and aging. It should be useful to ask whether manipulating O-GlcNAcylation can ameliorate inflammation and aging pathology in the CNS.
Mitochondrial integrity is critical for CNS recovery after injury or disease.41,73 Within the emerging paradigm of extracellular mitochondria transfer, our study suggests that post-translational modification via O-GlcNAcylation in astrocytes may regulate the secretion of functionally beneficial extracellular mitochondria in the CNS, and this mechanism may contribute to neuroprotection after stroke. Further studies are warranted to ask whether these mechanisms can be manipulated for therapeutic gain.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported by grants from NIH.
Acknowledgement
Cytometric findings reported here were performed in the MGH Department of Pathology Flow and Image Cytometry Research Core.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author contributions
J.H.P., Y.N., W.L., G.H. contributed to conducted experiments and data analysis. K.A., E.H.L., and K.H. contributed to experimental design, J.H.P., E.H.L., and K.H. contributed to manuscript preparation.
Supplemental material
Supplemental material for this article is available online.