
Abstract
Stroke is a major cause of death and disability in the United States and around the world with limited therapeutic option. Here, we discuss the critical role of mitochondria in stem cell-mediated rescue of stroke brain by highlighting the concept that deleting the mitochondria from stem cells abolishes the cells’ regenerative potency. The application of innovative approaches entailing generation of mitochondria-voided stem cells as well as pharmacological inhibition of mitochondrial function may elucidate the mechanism underlying transfer of healthy mitochondria to ischemic cells, thereby providing key insights in the pathology and treatment of stroke and other brain disorders plagued with mitochondrial dysfunctions.
Keywords
Accumulating evidence has implicated dysfunctional oxygen consumption and metabolic activity in stroke.1,2 We now recognize a close interaction between neuronal impairment of mitochondria, key organelles for energy production, and the onset and progression of secondary cell death in stroke. Many stroke symptoms may be the result of reduced oxygen availability and consequent energy production deficits. Accordingly, mitochondria-based regenerative medicine may represent as an appealing therapeutic approach for stroke.3,4
Mitochondrial dysfunction in stroke leads to deficits in bioenergetics, pointing to mitochondria as a primary exacerbating factor in stroke secondary cell death. Thus, restoration of mitochondrial function may have a critical role in preventing stroke progression. Our overarching hypothesis (Figure 1(a)) entails that following the primary ischemic insult, a cascade of secondary cell death events ensues, with dysfunctional mitochondria as major exacerbating factor, resulting in impaired cellular bioenergetics of the stroke brain. Multiple downstream pathways involve cell death processes that specifically target inflammation,
5
which in turn may compromise the neurovascular unit (neuronal, astrocytic, pericyte, and endothelial cell death). As impaired mitochondria play a critical role in pro-inflammatory signaling, the repair of mitochondrial function by stem cell therapy may sequester inflammation and facilitate central nervous system (CNS) homeostasis. The putative therapeutic action of stem cell in stroke may involve functional repair via healthy mitochondria transfer into the ischemic brain, fostering a therapeutic vasculome
6
capable of dampening the deleterious inflammation mediators leading to secondary cell death.
Characterization of mitochondrial transfer in stroke. (A) Our overarching hypothesis advances the concept that mitochondrial dysfunction is a major secondary cell death process in ischemic stroke, which is accompanied by impaired cellular bioenergetics. A key downstream pathway of dysfunctional mitochondria involves inflammation that compromises the neurovascular unit (neurons, astrocytes, and endothelial cells). Mitochondria-based regenerative medicine via stem cell therapy, combined with tissue-plasminogen activator (tPA) recanalization that restores blood flow and re-entry of nutrients and energy to the cerebral vasculature, is hypothesized to sequester inflammation and to aid in recapturing CNS homeostasis. We propose mitochondria repair as a novel stroke therapeutic pathway applicable to regenerative medicine; in particular, our envisioned mechanism of action entails functional restoration of bioenergetics through the transfer of healthy mitochondria into the ischemic brain, harnessing a therapeutic vasculome that robustly sequesters the toxic inflammation-plagued secondary cell death associated with stroke and other disorders characterized by mitochondrial deficits. (B) Mitochondrial morphology and distribution in normal and OGD-exposed cortical neurons. Representative pictures show cortical neurons stained for the neuronal marker MAP2 (red) and mitochondrial ATP5A (green). Scale bar = 10 µm. Cortical neurons (2 × 105) were plated and allowed to stabilize for five days. Cells were then exposed to control conditions (complete media, 5% CO2, 21% O2) or OGD (PBS, 5% CO2, 95% N2) for 90 min. Twenty-four hours later, cells were probed with MAP2 and ATP5A antibodies. Boxed areas are shown as magnified images of tubular and circular mitochondria. (C) Quantification of healthy tubular mitochondria in control and OGD neurons. Neurons exposed to OGD exhibited ∼35% less tubular mitochondria than controls (***p < 0.0001 by Student’s t test). (D) Calcein assay showing significantly reduced cell survival (∼55%; *p < 0.05) upon OGD exposure (b) relative to controls (a). Bar graphs represent mean ± SD of three individual experiments. Scale bar = 100 µM. (E) Bright-field images of neurons grown in ambient conditions, OGD, and OGD co-cultured with EPCs. Arrows indicate outgrowth processes, which were significantly reduced in OGD conditions and rescued when neurons were co-cultured with EPCs. (F) Oxygen uptake in digitonin-permeabilized neurons (2 × 106) was recorded with a Clark’s oxygen electrode (Oxygraph Plus System, Hansatech, UK) under phosphorylating conditions as described.
11
The activity of citrate synthase was evaluated by spectrophotometry (EnSpire plate reader, Perkin Elmer, USA) as described.
11
Data are shown as mean ± SD. Statistical analysis was performed with one-way ANOVA followed by Bonferroni’s post-hoc test. (G) Oxygen consumption in mitochondrial-enriched fractions from naive and stroke animals. Adult Sprague-Dawley rats were exposed to stroke via the middle cerebral artery occlusion. After 4.5 h, animals received stereotaxic transplants of either human EPCs (4 × 106 viable cells in 9 µl sterile saline) or vehicle (9 µl sterile saline), delivered into the cortex corresponding to the anticipated penumbra area. At 72 h post-transplantation, animals were euthanized, and the cortical region corresponding to the transplant site was harvested. Enriched mitochondrial fractions were obtained by mechanical tissue disruption and differential centrifugation.
12
Mitochondrial bioenergetics was assessed using the Seahorse XFe96 analyzer. Real-time measurements (mean ± SD, n = 6 samples per condition) of OCR (upper panel) and ECAR (lower panel) were recorded in the presence of ADP (40 mM), followed by the addition of oligomycin (25 µg/ml), FCCP (40 µM), and rotenone (2 µM)/antimycin A (40 µM) injected sequentially as shown. OCR and ECAR of mitochondria from stroke + vehicle and stroke + EPCs illustrate the difference in metabolic profiles between the two conditions. Data are shown as mean ± SD. Asterisks represent significant differences between the two groups (*p < 0.01) computed by one-way ANOVA followed by Bonferroni’s post-hoc test. (H) Co-localization of EPCs’ mitochondria in stroke rats’ neurons. Sprague Dawley male rats (∼250 g) were subjected to 60 min MCAO, and 3 h later, EPCs (0.4 × 106 cells in 3 µl) were stained with MitoTracker Deep Red FM (Invitrogen) at 500 nM and then transplanted into the ischemic penumbra. At day 1 after transplantation, the animals were sacrificed, perfused, and brains were harvested, cryosectioned and processed for NeuN (Abcam) IHC staining. Confocal images were captured at 60 × and 180 × (Green: NeuN; Red: EPCs’ mitochondria; Blue: DAPI). Results revealed that mitochondria transfer occurs in vivo from EPCs to neurons. The transfer of mitochondria can be seen in few cells adjacent to the original implantation site (a, magnified in a′), while higher number of neuronal cells with EPC mitochondria (white arrows) are more apparent within the transplant site (b). (I) Mitochondrial mass in EPCs and EPC-derived Rho0 cells. EPCs were cultured in MEM alpha (Gibco) supplemented with 20% FBS, 1%NEAA, 1% GlutaMax and 1% Penicillin/Streptomycin. Rho0 cells were obtained by treating EPCs with 0.1 µM ethidium bromide and supplemented with 1 mM pyruvate and 0.2 mM uridine for three weeks. Cells were stained with 500 nM MitoTracker Deep Red FM (Life Technologies), fixed and probed with an anti-β-tubulin antibody (Abcam, Cambridge, MA). Confocal images were captured at 180 × . Blue: DAPI; red: β-tubulin; cyan: mitochondria. Mitochondrial DNA copy number was evaluated by qRT-PCR. Total DNA (gDNA and mtDNA) was prepared by using Nucleospin tissue kit (TaKaRa, Cat. #740952.50) starting from Rho0 EPCs (4 × 106 cells) and EPCs (6 × 106 cells). Rho0 cells showed less than 1% mtDNA copy number relative to EPCs.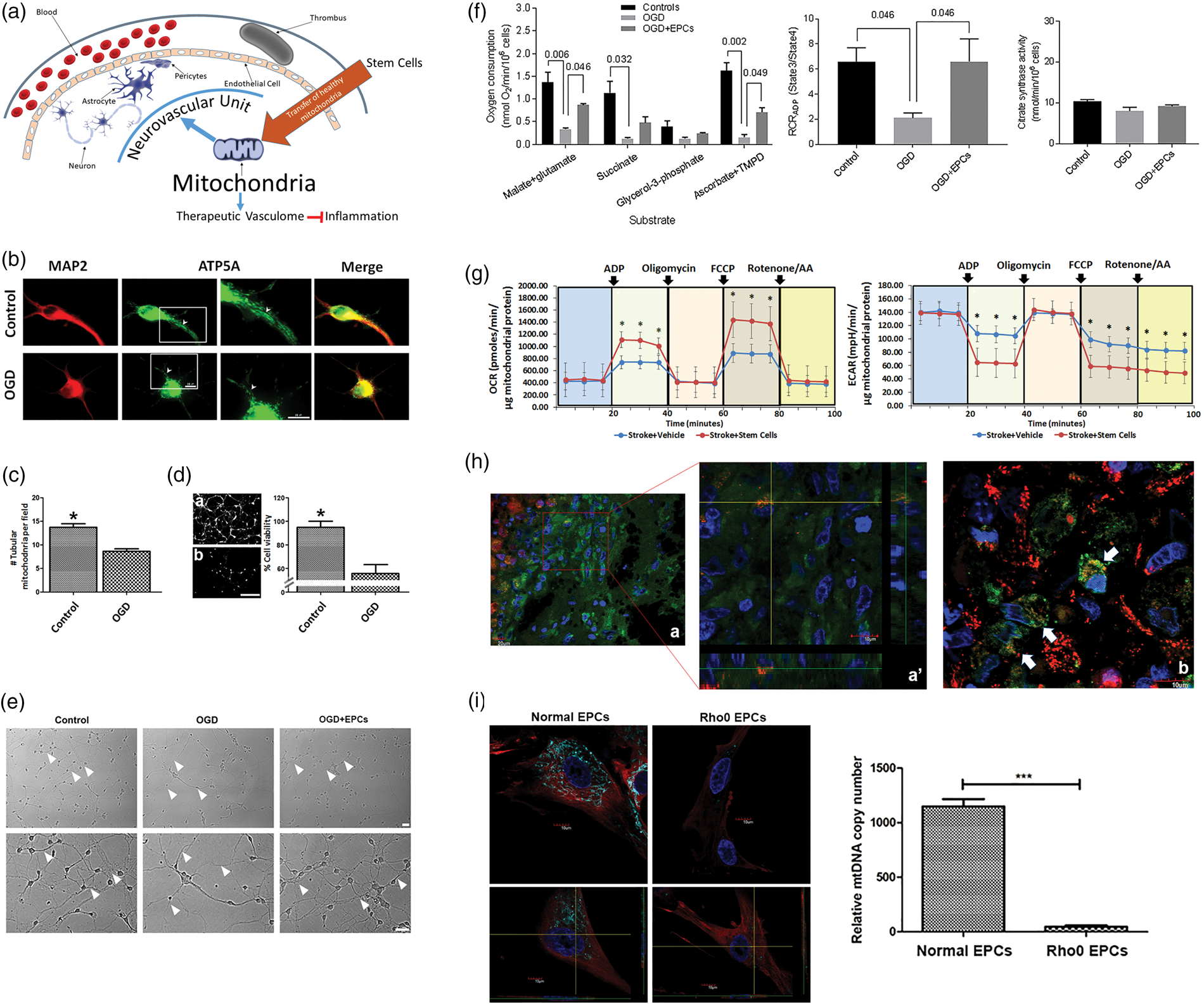
Recently, the pioneering discovery of mitochondrial transfer from healthy astrocytic cells to ischemic neuronal cells 7 provided a solid rationale to pursue therapeutic applications of mitochondria-based regenerative medicine in stroke. The successful extrapolation of mitochondrial transfer to a stem cell setting was recognized by transfer of healthy mitochondria from endothelial progenitor cells (EPCs) into ischemic brain endothelial cells, 8 as revealed by increased ATP levels and oxygen consumption coupled with enhanced angiogenesis and decreased transcellular permeability of brain endothelial cells. Transfer of EPC-derived extracellular mitochondria was further tested in oxygen-glucose deprivation (OGD) stroke model and showed increased transcellular endothelial permeability, accompanied by increased mitochondrial biogenesis, mitochondrial DNA copy number, and intracellular ATP levels, and reinstatement of endothelial tightness. 8 However, evidence of in vivo demonstration of mitochondrial transfer from stem cells into the ischemic brain is limited and warranted for translational guidance of this stem cell-mediated mitochondria therapy.
Here, we show evidence that stem cell therapy attenuates experimental stroke deficits in cultured neuronal cells by restoring mitochondrial function (Figure 1(b) to (f)). Neuronal mitochondrial morphology and distribution were altered following OGD exposure, with reduced number of tubular mitochondria and higher density of mitochondria in the soma relative to projections accompanying decreased cell viability (Figure 1(b) to (d)). OGD also reduced neurite outgrowth processes which were rescued by EPC co-culture (Figure 1(e)). Assessment of mitochondrial function showed decreased respiration in OGD-exposed neurons compared to controls, which was partially restored with EPC co-culture (Figure 1(f)). Next, in line with our long-standing goal of translating experimental therapeutics from the laboratory to the clinic, we discuss the safety and efficacy of stem cell therapy in restoring mitochondrial structure and function in ischemic neurons, resulting in behavioral recovery and reduction of histopathological deficits in stroke animals. We showed here that oxidative phosphorylation (OXPHOS) capacity was rescued in mitochondria isolated from the brains of stroke animals transplanted with EPCs compared to those stroke animals that received vehicle infusion (Figure 1(g)). Confocal microscopy confirmed transfer of mitochondria from EPCs to ischemic neurons in transplanted stroke animals (Figure 1(h)).
The use of Rho0 cells, devoid of functional mitochondria (Figure 1(i)), and specific pharmacologic mitochondrial inhibitors, combined with our stem cell technology for generating EPCs, as well as our established stroke paradigms of in vitro oxygen-glucose deprivation and in vivo thromboembolic animal model, allow us to directly probe the contribution of mitochondria in stroke pathology and its treatment. The concept that mitochondrial function reflects stroke disease states, as well as therapeutic readouts of neurobehavioral recovery, should impact on our understanding of cell death mechanisms and regenerative medicine. Our immediate goal is to evaluate the status of the mitochondria in stroke before and after stem cell treatments. Our long-term goal is the therapeutic application of stem cells, and the use of mitochondrial readouts as a robust biomarker in stroke patients. The demonstration that stem cell transplantation exerts therapeutic effects against stroke may similarly benefit other brain diseases characterized by mitochondrial deficits. Abnormalities in mitochondrial function have been implicated in the progression of hallmark pathological symptoms of Alzheimer’s disease and Parkinson’s disease.9,10 Acute ischemic stroke shares some pathological features typical of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes or MELAS; in particular, these patients display mitochondrial dysfunction that triggers a cascade of cell death events, such as oxidative stress, apoptosis, inflammation, blood–brain barrier breakdown altogether reminiscent of stroke, further supporting the notion that mitochondrial disruption plays a key role in stroke pathology. Elucidating the role of mitochondria in stroke-associated cell death processes will likely provide not only a better understanding of the disease pathology but equally offer innovative insights into mitochondria-based cerebroprotective therapies, thereby guiding both the diagnosis and the treatment of stroke. Paramount to these investigations is our pursuit to reveal how extracellular mitochondria can penetrate into the cell, which should aid in the development of novel treatment strategies designed to transfer healthy mitochondria from stem cells into ischemic cells.
Footnotes
Data Access
All data are stored in USF Center of Excellence for Aging and Brain Repair and freely distributed by Dr. Cesar V. Borlongan upon request.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Cesar V. Borlongan is funded by NIH and VA grants.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.