
Abstract
Local cerebral blood flow (CBF) responses to neuronal activity are essential for cognition and impaired CBF responses occur in Alzheimer’s disease (AD). In this study, regional CBF (rCBF) responses to the KATP channel opener diazoxide were investigated in 3xTgAD, WT and mutant Presenilin 1(PS1M146V) mice from three age groups using Laser-Doppler flowmetry. The rCBF response was reduced early in young 3xTgAD mice and almost absent in old 3xTgAD mice, up to 30%–40% reduction with altered CBF velocity and mean arterial pressure versus WT mice. The impaired rCBF response in 3xTgAD mice was associated with progression of AD pathology, characterized by deposition of intracellular and vascular amyloid-β (Aβ) oligomers, senile plaques and tau pathology. The nitric oxide synthase (NOS) inhibitor Nω-nitro-L-arginine abolished rCBF response to diazoxide suggesting NO was involved in the mediation of vasorelaxation. Levels of phosphor-eNOS (Ser1177) diminished in 3xTgAD brains with age, while the rCBF response to the NO donor sodium nitroprusside remained. In PS1M146V mice, the rCBF response to dizoxide reduced and high molecular weight Abeta oligomers were increased indicating PS1M146V contributed to the dysregulation of rCBF response in AD mice. Our study revealed an Aβ oligomer-associated compromise of cerebrovascular function in rCBF response to diazoxide in AD mice with PS1M146V mutation.
Introduction
Regional cerebral blood flow (rCBF) responses to local brain activity are critical to cognitive function. The rCBF response, which is coordinated with cerebrovascular reactivity, is reduced in Alzheimer’s disease (AD),1–4 albeit the underlying pathogenesis is not fully understood.5–7 Impaired cerebral vasoreactivity is associated with reduced CBF response, cerebral hypoperfusion and a deficit of brain energy metabolism, as well as aberrant neuronal excitability.8–10 Reduced cerebrovascular reactivity disrupts neurovascular coupling function, which could occur early in the disease, and play a pathogenic role on cognitive decline in AD. 11 , 12 Moreover, insufficient rCBF decreased the capacity on clearance of central nervous system amyloid-β peptide (Aβ), potentially resulting in the accumulation of Aβ aggregates in AD brains and development of inflammation reaction. 13 , 14 The rCBF response to neuronal activity involves complex signaling mechanisms, including ion channel network regulation and vasoactive mediators such as nitric oxide (NO).15–17 The neurovascular coupling function involves multiple cell types in the neurovascular units (neurons, endothelium, smooth muscle cells, pericytes and astrocytes).18,19 Studies of transgenic mice that express mutant forms of human β-amyloid precursor protein (APP) resulting in early-onset AD have demonstrated age-dependent reductions in rCBF that are correlated with deposition of Aβ pathology. 20 Notably, increases in rCBF that normally occur in response to increased neuronal activity and vasodilatory mediators, such as bradykinin and acetylcholine, are diminished in APP mutant transgenic mice 21 and may drive pathological process.
To investigate the roles of neurovascular functional coupling in the pathogenesis of AD, the rCBF response to diazoxide, an opener of the ATP-sensitive K+ channels (KATP) that coordinate brain energy state, neuronal excitability, vascular tone and CBF dynamics 15 ,22–24 was studied in the 3xTgAD, wild type and PS1m146v mutant mice. KATP channels are present in high amounts in neurons and cerebrovascular system. These channels open when cellular ATP/ADP ratio falls and thereby increases membrane permeability to K+ ions and hyperpolarizes the membrane potential (ΔΨs) in capillaries and vascular smooth muscle cells and result in vasorelaxation and CBF increase. 24 , 25 Membrane hyperpolarization also reduces calcium influx through voltage-gated calcium channels in smooth muscle cells thereby inhibiting vasoconstriction. 26 , 27 The activation of KATP channels in neurons can protect them against excitotoxic and ischemic injury in neuropathological conditions.28–30
We previously reported that diazoxide can protect cortical neurons against Aβ toxicity. 29 Diazoxide application improved stroke outcome in an experimental cerebral ischemia model, 30 and long-term administration of diazoxide started at early age lessens Aβ pathology and cognitive impairment in 3xTgAD mice. 27 These findings suggest a therapeutic potential for drugs that activate K+ channels in AD. However, it is unclear whether rCBF responses to diazoxide are altered with age advance in WT and AD mice; clearly a relevant question for an age-related neurodegenerative disorder. To evaluate this, we measured rCBF responses to diazoxide in young (6–8 months), middle-age (12–14 months) and old (24–28 months) 3xTgAD and wild type (WT) mice. We found that the rCBF response to KATP channel opener diazoxide declined early in 3xTgAD mice compared to age-matched WT mice and exacerbated with age advance. The age-related reduction on rCBF response was correlated to the progression of Aβ pathology in 3xTgAD mice. We found that NO, the active vasodilator, was involved in the mediation of rCBF response to diazoxide, and levels of active eNOS (pSer1177) were reduced in 3xTgAD mice with age, suggesting a role of deficient endothelium function in the compromised cerebrovascular reactivity. As 3xTgAD mice harbor a PS1M146V mutation, the rCBF response was further examined in PS1M146V knock in only mice without the human mutant form of APP or mutant tau, which revealed an age-related decline in rCBF response to diazoxide as well. Moreover, Aβ oligomers at high molecular weight (HMW) were elevated in brain soluble fractions of PS1M146V mice, indicating the PS1M146V mutation contributes to the neurovascular dysfunction in in 3xTgAD.
Material and methods
Animals
The generation and characterization of the original lines of PS1m146v mutant knock in mice and 3xTgAD mice were reported previously. 31 , 32 The 3xTgAD mice and PS1KI mice used in the present study were on a congenic C57BL/6 background. All mice used in this study are male, in the light of concerns that female hormone, such as estrogen, could influence CBF, and their levels would be expected to change from young to old age. Furthermore, a different estrogen cycle among individual female mice might increase variance within groups. Genotyping confirmed the three integrated mutant genes (APPKM670/671NLSwedish double mutation, human tauP301L(4R0N) and PS1M146V mutation) in 3xTgAD mice. The Aβ and human tau pathology were confirmed by immunohistology evaluation as described previously. 27 All procedures were approved by the National Institute on Aging Animal Care and Use Committee and complied with NIH guidelines. All in vivo studies on animals were reported with compliance to the ARRIVE guidelines (Animal Research: Reporting in Vivo Experiments).
Measurement of rCBF response
The rCBF responses to treatments were recorded and assessed using laser-Doppler flowmetry (LDF, PeriFlux System 5000, Perimed, Stockholm, Sweden) as described and modified.27,33 Mice were anesthetized lightly with isoflurane in oxygen gas (1.5%–2% for induction in anesthesia chamber, and 1%–1.5% for maintenance during the experimental procedure via a vapor mask). The regimens were consistent through the experiments to avoid unintended impact of anesthesia on rCBF responses across different groups of mice. Animal core body temperature was maintained at 37°C by the use of a thermo-controlled heating plate throughout the experiment and recovery from anesthesia. The mouse head was immobilized in a stereotactic frame, and an incision and a closed cranial window with thinned skull were created over the neocortex territory post and left to Bregma, the region supplied by the left middle cerebral artery. A flexible 0.5 mm diameter fiber optic LDF probe (Perimed; Probe 418, Stockholm, Sweden) was fixed on the thin surface of window. Drugs were infused into the femoral artery in a volume (100–200 µl) adjusted to body weight at the desired concentration and circulated via leptomeningeal/pial arteries, intracerebral arterioles and capillaries under the window following system administration. A catheter line was maintained if repeated drug administrations needed. The resting rCBF was recorded for at least 5 min prior to drug administration to ensure a stable baseline. The dynamic rCBF trace was, therefore, acquired and recorded for up to 1 h at 5 min intervals. After assessment on rCBF response to intervention, the skin incising was closed, and buprenorphine was administrated (1.0 mg/kg, subcutaneous) to relieve post-operation pain. Data on CBF responses were analyzed by PeriFlur software for hemodynamic values (Perimed) and expressed as percent change on rCBF (% ΔCBF) related to the baseline level.
Measurement of mean arterial pressure (MAP)
Mice underwent terminal invasive hemodynamic measurements under anesthesia with isoflurane (2% in oxygen) via a nosecone. Mice were placed in the supine position and kept at 37°C using a heating pad. After shaving skin hair in the frontal neck region, a small incision was made to access the right carotid artery. The right carotid artery was isolated and ligated. A 1F Mikro-Tip catheter (PVR-1045, AD Instruments, Colorado Springs, CO) was inserted into the carotid artery, and its tip was placed 0.5 mm above the aortic valve in the ascending aorta. Standard electrocardiogram (ECG) electrodes were placed on the limbs. Aortic blood pressure (BP) and Lead II ECG signals were recorded continuously throughout the experiment at a 1 k/s speed using a PowerLab System (AD Instruments, Colorado Springs, CO). After 10 min of recording, diazoxide (5 mg/kg) in saline was injected into the right jugular vein using a 30G needle; BP and ECG were recorded for an additional 60 min. BP was calculated as an average of every 1 min of recording. Normal BP was an average of 10 min recordings prior to diazoxide administration and was used to calculated percent changes of BP after diazoxide administration. All data are expressed as mean ± SD (standard deviation). Differences between two groups were determined by two tailed student’s t test. Statistical significance was set at p < 0.05.
Pharmacological reagents
Diazoxide, Nω-nitro-L-arginine methyl ester (L-NAME), sodium nitroprusside (SNP) and rhodamine B dextran (MW 200,000) were purchased from Sigma–Aldrich (St. Louis, MO). Glibenclamide was purchased from RBI (Natick, MA).
Immunoblot analysis
Mouse brain cortex tissues were prepared for western blot analysis as described previously. 27 Brain tissues were homogenized and solubilized in sample buffer (Invitrogen) containing cOmplete™ protease inhibitor cocktail and phosphatase inhibitors (Roche Diagnostics, Indianapolis, USA). The supernatants were collected after centrifuge (10,000 r/min for 30 min at 4°C) as the soluble protein fractions and subjected to concentration assay using a BCA kit (Pierce, Waltham, MA, USA). Protein samples (30 µg/each) were separated on precast 4%–12% NuPAGE gradient gels (Invitrogen, USA). Following electrophoretic transfer to a nitrocellulose membrane, blots were blocked in 5% nonfat milk in Tris-buffered saline (TBS-T) with 0.05% Tween 20 and incubated with the primary antibodies overnight at 4°C. The primary antibodies used were eNOS or nNOS (1:1000; BD Trasduction Lab, Le Pont de Claix, France), phospho-eNOS Ser1177 (1:1000; Cell Signaling Technology, Beverly, MA, USA), Kir6.1(1:1000; Santa Cruz BioTech Inc, Santa Cruz, CA, USA), β-amyloid antibody 82E1 (1:500; IBL, Minneapolis, MN, USA), PS1 antibody (1:500; which recognizing amino acid of the loop region of human presenilin 1 protein, generated by Guo et al. 31 ), and β-actin (1:2000; Sigma–Aldrich). After washing in TBS-T, the membranes were incubated with an HRP-conjugated secondary antibody (Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature. Blots were developed using an ECL chemiluminescence reagent (Pierce, Rockford, IL, USA). The integrated density of protein bands was quantified using Image J software (NIH) and normalized to the β-actin band density on the same blot.
Immunohistochemistry and rhodamine B dextran imaging
Mice were anesthetized with isoflurane and perfused intracardially, saline first and then 4% paraformaldehyde in phosphate buffered saline (PBS). Following euthanasia, the brains were removed immediately, post-fixed in 4% paraformaldehyde (PFA) in PBS overnight, cryoprotected by soaking in a solution of 30% sucrose in PBS at 4°C for 1 day, and then stored at −80°C. For immuno-histological assessment, serials coronal or sagittal brain sections were collected using a cryostat at 25 µm thickness. Endogenous peroxidases were quenched by incubation of sections in 0.3% hydrogen peroxide in methanol, and then sections were blocked with 5% normal goat serum or albumin in TBS buffer for 1 h before incubation with primary antibody overnight at 4°C. Primary antibodies used in our study included: for Aβ oligomers 82E1 (IBL America), β-amyloid 1-16 (6E10, Covance, Dedham, MA), β-amyloid peptide antibody (Cell Signaling, Danvers, MA), antibodies recognizing Phospho-Tau (Thr181) and human Tau (HT7) (Thermo Fisher Scientific) and antibodies to recognize astrocytes (GFAP, Chemicon) and microglial cells (Iba-1, Wako). Sections were then incubated for 1 h with Alexa-conjugated secondary antibody (Invitrogen, CA). After washing, sections were mounted in anti-fade solution with DAPI (Vector). Images were acquired using a confocal microscope (Zeiss LSM710). The rhodamine B dextran (MW 200,000, Sigma–Aldrich, St. Louis, MO) was prepared in buffered saline (1 mg/ml) and infused into mice intravenously. Mice were euthanized afterward, and brain sections were collected by cryostat and cerebral vascular labeling were acquired by confocal microscope.
Statistical analysis
Data are presented as mean ± SD (standard deviation). “R” software was used for some graphs and analysis. One-way analysis of variance (ANOVA) was performed for intergroup differences within three different mouse strains including WT, 3xTgAD and PS1m146V KI mice, and within three different age groups (young, middle and old age) of each mouse strain. Tukey’s post-hoc tests were performed for comparisons between two groups. Equality of data variance was assessed by F test. Two-tail student t tests were performed for normally distributed data from two groups, and probability p value <0.05 was considered statistically significant. Data on rCBF response were present as percent change of rCBF related to baseline level (% ΔCBF) in graphs at each time point of interval, with any statistically significance of difference indicated.
Results
The rCBF response to the KATP channel opener diazoxide was impaired early and progressively lost with age in 3xTgAD mice
Regional CBF responses to the KATP channel opener diazoxide (5 mg/kg) were evaluated in 3xTgAD and WT mice in three age groups (young, middle and old; n = 6–11 mice/group). Representative rCBF recording traces from mice in different age groups of 3xTgAD and WT mice were shown (Figure 1(a)). The administration of diazoxide via the intra-femoral artery induced a significant elevation of rCBF in young, middle-aged and old WT mice (Figure 1(a)–(d). The average percentage changes of rCBF (% ΔrCBF) from the three age groups of 3xTgAD and WT mice were presented as mean and SD at 5 min intervals (Figure 1(b) –(d)). The rCBF response to diazoxide was diminished and delayed in 3xTgAD mice compared to age-matched WT mice in all three age groups, up to 30%–40% reduction at the end of recording. The genotype difference appeared early in the young age groups (Figure 1(b)) and was exacerbated in older mice (Figure 1(c)–(d)). *p < 0.05; **p < 0.01, two tailed t test. In the old 3xTgAD mice, the rCBF to diazoxide was unstable and almost no significant increase within 1-h recording (Figure 1(d)). Figure 1(e) showed the rCBF dose-response to diazoxide in WT mice (∼1 year). The time response of rCBF to diazoxide (5 mg/kg) was plotted in in young mice (Figure 1(f), n = 8, 4–6 months age).
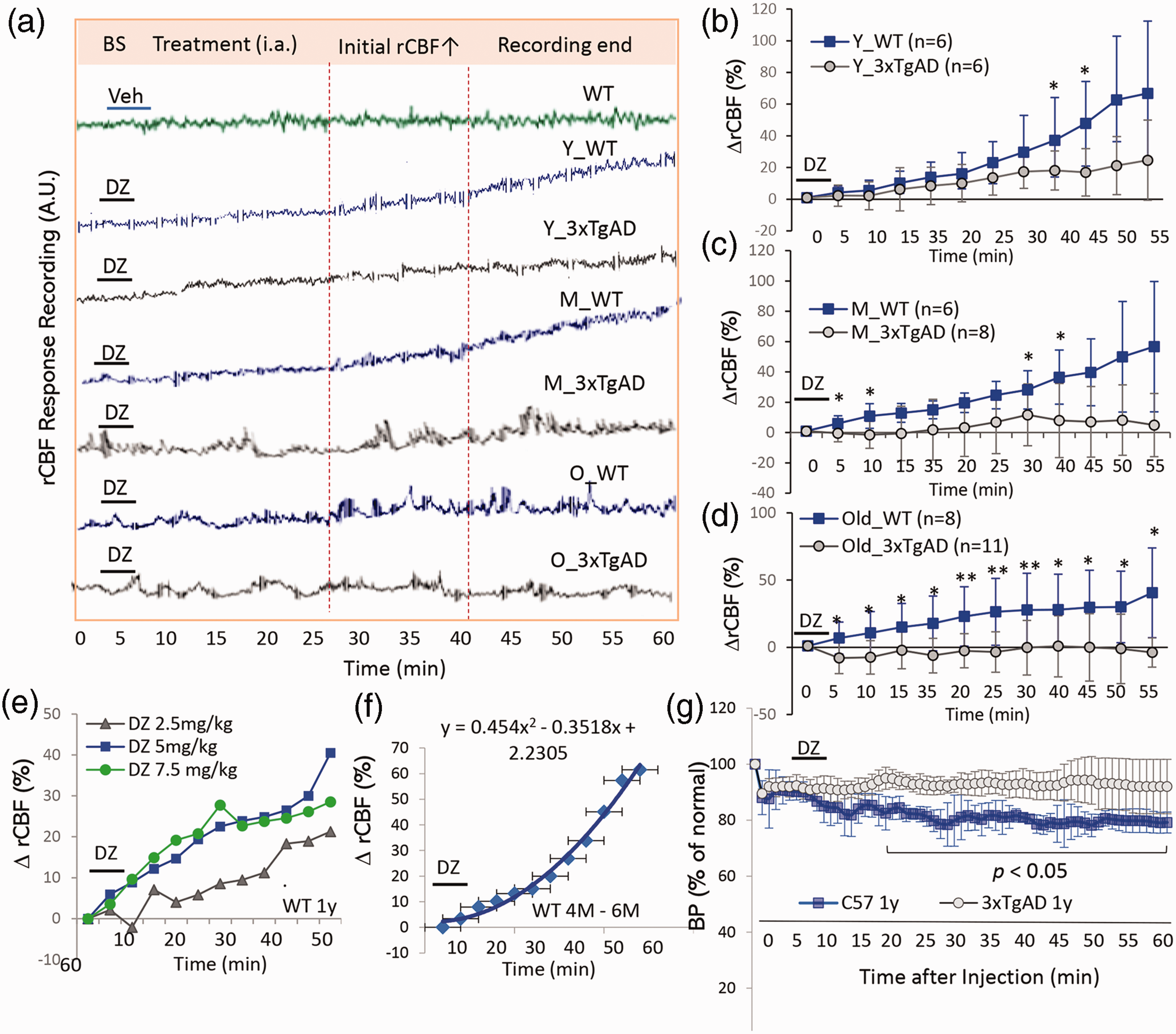
Age-related impairment of rCBF response to diazoxide in AD mice. (a) Representative rCBF traces recorded from young (Y), middle-aged (M) and old (O) WT and 3xTgAD mice after administration of diazoxide (5 mg/kg, i.a.). (b, c, d) Average percentage changes of rCBF (% ΔrCBF) from baseline in response to diazoxide in three age groups of WT and 3xTgAD mice. Two tailed t test used to analyze difference between 3xTgAD and WT mice in each age group; Data are mean ± SD; **p < 0.01, *p < 0.05, n = 6–11 mice/group. (e) The rCBF response to three different doses of diazoxide in WT mice (1 year). (f) Kinetics of rCBF response to diazoxide in young WT mice (5 mg/kg, 3–6 months, n = 8). (g) Mean aortic blood pressure (MBP, in mmHg) in response to diazoxide administration (5 mg/kg, i.v.) in 3xTgAD and age-matched WT mice (n = 4 mice/group, 1 year). Data expressed are the mean ± SD of % change of MBP to baseline. *p < 0.05, two tailed Student t test between 3xTgAD and WT mice.
As described in Poiseuille’s law on CBF, in our model, the intra-cranial pressure (ICP) and velocity (η) are not expected to change significantly before and after drug treatment during the recording period. The diameter of resistance artery (r) and the MAP will be the determinants to affect CBF. The mean blood pressure (MBP) was measured in separate groups of WT and 3xTgAD mice (1 year old) to determine whether diazoxide reduced systemic BP in mice (Figure 1(g)). The MBP was reduced in response to diazoxide (5 mg/kg, i.v.) in both WT and 3xTgAD mice, with a significantly greater effect in WT mice (∼20% decrease in MAP) compared to 3xTgAD mice (<10% decrease in MAP). Two tail t test was performed on percent change of MAP from baseline levels at each time point, n = 4 mice/group, *p < 0.05 as indicated at time points with significant changes. The MAP study supports the reduced arterial vaso-relaxation in response to diazoxide in 3xTgAD versus age-matched WT mice.
The age impact on rCBF responses to diazoxide in WT and 3xTgAD mice
In WT mice, the rCBF responses to diazoxide were not significantly different in young compared to middle-age mice (Figure 2(a)), while rCBF response in old WT mice showed reduced tendency compared to young WT mice (#p = 0.05, n = 6–8 mice/group). One-way ANOVA of % ΔrCBF of WT mice from three age groups were p = 0.13, df =19, F = 2.24 at the end of recording. In contrast, within 3xTgAD mice, the rCBF responses were reduced early in the young age group (up to 30% reducing compared to young WT mice). There were further age-related reductions in rCBF response to diazoxide among the two further aged groups of 3xTgAD mice (Figure 2(b)). Particularly notable, the % ΔrCBF declined significantly in old 3xTgAD mice compared to young 3xTgAD mice (*p < 0.05, two tail t test, n = 7–11 mice/group). One-way ANOVA of the end of recording from the three age groups of 3xTgAD mice were p = 0.06, df = 24, F = 2.24. The ranges of % ΔrCBF from individual mouse at the end point of recording from WT or 3xTgAD mice of three age groups are presented in Figure 2(c). The average % ΔrCBF was significantly lower in 3xTgAD mice compared to age-matched WT mice in all age group (*p < 0.05; **p < 0.01).
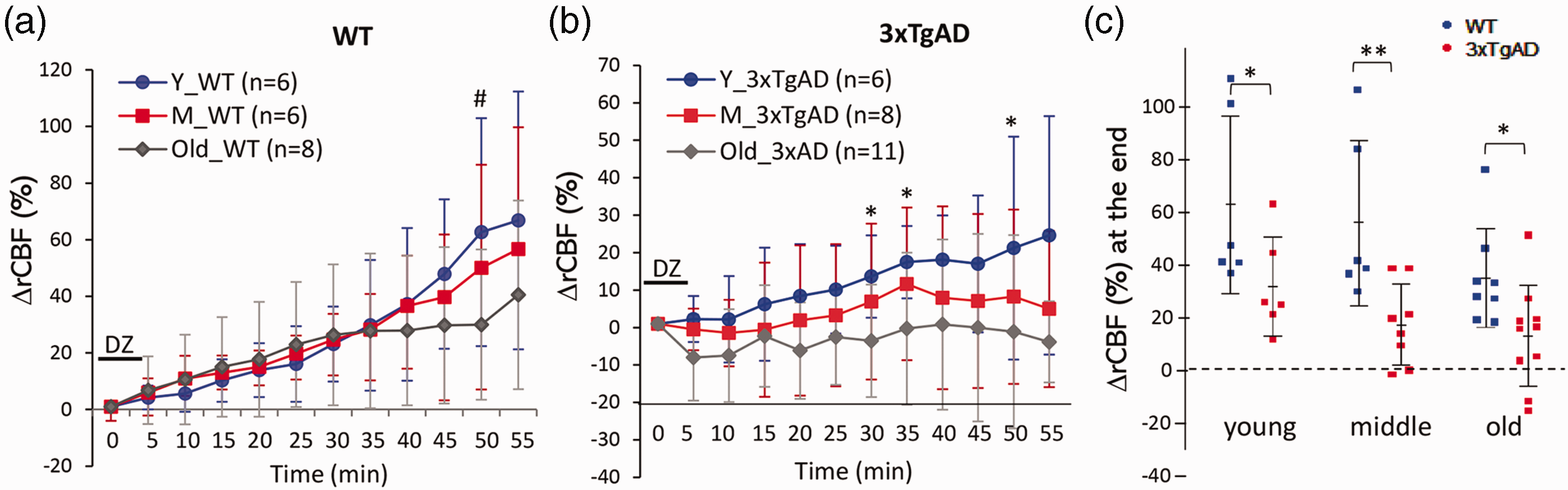
Age impact on rCBF responses to diazoxide in WT and 3xTgAD mice separately. (a) The % ΔrCBF in response to diazoxide within three age groups of WT mice (one-way ANOVA, p = 0.13; df = 19; F = 2.24). There was no significant difference between young and middle-aged WT mice, though % ΔrCBF reduced in old WT mice compared to young WT mice (#p = 0.05, t test; n = 6–8 mice/group). (b) The % ΔrCBF to diazoxide in three age groups of 3xTgAD mice (one-way ANOVA, p = 0.06, df = 24; F = 2.24). There was an age-related reduction of rCBF response in young versus old 3xTgAD mice; *p < 0.05, two-tail t test; n = 6–11 mice/group. (c) Summary on the difference and distribution of % ΔrCBF at the end of the recording from animals between WT and 3xTgAD mice in three age groups *p < 0.05; **p < 0.01, two-tail t test; n = 6–11 mice/group.
We also analyzed the kinetics of rCBF responses to diazoxide. The rCBF velocity was characterized by increased latency (delayed initial time) on rCBF response (time to reach 10% rCBF increase), reduced peak amplitude; and slow rate of rCBF elevation (time to 20% increase) in middle and old aged 3xTgAD mice compared to age matched WT mice (Table 1). There was a delay in the onset of the rCBF response to diazoxide in all three age groups of 3xTgAD mice (Table 1).
The rCBF velocity in response to diazoxide in 3xTgAD and WT mice from three age groups.
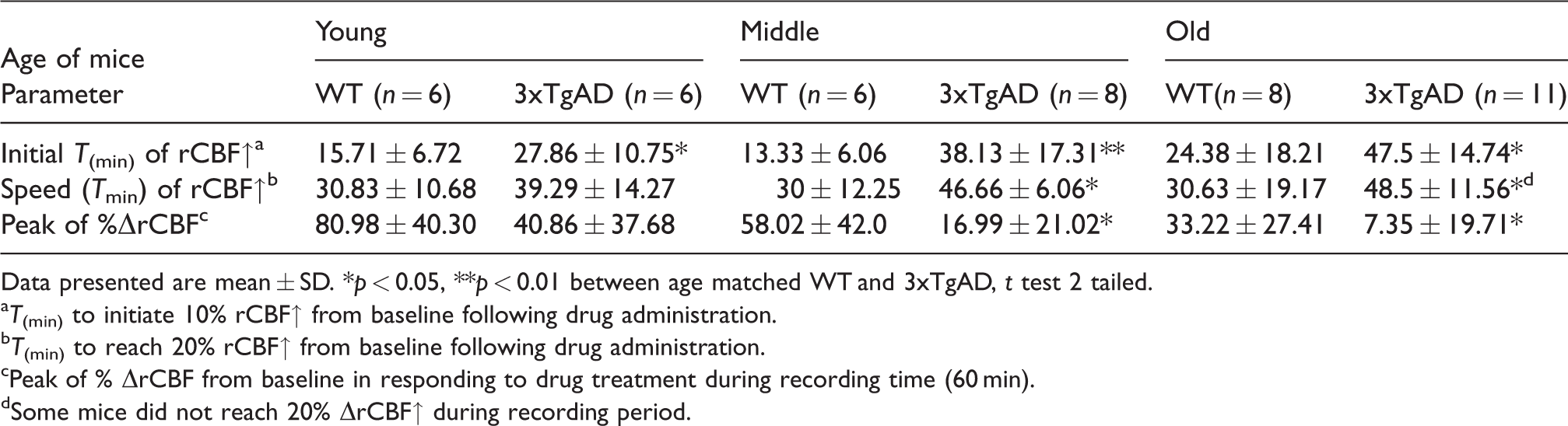
Data presented are mean ± SD. *p < 0.05, **p < 0.01 between age matched WT and 3xTgAD, t test 2 tailed.
T(min) to initiate 10% rCBF↑ from baseline following drug administration.
T(min) to reach 20% rCBF↑ from baseline following drug administration.
Peak of % ΔrCBF from baseline in responding to drug treatment during recording time (60 min).
Some mice did not reach 20% ΔrCBF↑ during recording period.
Age-dependent progression of Aβ-peptide and mutant hTau pathology in 3xTgAD mice
We next examined AD-related brain pathology biomarkers, Aβ-peptide and mutant human tau (hTau) in 3xTgAD mice. There was an age-related progression of Aβ pathology in the hippocampus and cerebral cortex of 3xTgAD mice. In young 3xTgAD mice (6–8 months), no obvious extracellular Aβ plaques were observed (Figure 3(A-a)), although intracellular Aβ was detected in some neuronal cell bodies within the hippocampus (b). Extracellular Aβ plaques were, however, apparent in middle-aged 3xTgAD mice and observed early in brain regions of subiculum (c) and amygdala (not shown). The accumulation of plaques was spread throughout the cerebral cortex and hippocampus in old 3xTgAD mice (d). Mutant human tau pathology (hTau) appeared early (8–9 months) in the isocortex, hippocampus (e) and entorhinal cortex (f). Figure 3(b) demonstrates that Aβ immunoreactivity was also detected in cerebral vessels (arrow indicated) in brain parenchyma (b–d) and choroid plexus (c) with antibodies recognizing Aβ peptides or oligomers. Representative inflammation, as evidenced by the presence of activated astrocytes (GFAP) around amyloid plaques (e) and microglia (Iba1) in regions with phospho-tau (Thr181) stained vessels (f) was present in images from middle aged (16 M) and old (24 M) 3xTgAD mouse brains.
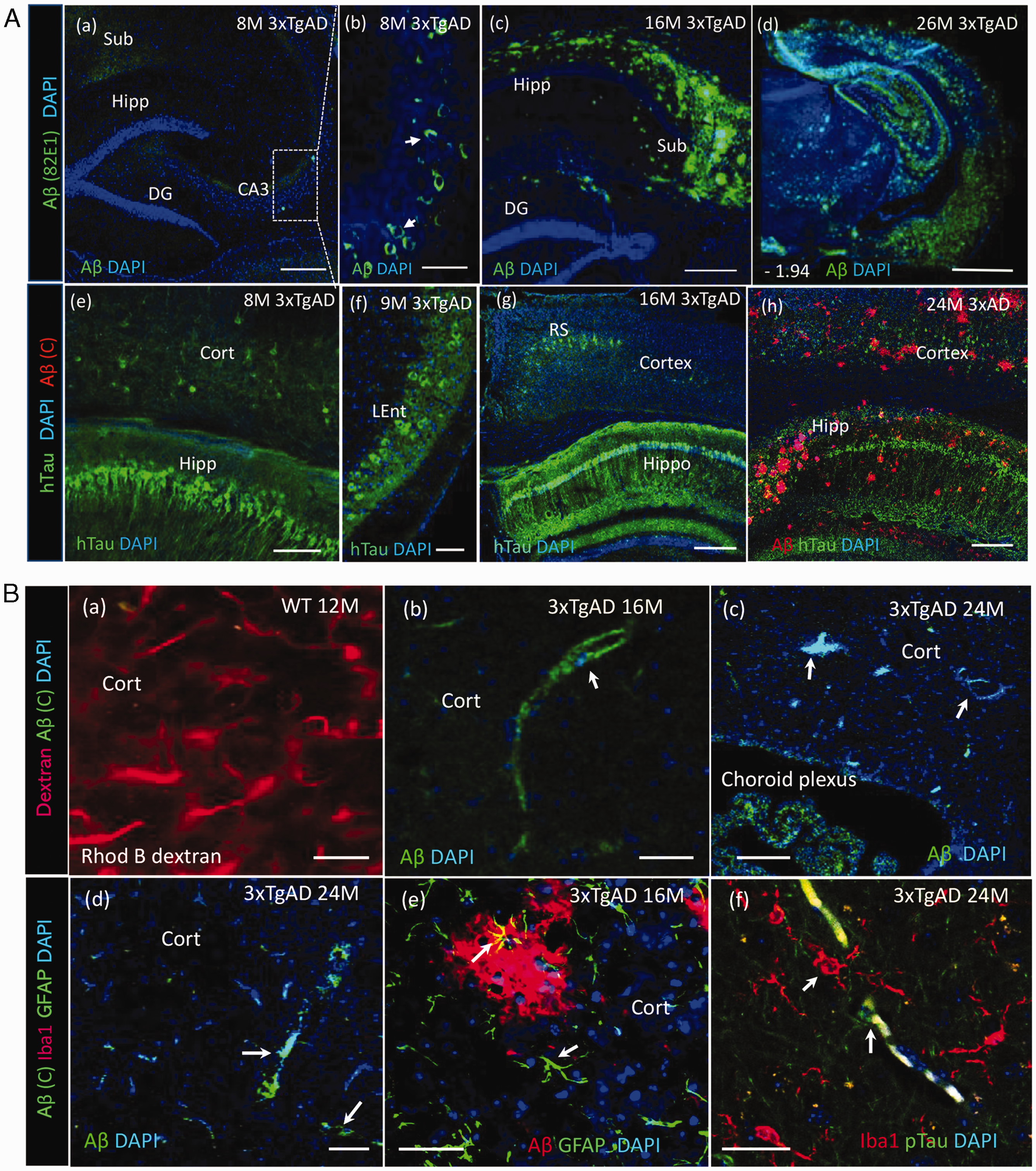
Age-related progression of Aβ and tau pathology in 3xTgAD mice. (A). Confocal images showed deposition of Aβ and human mutant tau (hTau) with age advance in 3xTgAD mice. (a) Representative image showed extracellular Aβ plaques were not evident in hippocampus of a representative young 3xTgAD mouse (8 months). Scale 100 µm. (b) Whereas intracellular Aβ oligomers were observed in some cells at CA3 region, scale 20 µm. (c) Extracellular Aβ plaques appeared abundantly at the subiculum of a middle-aged 3xTgAD mouse (16 months). (d) Widespread Aβ plaques in an old 3xTgAD mouse, scale 250 µm. (e, f) hTau pathology appeared early at neocortex, hippocampus, scale 100 µm and entorhinal cortex, scale 25 µm from a young 3xTgAD mouse. (g) The positive staining of hTau in RS and hippocampus of a middle aged 3xTgAD mouse, and (h) Co-staining of hTau (green) and Aβ (red, Cell Signaling) in hippocampus of an old 3xTgAD mouse (24 months). (B). Cerebral amyloid angiopathy in 3xTgAD mice. (a) Rhodamine B dextran in vivo labeling showed cerebral vasculature in a WT mouse, scale 20 µm. (b, c, d) Representative Aβ immunoreactivity in cerebral vessels and choroid plexus from middle-aged and old 3xTgAD mice. Scale 20 µm. (e) Activated astrocytes (GFAP, green) around amyloid plaques (6E10, red) in brain parenchyma of a middle aged 3xTgAD mouse. (f) Activated microglial cells (Iba1, red) with phospho-tau (Thr181, green) co-staining within the parenchyma from an old 3xTgAD mouse (arrows indicated). Scale 20 µm.
NO is involved in rCBF response to diazoxide and active eNOS (p-eNOS Ser1177) is reduced in 3xTgAD mice
To examine the signaling pathways and determine whether NO is involved in the mediation of rCBF response to diazoxide, WT mice were treated with the nitric oxide synthase (NOS) inhibitor Nω-nitro-L-arginine (L-NAME) before or after diazoxide administration. L-NAME treatment completely abolished the rCBF response to diazoxide (Figure 4(a) and (b), rCBF recording and % ΔrCBF to diazoxide without L-NAME (59.64 ± 34.63) compared to % ΔrCBF with L-NAME (6.4 ± 17.43); p = 0.017, n = 4–8 mice/group), indicating that NO was involved in the mediation of diazoxide-induced vasorelaxation. Pre-treatment with KATP channel blocker glibenclamide also blocked the rCBF responses to diazoxide (Figure 4(a)), supporting that the rCBF response was associated with vascular KATP channel activation. To determine whether cerebral vessels in AD mice were still responsive to NO, NO donor SNP was administrated (0.5 mg/kg) and rCBF was measured. SNP treatment induced a rapid but not long-lasting elevation of rCBF in both WT and 3xTgAD mice (Figure 4(c)). Notably not lower on % ΔrCBF in response to SNP in 3xTgAD mice compared to WT mice (Figure 4(c)), probably even basal perfusion level of rCBF was lower in 3xTgAD mice, a relatively intact vascular reactivity (vasorelaxation) retained in response to NO induced by SNP (Figure 4(d)). The conserved cerebrovascular reactivity to SNP, but impaired response to diazoxide suggested the possibility of a deficit in endothelium eNOS and bioavailability of endothelial derived NO in cerebral vessels of 3xTgAD mice. The levels of eNOS and phospho-eNOS (Ser1177) were hence examined in cortex tissues from middle-aged and old WT and 3xTgAD mice. There was a non-significant trend of reduced eNOS and p-eNOS levels in cortex samples from middle-aged 3xTgAD mice compared to middle-aged WT mice (Figure 4(e) and (g)). Levels of eNOS were moderately lower (p = 0.06) and p-eNOS (Ser1177) were lower significantly (*p = 0.01, n = 5, two tailed t test) in cortex of old 3xTgAD mice compared to age-matched WT mice (Figure 4(e) and (g)). In contrast, levels of nNOS were elevated in cortex samples from middle-aged or old 3xTgAD mice compared to age-matched WT mice (Figure 4(f) and (h)). To determine whether the reduced rCBF response to diazoxide in 3xTgAD mice might result from a reduction in levels of KATP channel component, we measured relative levels of the KATP channel protein Kir6.1, 35 a subunit critical in the control of CBF by immunoblot analysis. There was a nonsignificant trend towards reduced Kir6.1 level in cerebral cortical samples from aged 3xTgAD mice compared to WT mice (Figure 4(i) to (j)). Others have reported aberrant expression of Kir6.2 but no significant change of Kir6.1 in old 3xTgAD mice. 36
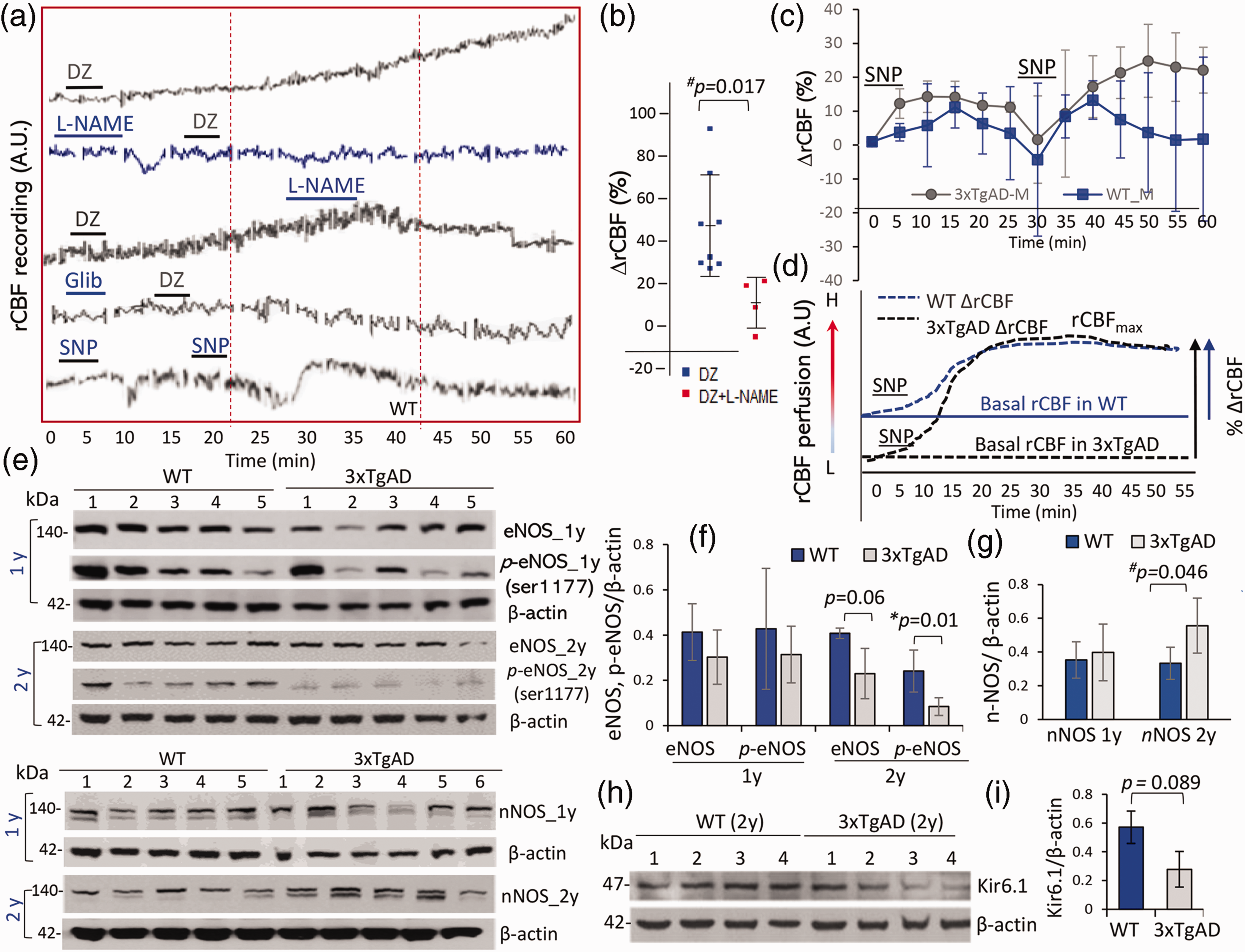
NO involved in the mediation of rCBF response to diazoxide. (a,b) Representative rCBF recording traces in responding to treatments. The NOS inhibitor L-NAME (0.5 mmol/l, administered before or after diazoxide) abolished the rCBF response to diazoxide in WT mice (p < 0.05, two-tail t test; n = 4–8 mice/group, ∼1 year). KATP channel blocker glibenclamide (20 µM) blocked rCBF response to diazoxide (n = 2). (c) Administration of the NO donor SNP (0.5 mg/kg) induced a rapid elevation of rCBF in both WT and 3xTgAD mice (n = 3 mice/group, 1 year). The difference on % ΔrCBF between 3xTgAD and age-matched WT mice was not significant. (d) An illustration that SNP induced rCBF response (arbitral unit on CBF level) bypassed deficit in endothelium function as vascular reactivity preserved in AD mice at the age even baseline CBF was lower than WT. (e, g) Western blots showed reduced levels of eNOS and phosphor-eNOS (Ser1177) in cortex tissues of 3xTgAD mice compared to age-matched WT mice from two age groups (*p = 0.01 in 2 year mice, n = 5 mice/group). (f–h) Levels of nNOS in 3xTgAD mice compared to WT mice from two age groups, which was higher in 2 years mice (#p = 0.046; n = 5 mice/group). (h–i) Levels of Kir6.1, the subunit of KATP channels in vascular cells, were moderately lower in 3xTgAD than WT mice (p = 0.089; n = 4 mice/group, two-tail t test). Data were mean ± SD.
Diminished rCBF response to diazoxide and elevated brain Aβ oligomers in PS1M146V mutant mice
Since 3xTgAD mice harbors the mutant presenilin-1 (PS1M146V), in which the wild-type PS1 allele is replaced with the mutant allele under its own promotor, the rCBF response to diazoxide was further examined in mice only expressing mutant PS1M146V (PS1 KI) in the absence of mutant human APP and Tau, to clarify the possible role of PS1 mutation on cerebrovascular function of AD mice. The rCBF response in young PS1 KI mice showed a tendency to decline but not significantly compared to young WT mice, whereas the rCBF response in young WT was lower than young 3xTgAD mice (
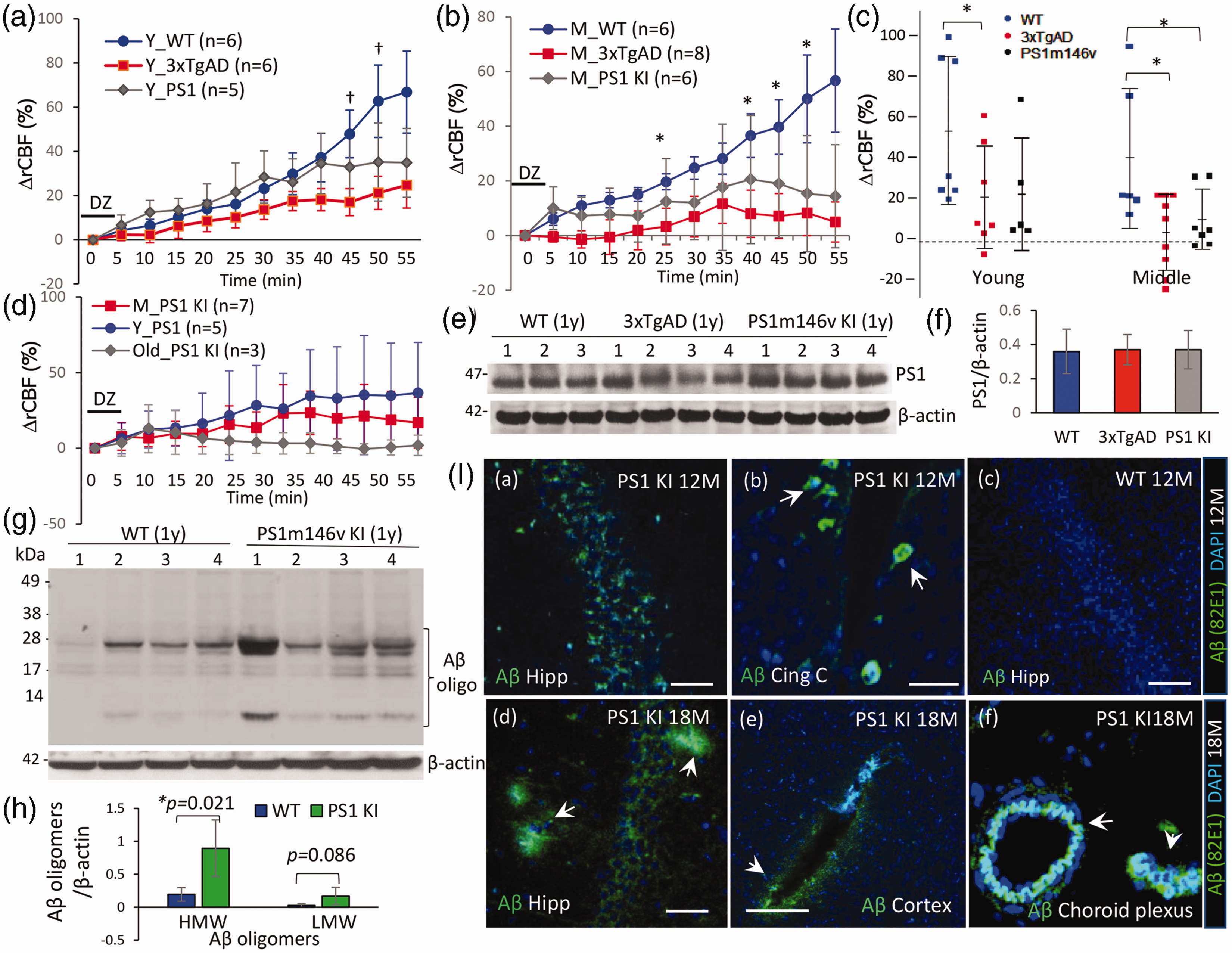
Reduced rCBF response to diazoxide and increased Aβ oligomers in mutant PS1M146V mice. (a) In young PS1M146V mice the rCBF response to diazoxide was lower but not significantly versus young WT mice, whereas the rCBF responses in young 3xTgAD mice were reduced significantly versus young WT mice (†p < 0.05). (b) Middle-aged PS1M146V mice demonstrated a significant decline in their rCBF response versus WT mice (*p < 0.05, n = 6 mice/group). (c) The % ΔrCBF to diazoxide at the end of recording from WT, 3xTgAD and PS1M146V mice from two age groups. One-way ANOVA *p = 0.010; df =14, F = 5.95 within three groups; n = 5–8 mice/group. Difference between each two groups was analyzed by unpaired two-tail t test, *p < 0.05 as indicated. (d) The age impact on % ΔrCBF to diazoxide in PS1M146V mice. (e–f) Western blot showed presenilin 1 protein levels were not different in cortex tissues from WT, 3xTgAD and PS1M146V mice (∼1 year, n = 3–4 mice/group). (g–h) Western blot detected elevated Aβ oligomers (82E1) at high molecular weight (HMW) in soluble fractions of cortex tissue from PS1M146V mice (*p = 0.021, two-tail t test; n = 4 mice/group). (i) Immunohistochemistry revealed intracellular Aβ oligomer deposition at hippocampus (a), and cingulate cortex (b) of PS1M146V mice (1 year, scale 100 µm), but no evident extracellular plaques at this age. In older PS1M146V mice (18 months), occasional small amyloid plaques were observed at hippocampal region (d), and Aβ immunoreactivity was also observed in cerebral vessels (e) and vessel wall at choroid plexus (f). Scale 50 µm (I-b,e,f).
Immunohistochemistry detected an increased deposition of intraneuronal Aβ oligomers in the hippocampus and cingulate cortex regions of one-year-old PS1M146V mice (Figure 5(I-a,b)), without evident extracellular Aβ plaques observed at this age. In older (18 months) PS1M146V mice, occasional small extracellular amyloid deposits/plaques were evident (Figure 5(I-d) and Aβ positive staining detected in cerebral vessels and vessel wall at choroid plexus with amyloid oligomer antibody (Figure 5(I-e) and 5(I-f)). Together, these results revealed that the mutant presenilin-1 (PS1M146V) contributes to the impaired cerebrovascular function in AD mice with elevated levels of Aβ oligomers and cerebral amyloid angiopathy (CAA) in brain, even without significant deposition of extracellular β-amyloid plaques at brain parenchyma.
Discussion
KATP channels are expressed in cells that comprise the brain neurovascular unit which mediates rCBF responses to neuronal network activity including neurons and vascular endothelial cells and smooth muscle cells. 23 , 24 , 27 We previously reported that diazoxide can protect cultured neurons against excitotoxicity, metabolic stress and Aβ toxicity, 29 , 30 each of which are considered key elements in mechanisms underpinning AD progression. Long-term treatment of 3xTgAD mice with diazoxide initiated at an early stage ameliorates cognitive deficits and reduces brain pathologies. 27 In the present study, we show that diazoxide increases rCBF in young, middle-aged and old WT mice, as well as young 3xTgAD mice. However, the rCBF response to diazoxide is greatly reduced in middle-aged and old 3xTgAD mice, without treatment. The age-exacerbated impairment of the rCBF response to diazoxide in 3xTgAD mice is associated with the progression of Aβ pathology in AD brains, as evidenced by increased levels of Aβ oligomers in brain tissues, deposition of senile plaques and amyloid angiopathy. The rCBF response to diazoxide is abolished by the NOS inhibitor L-NAME, thereby indicating that NO, the active vasodilator, was involved in the vaso-relaxation induced by diazoxide. Notably, levels of eNOS were lower in both one- and two-year-old 3xTgAD versus WT mice, and active p-eNOS (Ser1177) was significantly lower in aged 3xTgAD mice, in line with the finding that phosphorylation of eNOS at Ser1177 is reduced in aortas of APP mice. 38 Our study demonstrated an early functional changes on cerebral vascular reactivity in AD mice, the underlying mechanism linked to Aβ oligomer accumulation associated deficit of endothelial function on signaling and regulation of CBF response. Whereas 3xTgAD mice still exhibit a rCBF response to the NO donor, SNP indicating rCBF responsivity to NO is retained and potentially accessible.
The accumulation of Aβ oligomers in AD brain has the potential to impair vascular endothelial function, compromise the regulation of vascular reactivity and CBF response, even before the apparent deposition of extracellular Aβ plaques. 20 , 39 , 40 Deposition of Aβ oligomer aggregates can trigger cerebral inflammation as well as induce vascular neutrophil adhesion and inflammation cell infiltration. 14 Aβ oligomers can also induce constriction of cerebral capillaries that can further reduce CBF and decrease Aβ peptide clearance; thus exacerbating Aβ oligomer formation and plaque accumulation in AD bains. 41 In 3xTgAD mice, mutant tau pathology is evident early (Figure 3(a)), and its potential role in the early diminished rCBF response in 3xTgAD mice hence warrants further investigation. Previous studies have linked tauopathy to neurovascular changes and CAA in AD. 42 Our studies show that the rCBF response to diazoxide was reduced in PS1M146V mutant mice as well, even in the absence of mutant human APP or Tau expression. Moreover, levels of HMW Aβ oligomers were substantially elevated in the soluble fraction of brain tissues obtained from PS1M146V mice, even in the absence of observable accumulation of extracellular Aβ plaques (Figure 5(g) to (i)). PS1 is a critical component of the ϒ-secretase complex involved in the cleavage pathway of APP. Key mutations in PS1 can contribute and exacerbate Aβ pathology in AD preclinical models, such as in 3xTgAD or APP/PS1 mice that both harbor a PS1 mutation relevant to AD onset. 34 , 37 The impaired rCBF response in PS1M146V mice indicates that such a specific PS1 mutation can, at least, partially contribute to the impaired rCBF response to diazoxide in AD mice, which provides a connection between the exacerbated formation of toxic Aβ oligomers and disturbed cerebrovascular regulation evident in human AD.39–41 Mutations in the PS1 and PS2 genes cause the most common form of early onset dominantly inherited familial Alzheimer’s disease (FAD) and previous studies have reported that PS1 mutation alters intracellular Ca2+ regulation and lysosomal function as well. 43 , 44 Our study’s revelation that PS1M146V mutation can disrupt cerebrovascular reactivity, and the CBF response suggests potential avenues for future investigations to further evaluate the role of presenilin mutation on cerebrovascular function in AD pathogenesis.
Previous studies have reported altered cerebrovascular reactivity in AD patients with advanced CAA, as well as compromised rCBF regulation and reduced glucose utilization in response to neural activity in mice with APP mutations.20,21,45 Other studies have demonstrated that Aβ can damage both neurons and vascular endothelial cells by mechanisms involving oxidative damage and impaired cellular Ca2+ handling. 46 , 47 Our findings suggest that a reduced cerebral vascular reactivity and rCBF response to selected drugs, such as diazoxide, could provide an early functional biomarker and possible mechanism involved in AD pathogenesis, as these additionally provide connections to vascular risk factors, impaired neurovascular coupling and cognitive function that are known to occur in AD. 48 It is notable that cerebral blood vessel in the 3xTgAD mouse model retain their ability of responding to NO even in the presence of Aβ and tau pathology. NO mediates vasorelaxation by activating soluble guanylate cyclase in vascular smooth muscle cells, an endothelium-dependent NO-cGMP pathway that generates cyclic GMP. 49 , 50 Our findings therefore suggest that drugs that elevate cGMP levels, such as by inhibiting phosphodiesterases that regulates cGMP levels via hydrolysis, might be effective in improving CBF perfusion and cognition in AD patients. Support for this notion, derives from preclinical studies showing that the phosphodiesterase 5 inhibitor sidenafil ameliorates cognitive deficits in APP mutant and APP/PS1 double-mutant transgenic AD mice. 51 , 52 Sildenafil also ameliorated a cognitive deficit resulting from NOS inhibition in rats. 53 Moreover, it was recently reported that a single dose of sildenafil increased CBF in AD patients. 54 Whether drugs that elevate cyclic GMP levels also improve cognition in AD patients remains to be determined.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Intramural Research Program of the National Institute on Aging (NIA), National Institutes of Health.
Acknowledgments
The authors are grateful to Drs. S. Camandola and M. Wilson, Laboratory of Neuroscience, NIA, for advice and technical support. Dr. Z. Zhou, Department of Neurology, Xinqiao Hospital, Chongqing, China, and Dr. C. Maharana in LNS NIA provided technical support and Dr. B. Zamora, Comparative Medicine Section, NIA, for veterinarian regulation.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author contributions
D.L. planned and performed LDF experiments and data analysis, histology and co-wrote and revised the manuscript. I.A. planned and performed the measurement on mean arterial pressure and data analysis. B.G. performed biochemistry assays and data analysis. M.P.M planned and co-wrote the manuscript. T.D. and N.H.G. responded to reviewer’s comments and substantially revised the manuscript.