
Abstract
Modulator of apoptosis 1 (MOAP-1) is a Bax-associating protein highly enriched in the brain. In this study, we examined the role of MOAP-1 in promoting ischemic injuries following a stroke by investigating the consequences of MOAP-1 overexpression or deficiency in in vitro and in vivo models of ischemic stroke. MOAP-1 overexpressing SH-SY5Y cells showed significantly lower cell viability following oxygen and glucose deprivation (OGD) treatment when compared to control cells. Consistently, MOAP-1−/− primary cortical neurons were observed to be more resistant against OGD treatment than the MOAP-1+/+ primary neurons. In the mouse transient middle cerebral artery occlusion (tMCAO) model, ischemia triggered MOAP-1/Bax association, suggested activation of the MOAP-1-dependent apoptotic cascade. MOAP-1−/− mice were found to exhibit reduced neuronal loss and smaller infarct volume 24 h after tMCAO when compared to MOAP-1+/+ mice. Correspondingly, MOAP-1−/− mice also showed better integrity of neurological functions as demonstrated by their performance in the rotarod test. Therefore, both in vitro and in vivo data presented strongly support the conclusion that MOAP-1 is an important apoptotic modulator in ischemic injury. These results may suggest that a reduction of MOAP-1 function in the brain could be a potential therapeutic approach in the treatment of acute stroke.
Keywords
Introduction
Ischemic stroke occurs when a blood vessel is occluded by a blood clot leading to disruption of blood supply to the brain areas served by this occluded vessel. The effects of a stroke can be catastrophic because of widespread cell death in the affected brain regions. It is generally accepted that cell death in the ischemic core occurs predominantly through necrosis.1,2 In contrast, cell death in the surrounding penumbra is predominantly through apoptosis that occurs less rapidly.3,4 During the progression of a stroke, the loss of functional neurons through apoptosis leads to a gradual loss of brain functions. 5 Therefore, apoptotic cell death mechanism is of intense interest in stroke research, as it presents as a potential target for therapeutic intervention to limit ischemic injuries in the event of a stroke.
Apoptosis may be induced via both the extrinsic and intrinsic pathways. 6 The Bcl-2-associated X protein (Bax) is a well-known proapoptotic regulator involved in both pathways. 7 More recently, modulator of apoptosis (MOAP-1) has been identified as a Bax-associating protein. 8 It is a mitochondria-enriched protein of 39.6 kDa molecular weight 9 with a half-life of only about 25 min and is constitutively degraded by the ubiquitin-proteasome system (UPS). 10 When tumor necrosis factor (TNF)-α binds to its receptor TNF-R1 in response to a death stimulus, TNF-R1 internalizes and complexes with MOAP-1 and Ras association domain family 1A (RASSF1A). This triggers MOAP-1 to expose its BH3-like domain (an “open state”) and bind to cytosolic Bax, which then becomes activated and translocates into mitochondria. 9 Insertion of Bax to the mitochondrial membrane promotes the release of cytochrome c (cyt c), facilitates the activation of downstream caspase-dependent pathways and subsequently results in cell death. 9 MOAP-1 has also been shown to mediate Fas-induced apoptosis in the liver and MOAP-1−/− mice are protected from Fas apoptotic signaling-induced acute liver injury. 11
To our knowledge, the pathophysiological role of MOAP-1 has only been studied in the liver. 11 Interestingly, MOAP-1 is highly abundant in the brain relative to other major organs which may suggest that it plays an important role in the CNS. 12 Moreover, Takaji et al. 13 suggested that such high expression makes it unlikely for MOAP-1 to induce apoptosis in the adult brain under normal physiological conditions. In this study, we sought to explore the prospective role of MOAP-1 in cerebral ischemia, employing gain or loss of function strategies in both in vitro and in vivo acute ischemic stroke models.
Material and methods
All animal procedures were carried out with evaluation and oversight from the Institutional Animal Care and Use Committee (IACUC) of the National University of Singapore or National Institute of Health guidelines with United States Public Health Service's Policy on Human care and Use of Laboratory Animals and conducted in compliance with the National Advisory Committee for Laboratory Animal Research (NACLAR) guidelines. Randomization, blinding and statistical criteria that we used in the experiments are in compliance with the ARRIVE guidelines (Animals in Research: Reporting In vivo Experiments).14,15 For tMCAO model, infarct volume measurements, and functional studies, WLT performed the blinding and randomization, SJC and JH were blinded for mice groups throughout the study. SJC performed the tMCAO, SJC and JH performed the rotarod experiment, SJC performed the infarct volume measurement and TVA performed the laser speckle contrast imaging.
Generation of MOAP-1−/− mice
The generation of MOAP-1−/− mice was previously described. 11 Briefly, the targeting vector was designed to replace the entire coding region of MOAP-1 Exon2 gene with the neomycin cassette via homologous recombination. The linearized targeting vector was introduced into murine embryonic stem cells via electroporation. The heterozygous MOAP-1+/− mice (129 × C57/BL6) were backcrossed with the C57/BL6 strain for more than 10 generations to generate homozygous MOAP-1−/− mice. Genotypes of the mice were confirmed by PCR and Western blot. Both MOAP-1+/+ and MOAP-1−/− mice used for experiments were from the same heterozygous founders in pure C57/BL6 genetic background and were in-bred for no more than six generations.
Genotyping
Routine genotyping was done on littermates. Briefly, around 2 mm of tail was snipped from both wild-type and MOAP-1−/− mice and incubated in lysis buffer (50 mmol L−1 Tris, pH 8.8; 100 mmol L−1 NaCl; 100 mmol L−1 EDTA; 1% SDS) with proteinase K (Sigma-Aldrich) at 55℃ overnight. Saturated NaCl solution was added to the digested tail and agitated for 5 min followed by centrifugation at 14,000 r/min at 4℃ for 20 min. Isopropanol was then added to the supernatant and the samples were centrifuged for 12 min. Finally, DNA was extracted, resuspended in 70% ethanol, and dried by incubating at 37℃ for 30 min. Both quality and quantity of the final DNA product were determined by NanoDrop Spectrometer (Thermo scientific, USA) and stored at −20℃ until use. Gene expression analysis was done by PCR with primers (for wild-type: 5′-ATCCATCCCATCCGCTGACG-3′ and 5′-GACTCTTGCTCGCTTGCAGG-3′; MOAP-1−/−: 5′-ATCCATCCCATCCGCTGACG-3′ and 5′-AGTAGAAGGTGGCGCGAAGG-3′) and Qiagen HotStarTaq Master Mix Kit (Qiagen, USA) under the following conditions: Initial incubation at 95℃ for 15 min, followed by 40 cycles of amplification (95℃ for 40 s, 58℃ for 40 s and 72℃ for 2 min), ending with a final extension step of 72℃ for 5 min. Equal amount of PCR product was electrophoresed on 1% agarose gel containing SafeviewTM (Applied Biological Materials, Canada) and finally visualized on a Gel Doc XR+ system (Biorad, USA).
Mice used in in vivo studies
Thirty-two each of MOAP-1+/+ and MOAP-1−/− mice (6–8 weeks old, males) were used in the study. Five each were used for laser speckle contrast imaging and NeuN immunohistochemistry. Nine and 11, respectively, were used for infarct volume and functional (rotarod) assessments. Fifteen each were used to evaluate NeuN expression. The remaining four mice were excluded for not reaching the inclusion criteria.
Transient middle cerebral artery occlusion model
Unilateral transient middle cerebral artery occlusion model (tMCAO) was performed as described previously. 15 Studies were done in a randomized and blinded manner. Laser-Doppler sonography probe was mounted directly on the skull, and only tMCAO mice with relative cerebral blood flow (CBF) of less than 20% of pre-MCAO baseline were included in the study. Mice were anesthetized with isoflurane (1.5% in 30% O2/70% N2O) throughout the 90 min tMCAO procedure. Body temperature was monitored and maintained at 37℃ throughout the entire operation and occlusion procedure until mice regained consciousness and mobility. Briefly, a midline neck incision was made. Trachea was exposed by dissecting the soft tissues. Common carotid artery (CCA) and external carotid artery (ECA) were carefully dissected without harming the vagal nerve. The ECA was ligated with 7/0 suture and a second knot was made on the ECA at the bifurcation of the ECA with the internal carotid artery (ICA). The CCA was temporarily ligated using a 4-0 silk suture and a small vascular clip was applied on the ICA. A small cut was made in the ECA for the insertion of a modified silicon-coated 7-0 polypropylene monofilament. The silicon-coated filament was further advanced into the ICA until a significant drop of relative CBF from 100% to around 15% was observed by means of a laser-Doppler sonography probe mounted directly on the skull. After 90 min, the filament was withdrawn and full reperfusion was achieved as indicated by laser-Doppler flowmetry. Mice were injected subcutaneously with buprenorphine (0.1 mg/kg) for pain relief and an antibiotic anrofloxacin (Baytril, 5 mg/kg) after tMCAO. For sham-operated mice (Western blot experiment), the filament was inserted only partially without affecting blood flow. Three MOAP-1+/+ and one MOAP-1−/− mice were excluded from the study due to excessive bleeding and reduction of relative CBF greater than 20%.
CBF measurements
MOAP-1+/+ and MOAP-1−/− mice were anesthetized with isoflurane (1.5% in 30% O2/70% N2O), placed in a flat position on a warming pad thermostatically controlled at 37℃. The skull was exposed through a cut in the skin along the midline. Images were acquired through the Laser Speckle Contrast Imager (PSI system, Perimed Inc.) at a working distance of 10 cm from the skull surface. CBF measurements were performed repeatedly at 15 min before onset of MCAO (pre-MCAO), 5 min before reperfusion (MCAO), 3 h, and 24 h after reperfusion. Mice were allowed to recover from anesthesia between time points. CBF was calculated using a blood perfusion imaging software from the region of interest (ipsilateral area). Mice were killed after the last measurement, and the brains were collected for NeuN histochemistry.
Infarct volume assessment
Whole brains were cut into 1 mm coronal sections and immediately stained with 2% tetrazolium chloride (TTC, Sigma Aldrich, USA) for 15 min. The infarct volume was measured using Image-J (NIH, USA). Infarct volume was calculated according to Swanson et al., 16 by subtracting the area of non-infarcted area in the ischemic hemisphere from the area of the respective contralateral hemisphere. Based on preliminary data, the infarct volume obtained in control mice was approximately 130 ± 30 (SD) mm 3 . Therefore, to detect a 30% change in infarct volume at 80% power and α = 0.05, the minimum sample size required is 9.
Primary cortical neuron culture
Cultures of wild-type (MOAP-1+/+, C57/BL6) and MOAP-1 knockout (MOAP-1−/−, C57/BL6) mice embryonic days 15–16 cortical neurons were prepared as previously described 17 with minor modification. Briefly, cortices were dissected aseptically and subjected to papain digestion (Worthington Biochemical Corporation, USA). The digested tissue was further dissociated by mechanical trituration and finally resuspended in neuron culture medium which contains neurobasal media (Gibco, Life Technologies, USA) supplemented with 2.5% B-27 supplement, 0.25% GlutaMAX-I supplement (Gibco, Life Technologies, USA), 100 U mL−1 penicillin and 100 mg mL−1 streptomycin (Gibco, Life Technologies, USA). Cells were seeded at a density of 1 × 106 or 0.25 × 106 per well in poly-D-lysine (0.1 mg mL−1, Sigma, USA) pre-coated 6-well or 24-well plates respectively. Cells were then incubated at 37℃ with 5% CO2. On day 4, half of the medium in each well was changed to fresh neuron culture medium with additional cytosine arabinoside (AraC, Sigma, USA) at a final concentration of 1 µmol L−1. Mature cortical neurons were used on days 9–12. Cell Purity was checked with MAP-2 (1:200, Sigma, USA) and GFAP (1:200, Sigma, USA) staining.
Human neuroblastoma SH-SY5Y cells
Neuroblastoma SH-SY5Y cell line was obtained from American Type Culture Collection. Cell were maintained in DMEM/F12 (Gibco, Life Technologies, USA) containing 10% fetal bovine serum (FBS, Gibco, Life Technologies, USA), 100 U mL−1 penicillin, and 100 mg mL−1 streptomycin (Gibco, Life Technologies, USA) at 37℃ and incubated in a humidified atmosphere with 95% air/ 5% CO2.
Construction of MOAP-1 lentiviral expression vectors
Lentiviral vector construction was performed as described previously. 18 MOAP-1 (NM_022323) cDNA with Myc-DDK-tagged was purchased from Origene (Catalogue no. MR205352). Briefly, MOAP-1 cDNA was amplified and subcloned into pL6mCWmycIE lentiviral vector to give pL6mCWmycMOAP-1IE. The vector, pLenti6mCWmycIE, was modified from pLenti6/V5-D-TOPO (Invitrogen) by reengineering the multiple cloning site, insertion of the cPPT and WPRE elements, and insertion of the N-terminal myc tag coding sequence and IRES-EGFP reporter cassette. Lentivirus packaging was performed in 293FT cells according to the protocol provided with the ViraPower™ Lentiviral Directional TOPO® Expression Kit (Invitrogen). Lentivirus particles were harvested from cell culture supernatant according to the protocol of Deiseroth Lab (http://www.stanford.edu/group/dlab/resources/lvprotocol.pdf). Lentivirus carrying the MOAP-1 expression constructs was used to infect undifferentiated SH-SY5Y cells. Prior to infection, cells were cultured to 90% confluence. Concentrated virus particle was added to cell culture medium containing 8 µg mL−1 of Polybrene (Sigma Aldrich). Where long-term expression of transgene was needed, antibiotic selection was applied by adding Blasticidin S (Invitrogen) at a final concentration of 10 µg mL−1 to the medium. Expression of transgene was visualized by EGFP fluorescence.
Oxygen glucose deprivation
Oxygen glucose deprivation (OGD) was achieved by incubating cells in glucose-free DMEM medium (Gibco, Life Technologies, USA) supplemented with 100 U mL−1 penicillin, and 100 mg mL−1 streptomycin (Gibco, Life Technologies, USA) in a hypoxia chamber (5% CO2, 5% H2, 90% N2) for 2, 4, or 8 h followed by 22, 20, or 16 h, respectively, incubation in DMEM/F12 complete medium for SHSY5Y cells or neurobasal medium for primary cultures at 37℃ in a humidified incubator.
Western blot
Control, sham-operated (8 h) and tMCAO (1, 3, and 8 h) mice were perfused with sterile PBS, and whole ipsilateral (infarcted) cortex or contralateral cortex were harvested and stored at −80℃ until used. Frozen samples were lysed by RIPA buffer (Cell Signalling Technologies, USA) supplemented with a protease inhibitor and phosphatase inhibitor cocktail (Roche, Manheim, Germany). Total protein was determined by using a BCA protein assay kit (Pierce, ThermoScientific, Rockford, USA). Proteins were separated by 10% SDS/PAGE electrophoresis, transferred onto a poly-vinylidene difluoride (PVDF) or nitrocellulose membrane (Life Technologies, USA), and blocked with 10% non-fat milk. The membrane was incubated with primary antibodies against MOAP-1 (1:1000, Sigma Aldrich, USA), NeuN (1:1000, Millipore Corporation, Billercia, MA, USA) and β-actin (1:1000, Cell Signalling Technologies, USA) at 4℃ overnight, washed, and incubated with HRP-conjugated anti-rabbit or mouse IgG at room temperature for 1 h. Visualization was carried out using Luminata Forte or Crescendo Western HRP substrate (Millipore Corporation, USA), and the chemiluminescence signals were detected using the UVIchemi (UVItec, Cambridge, UK) system.
Immunofluorescent
Mice were anesthetized and perfused with 0.1 mol L−1 phosphate buffer saline. Brains were harvested and post-fixed with 4% paraformaldehyde at 4℃ overnight, cryoprotected in 10% and then 20% sucrose at 4℃ until use. Coronal sections (20 µm) were mounted onto glass slides (Matsunami, Japan) and stored in −80℃ until use. Before staining, brain sections were air dried at room temperature for 30 min and permeabilized with 0.1% Triton-X (Sigma, USA, dissolved in 0.1 mol L−1 phosphate buffer saline) for 10 min. Brain sections were incubated at 4℃ overnight with primary antibodies against MOAP-1 (1:200, Sigma, USA), NeuN (1:400, Chemicon, Millipore or Abcam, UK), and β-actin (1:1000, Cell Signalling, USA), washed and then further incubated with anti-rabbit secondary antibody conjugated with Alexa Fluor 488 (Life technologies, USA) and/or anti-mouse secondary antibody conjugated with Alexa Fluor 555 (Life technologies, USA) for 1 h at room temperature. Non-specific binding was blocked by incubating the mice brain section in 5% goat serum for 1 h at room temperature. Nuclei were counterstained with diamidino-2-phenylindole dihydrochloride (DAPI, Sigma, 0.5 µg mL−1 in 0.1 mol L−1 phosphate buffer saline) for 5 min, followed by mounting with Pro-Long gold antifade fluorescent mounting medium (Life technologies, USA). Negative control was performed by probing sections with 0.1% Triton-X (Sigma, USA, dissolved in 0.1 mol/L phosphate buffer saline) overnight. All the fluorescent images were captured using an Olympus FluoViewFV1000 (Olympus, Japan) laser scanning confocal microscope and images captured was processed by FV10-ASW1.7.
Immunoprecipitation
At 24 h after tMCAO, protein samples were prepared separately from ipsilateral and contralateral striatum and cerebral cortex using Pro-PREPTM Protein Extraction Solution (BOCA SCIENTIFIC). According to the manufacturer’s instruction, anti-rabbit MOAP-1 antibody (Sigma, USA) and Dynabeads Protein G Immunoprecipitation kit (Life technologies) were used to precipitate MOAP-1 from each protein sample (100 µg). Following MOAP-1 immunoprecipitation, the association with Bax was assessed by Western blot using anti-mouse Bax monoclonal antibody (Santa Cruz).
Rotarod assessment
Mice were assessed in a blinded manner for neurological deficit by the rotarod (TSE system, USA) test 1 h before infarct volume assessment using an accelerating rotarod apparatus (Ugo Basile, Italy) which accelerated from 4 to 400 r/min over 300 s. Each mouse was assessed three times with 20 min intervals. The average latency to drop obtained in the last two trials was used.
Statistical analysis
Animal sample size was calculated based on Cohen's d number (1.73) and pooled sample size S.D. of two groups (82.4), with a desired statistical power level of 80% and a probability level p for significance at 0.05. We obtained a minimum sample size of 7 (total sample size of 14) for two-tailed t-test. All comparisons were performed by Student’s t-test or one-way ANOVA with Bonferonni correction using IBM SPSS Statistics 19. Two-way ANOVA was performed using GraphPad Prism 6. Data are expressed as mean ± SD. Statistical significance is reached when p < 0.05.
Results
Regional and cellular distribution of MOAP-1 in brain
Western blot analysis of tissue lysates showed that MOAP-1 was hardly detectable in all organs except the brain (Figure 1(a)), which is consistent with previous findings.
12
No gender differences were detected (data not included). Within the brain, the highest expression of MOAP-1 was observed in the cortex and cerebellum, while those in the striatum, hippocampus, thalamus, and midbrain were comparable but at an apparently lower level (Figure 1(b)). However, the small regional differences observed did not reach statistical significance. Immunofluorescent histochemical staining showed that MOAP-1 was highly associated with NeuN (a marker for neuron)-positive cells but only minimally with GFAP (marker for astrocytes)-positive cells (Figure 1(c)), indicating a predominantly neuronal distribution.
Regional and cellular distribution of MOAP-1 in the mouse brain.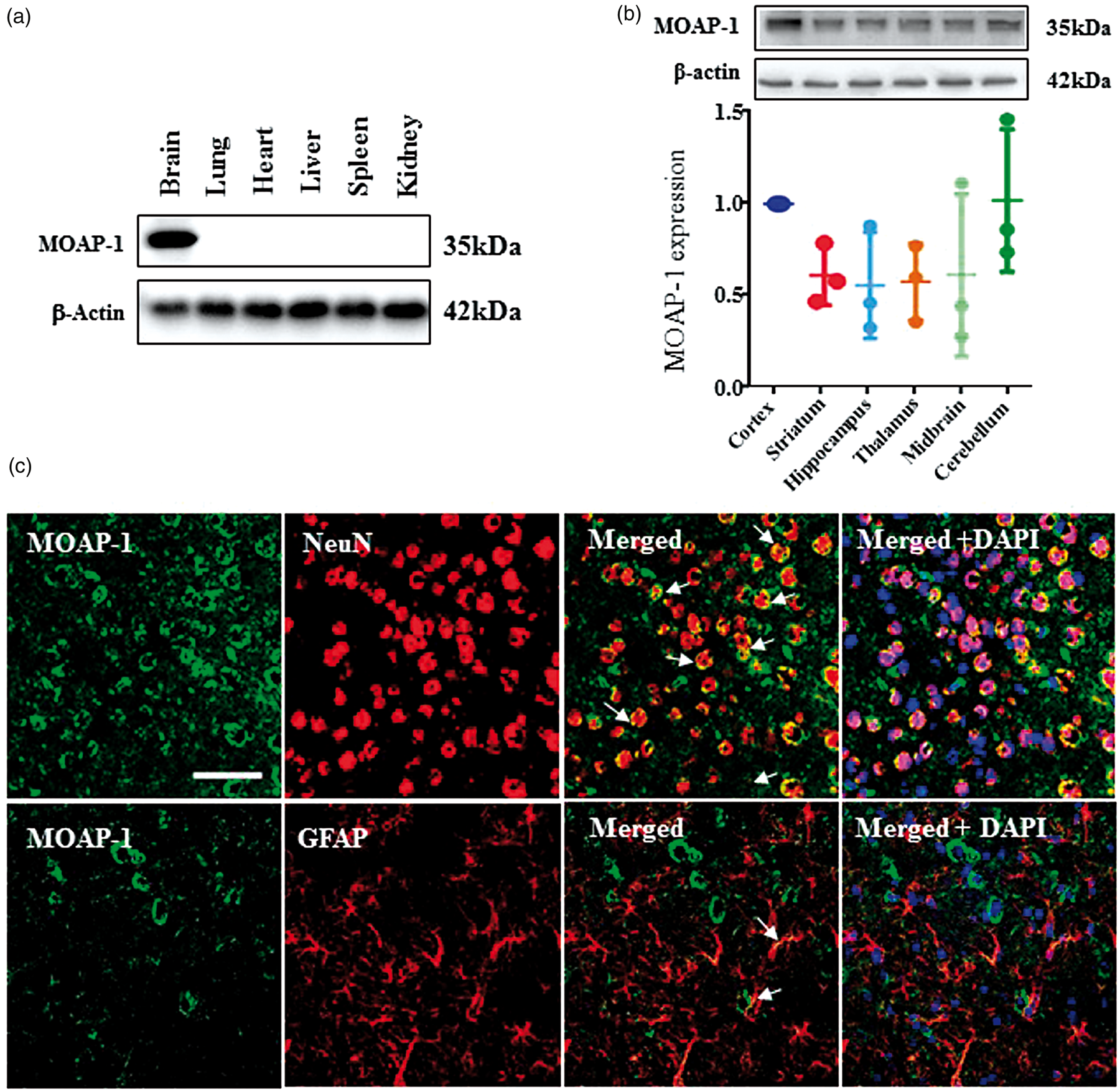
MOAP-1 overexpression increased cell death induced by OGD
MOAP-1-GFP fusion protein was successfully overexpressed in human neuroblastoma cells (SH-SY5Y) as demonstrated in both GFP expression (Figure 2(a)) and immunoblotting (Figure 2(b)). The overexpression of MOAP-1 has no effect on cell viability (Figure 2(c)). In the MTT assay, cell viability was reduced by 43 ± 5% in MOAP-1 OE cells vs. 24 ± 4% vector control after 4 h OGD, and 68 ± 1% vs. 36 ± 3% after 8 h OGD (Figure 2(d)). Thus, when cells were subjected to OGD, MOAP-1 overexpressing cells (MOAP-1 OE) showed significantly reduced level of cell viability than vector-transfected SH-SY5Y cells, suggesting increased cell death in MOAP-1 OE cells. This is supported by a significant increase in LDH release by MOAP-1 OE cells after 8 h OGD (Figure 2(e)).
MOAP-1 overexpression caused a reduction in viability in SH-SY5Y cell under OGD condition.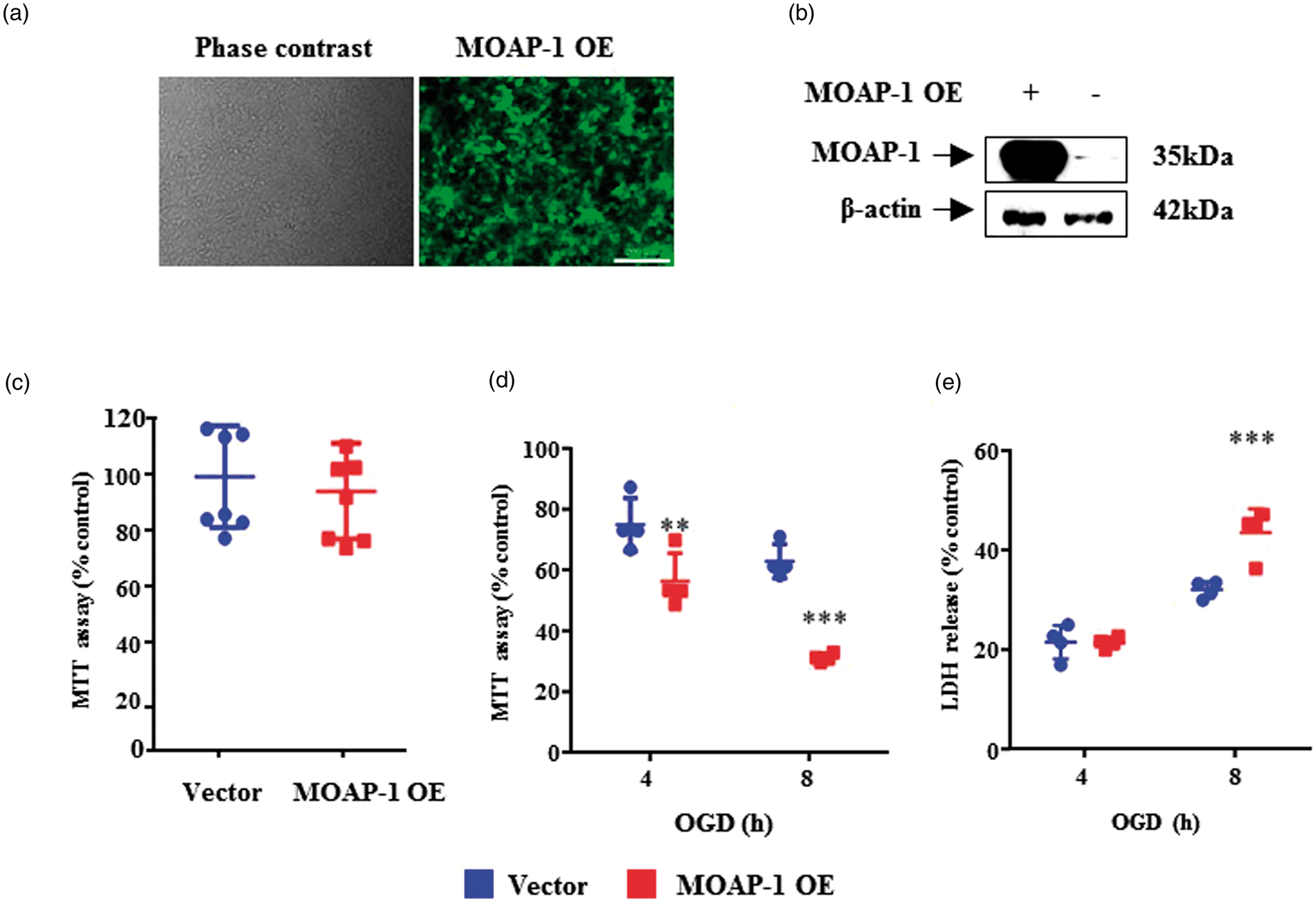
MOAP-1 deletion protected against cell death induced by OGD
Based on gain-of-function results described in Figure 2, we further investigated whether MOAP-1 deletion may afford protection against cell death in primary neurons. MOAP-1−/− mice were generated by deletion of Exon2 of MOAP-1 that covers the entire coding region as described in Figure 3(a).
11
PCR-based genotyping was done routinely in littermates to ensure MOAP-1 deficiency before use (Figure 3(b)). Primary cortical neuronal cultures of high purity (90–95% MAP-2 positive) with little to negligible contamination of GFAP-positive cells were prepared from both MOAP-1+/+ and MOAP-1−/− mouse brain (Figure 3(c)). Only mature neuron at days 9–12 was used in the study. When these cells were subjected to OGD, significant difference in cell viability was observed at 2 and 4 h (Figure 3(d) and (e)). MOAP-1−/− cells exhibited higher cell viability at 68 ± 1% and 56 ± 1% as opposed to 54 ± 3% and 30 ± 1%, respectively, for MOAP-1+/+ cells, the opposite of those effects observed in MOAP-1 OE cells. These observations thus reinforce the idea that MOAP-1 plays a role in neuronal loss induced by OGD.
MOAP-1−/− primary cortical neurons are more resistant to oxygen glucose deprivation (OGD)-induced cell death.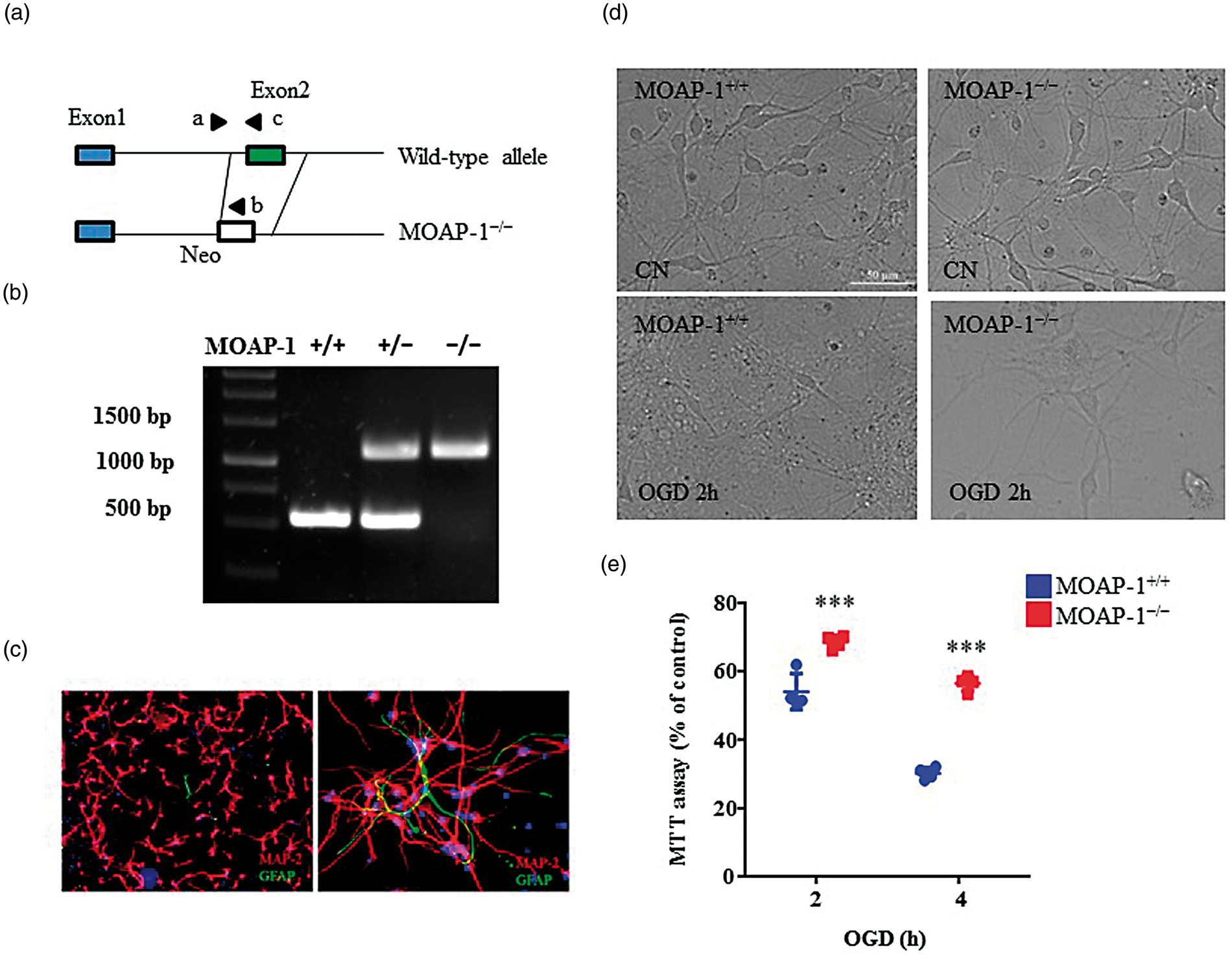
MOAP-1 deletion promoted neuroprotective effect in ischemic stroke condition
Figure 4(a) accounts for the animal usage in this study. Association of MOAP-1 with Bax was observed in the ipsilateral cortex and striatum of mice at 24 h after tMCAO (Figure 4(b)), which was not observed on the contralateral side. These observations suggest that MOAP-1 and Bax participate in tMCAO-induced apoptosis in the affected areas of the brain.
Demonstration of MOAP-1 and Bax association in the infarcted cortex and striatum following tMCAO but not in the contralateral control.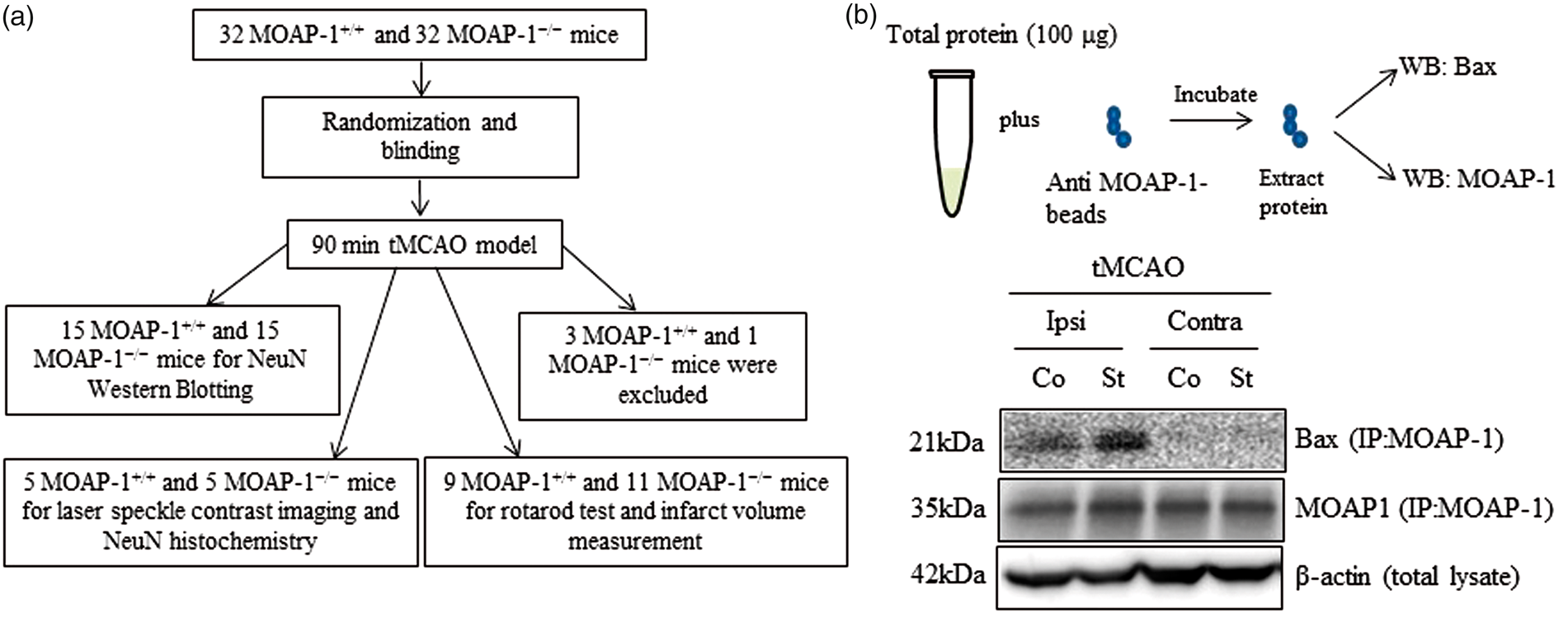
Using laser speckle contrast imaging, the cerebral cortical blood flow in the frontal and medial regions was compared between the MOAP-1+/+ and MOAP-1−/− mice before and after tMCAO. It can be seen from Figure 5(a) and (b) that MOAP-1 deficiency does not alter CBF. When subjected to tMCAO (90 min), blood flow was reduced drastically to about 20% pre-MCAO level on the ipsilateral side. This is consistent with the laser-Doppler measurement typically at 15–20%. Upon reperfusion, blood flow recovered to about 55 and 72% at 3 h and 24 h, respectively, in both groups of mice. In contrast, the resultant infarct volume was significantly smaller in the MOAP-1−/− mice than that observed in MOAP-1+/+ mice at 24 h after tMCAO by 55% (58 ± 9 vs. 129 ± 10 mm3, Figure 5(c) and (d)). Consistent with the reduced infarct size, MOAP-1−/− mice showed significantly reduced neurological deficits than the MOAP-1+/+ mice 23 h after tMCAO as demonstrated by their performance in the rotarod test (118 ± 28 vs. 33 ± 9 s, Figure 5(e)).
MOAP-1 deficiency protects against ischemic injuries in the mouse tMCAO stroke model. (a) Representative cerebral cortical blood flow distribution obtained by laser speckle contrast imaging. Unilateral tMCAO was performed on the left brain. Images were taken from the same mouse at 15 min before MCAO, 5 min before reperfusion, 3 h, and 24 h after the reperfusion. Color coding indicating normalized relative CBF speed (from the highest in dark red to the lowest in dark blue). (b) Ipsilateral cortical blood flow of MOAP-1+/+ and MOAP-1−/− mice at various time points after tMCAO. Data were captured from the medial and frontal regions and presented as % of the pre-MCAO CBF of WT mice. Statistical analysis was performed by two-way ANOVA with repeated measures followed by Bonferroni’s multiple comparisons test using GraphPad Prism 6: Time factor: F = 333.8, p < 0.0001; Group factor: F = 2.151, p = 0.1807; Time × Group interaction: F = 0.2210, p = 0.8809. ***p < 0.001 against respective Pre-MCAO group. (c) Representative TTC staining of MOAP-1+/+ and MOAP-1−/− tMCAO mice at 24 h after MCAO. Unilateral tMCAO was performed on the right brain. (d) Infarct volume of MOAP-1+/+ (n = 9) and MOAP-1−/− (n = 11) 24 h after tMCAO. Bars represent SD. ***p < 0.001 by two-tailed independent t-test. (e) Functional evaluation on neurological deficits. MOAP-1−/− (n = 10) mice showed longer fall latency than that of the MOAP-1+/+ (n = 8) mice at 24 h after tMCAO. Bars represent SD. ***p < 0.001 by two-tailed independent t-test.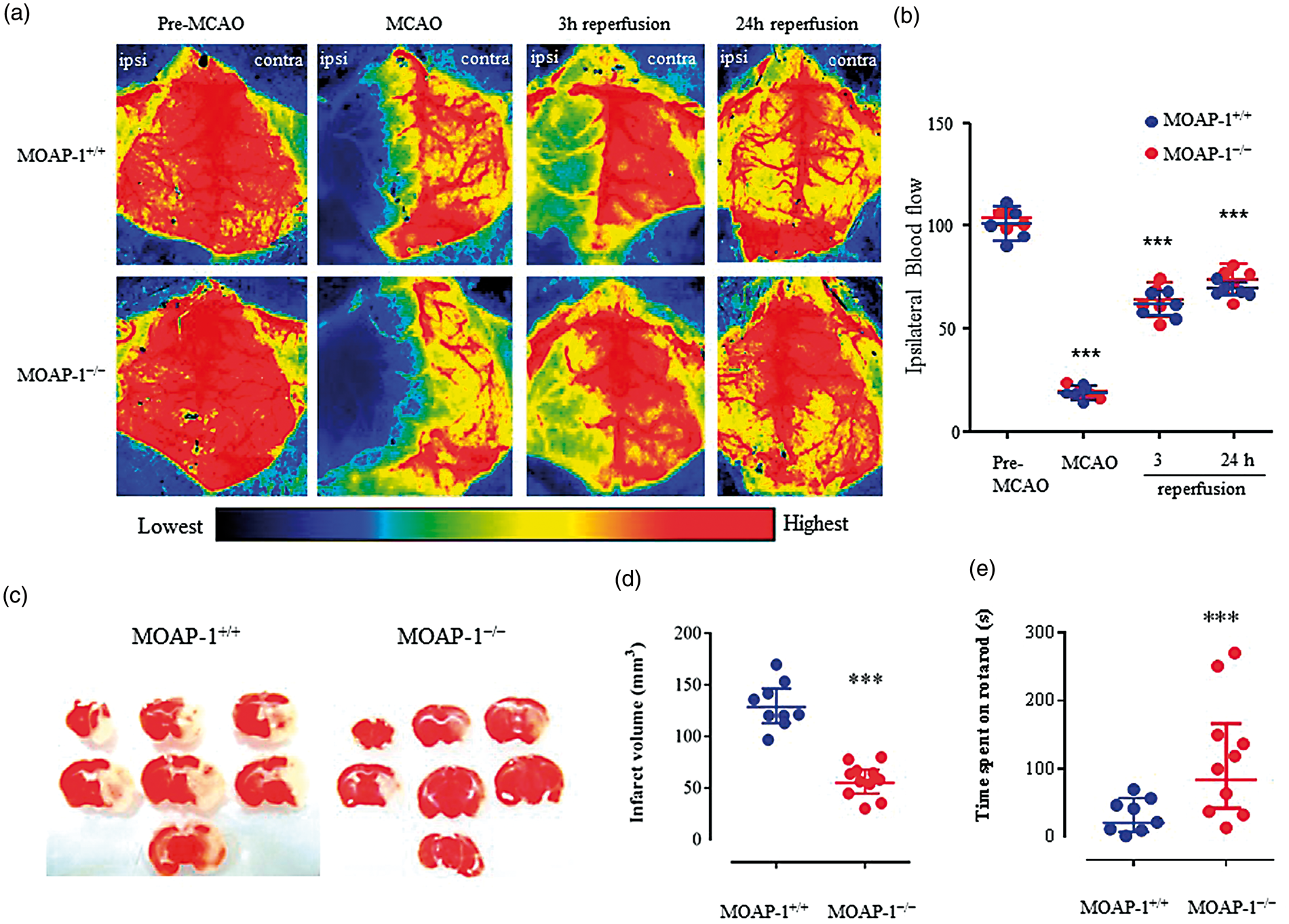
Using immunofluorescent histochemistry, neurons (NeuN-positive cells) were visualized in the infarcted cortex (Figure 6(a) and (b)). The number of neurons in the ipsilateral cortex of MOAP-1−/− mice was markedly greater than that of the MOAP-1+/+ mice. However, the numbers of neurons in both were much less when compared to those on the contralateral side. We further studied the time course of change in NeuN expression by Western blot. The expression of NeuN in the infarcted cortex of MOAP-1+/+ mice was found to be markedly reduced within the first 8 h following tMCAO indicating rapid neuronal loss. However, NeuN expression was not significantly changed in the MOAP-1−/− mice over the same 8-h period (Figure 6(c) and (d)).
MOAP-1 deficiency protects against neuronal loss in the mouse tMCAO stroke model. (a) Cresyl violet-stained sections showing the locations of the immunofluorescent staining images presented in b. (b) NeuN-positive cells in the infarcted cortex 24 h after MCAO indicating markedly more significant neuronal losses in the MOAP-1+/+ than MOAP-1−/− mice. (C) Representative images of NeuN and β-actin expression in ipsilateral cortex of naïve mice (c), sham-operated (S) or tMCAO cortex at 1, 3, and 8 h after tMCAO. (d) The densitometry measurement for NeuN of MOAP-1+/+ and MOAP-1−/− ipsilateral cortex at 1, 3, and 8 h after tMCAO. Data were normalized to its respective control (NeuN expression in ipsilateral cortex of naïve mice). Bars represent SD, n = 3. *p < 0.05 by two-way ANOVA with Bonferroni correction.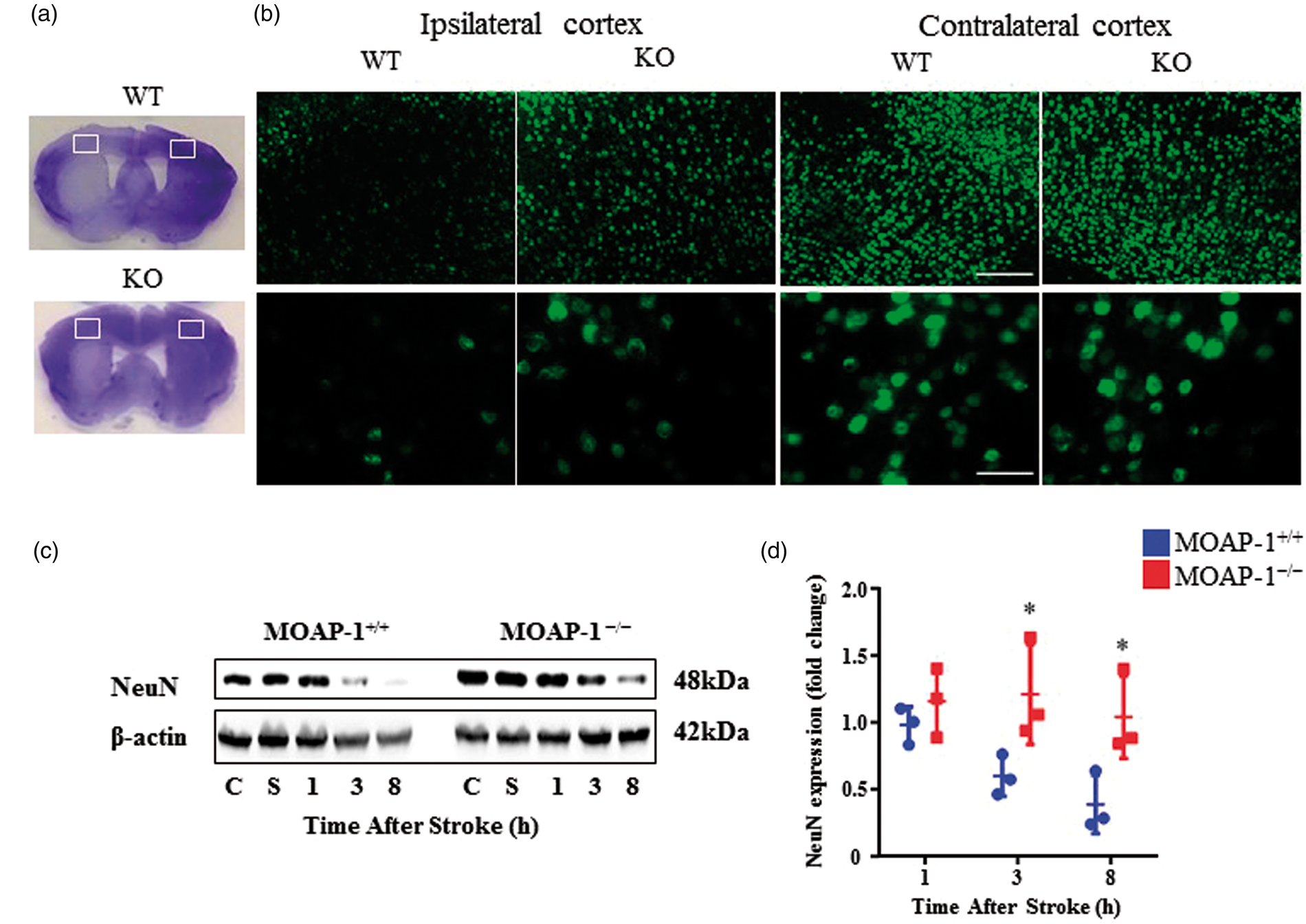
Discussion
MOAP-1 was first reported to be highly expressed in the adult human brain by Schüller et al. 12 using the Northern blot technique. Consistently, our current data obtained by Western blot demonstrated the high expression of MOAP-1 in the mouse brain as opposed to the low expressions in other major organs including liver, lung, spleen, kidney, and heart (Figure 1(a)). In the brain, MOAP-1 was observed to be ubiquitously distributed among the brain regions studied with perhaps marginally higher expressions in the cortex and cerebellum, but no statistical significance was obtained overall (Figure 1(b)). This is consistent with the results demonstrated by Takaji et al. 13 using Northern hybridization and RT-PCR. With regard to cellular localization, we focused on the cortex and the striatum as they are the regions most affected by tMCAO. Double immunofluorescent staining demonstrated substantive colocalization of MOAP-1 and the neuronal marker NeuN in the cortex of healthy adult MOAP-1+/+ mice (Figure 1(c)) and similarly in the striatum (not shown). In contrast, markedly less colocalization of MOAP-1 and the astrocytic marker GFAP was observed in both regions, especially in the cortex as there were more GFAP-positive cells in the striatum than cortex. These results indicate that MOAP-1 is predominantly expressed in neurons. In a recent study, Gokce et al. performed transcriptome analysis using microfluidic and fluorescence activated cell sorting (FACS)-based single-cell RNA sequencing of the mouse striatum. 19 Extracting from the data included in this report, MOAP-1 RNA is expressed in 60% of neurons, as opposed to 10% of astrocytes and 2% of microglia, consistent with our double staining results. Moreover, using real-time PCR, the relative expression of MOAP-1 RNA in rat primary neurons, astrocytes, microglia, and endothelial cell cultures was found to be approximately in the ratio of 19:4:1:1.5, respectively (supplementary data). Therefore, MOAP-1 appears to have a predominantly neuronal localization in both mouse and rat. As such, neuroblastoma and primary neurons were used as in vitro models in this study.
Using a lentiviral approach, we successfully induced the MOAP-1 transgene in SH-SY5Y cells to overexpress MOAP-1. As expected from previous findings, 10 these MOAP-1 OE cells did not show any significant change in cell viability when compared to vector-treated control cells (Figure 2(c)). When they were subjected to OGD which causes cell death, an accepted and widely used method to simulate pathophysiological conditions of stroke in vitro, 20 we observed significantly decreased cell viability suggesting increased cell death (Figure 2(d)). This was corroborated by measuring LDH release in these cells (Figure 2(e)). To further confirm this observation, we compared primary neurons isolated from MOAP-1−/− and wildtype mice subjected to the same OGD treatment and observed the opposite effects, whereby cell death was significantly reduced in the presence of MOAP-1 deficiency (Figure 3(d) and (e)). This is supported by previous findings that MOAP-1 knockdown cells were more resistant to various apoptotic stimuli including tumor necrosis factor α (TNF-α), UV irradiation, serum withdrawal, TNF-α related apoptosis-inducing ligand (TRAIL) in MCF-7 and HCT116 cell lines. 9 It may be concluded from these observations that MOAP-1 is competent in executing apoptosis in brain, but its activity is effectively restrained under normal physiological conditions only to be activated by suitable stimuli or conditions, in this case by OGD. It has been shown previously that MOAP-1 could recruit t-Bid in Fas-induced hepatocellular apoptosis without altering caspase 8 expression. 11 MOAP-1 could also be recruited by TNF-α, TRAIL and Fas-mediated extrinsic signaling in apoptosis-related cell death cascade.9,11,21 Evidence has shown that Fas, TNF-α, and TRAIL pathways were activated after cerebral ischemia and contributed to the pathophysiology of ischemic stroke.22–24 For instance, blockade of TNF-α and TRAIL signaling pathway promotes neuroprotection in brain ischemia,25,26 and mutation of the Fas gene led to smaller infarct volume in mice. 23 Interestingly, double knockout of FasL and TNF-α mice showed the greatest reduction of infarct volume (93%) than FasL−/− (54%) or TNF-α−/− (67%).24,27 Hence, knocking out MOAP-1 which simultaneously inhibits both pathways could potentially maximize neuroprotection against ischemia.
In order to test if the observed MOAP-1 involvement in post-ischemia apoptosis could be demonstrated under in vivo conditions, we employed the tMCAO model. Transient MCAO is considered to be more appropriate than the permanent occlusion model as it has been reported that caspase 3 becomes active within minutes following reperfusion but remained inactive for hours after pMCAO.28,29 Our results demonstrated the association of Bax and MOAP-1 at the ipsilateral cortex and striatum following the ischemic insult while no association was observed on the contralateral side (Figure 4(b)). This highlighted the role of MOAP-1 as a modulator of apoptotic cell death in ischemic stroke.
Using Laser speckle contrast imaging, it was demonstrated that MOAP-1−/− deficiency does not cause any changes in cortical blood flow distribution. When subjected to tMCAO, both MOAP-1−/− and MOAP-1+/+ mice responded in the same manner and magnitude (Figure 5(a) and (b)). However, the infarct volume was found to be markedly reduced by 55% in the MOAP-1−/− mice 24 h following tMCAO (Figure 5(c) and (d)). No consistent heterogeneity in the perfusion response was observed in different regions due to within group variations. This is reflected functionally in the rotarod test where the MOAP-1−/− mice out-performed the control mice (Figure 5(e)), indicating significantly reduced neurological deficits as a result of reduced ischemic damage. Marked neuronal loss was detected in the infarcted cortex of MOAP-1+/+ mice 24 h after tMCAO which was partially rescued in the MOAP-1−/− mice (Figure 6(a) and (b)). Interestingly, it can be seen from Figure 6(c) and (d) that NeuN expression in the initial 8 h after tMCAO indicated that neuronal loss in the MOAP-1−/− mice was minimal as opposed to the severe loss in the MOAP-1+/+ mice. In other words, neuron loss was attenuated through at least 24 h in the MOAP-1−/− mice following an ischemic insult. Taken together, the current in vitro and in vivo observations strongly suggest that deletion of MOAP-1 may significantly limit apoptotic events in ischemic neurons, thus delaying and reducing neuronal loss.
To conclude, the present study provides substantive evidence that MOAP-1 overexpression leads to increased cell death, while MOAP-1 deficiency leads to decreased cell death when cells were subjected to OGD. Moreover, MOAP-1−/− mice are more resistant to ischemic injuries when subjected to tMCAO. Thus, MOAP-1 appears to be actively participating in the ischemia-induced neuronal loss through apoptosis. This may signal the possibility of a novel therapeutic approach for stroke treatment via inhibition of MOAP-1 activity. The suggestion that MOAP-1 may not be involved in apoptosis under normal physiological conditions 13 gives added impetus to such an idea. One can envisage several theoretical approaches such as inhibiting the interaction between MOAP-1 and TNF-R1 and RASSF1A, inhibiting the association of MOAP-1 with Bax, and enhancing MOAP-1 degradation by inhibiting Trim39 (a RING domain E3 ligase), which inhibits the MOAP-1 degrading enzyme anaphase-promoting complex (APC/C(Cdh1)) ubiquitin ligase. 30 However, caution is required as outcome assessment after only 24 h is not sufficient for an informed decision whether the neuroprotective action of MOAP-1 inhibition would provide beneficial outcome measure in the long-term after stroke. Further work focusing on longer periods of post-stroke recovery and detailed study to dissect the mechanism of action of MOAP-1 in inducing apoptosis in the ischemic brain are warranted.
Supplemental Material
Supplemental material for Modulator of apoptosis-1 is a potential therapeutic target in acute ischemic injury
Supplemental material for Modulator of apoptosis-1 is a potential therapeutic target in acute ischemic injury by Su Jing Chan, Hui Zhao, Kazuhide Hayakawa, Chou Chai, Chong Teik Tan, Jiawen Huang, Ran Tao, Gen Hamanaka, Thiruma V Arumugam, Eng H Lo, Victor Chun Kong Yu and PeterTsun-Hon Wong in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Ministry of Education (MOE) of Singapore (Grant Nos. MOE2012-T2-1-132 to PTHW and VCHY and MOE-T1-148-000-214-112 and 148-000-235-114 to VCHY).
Acknowledgements
The authors are grateful for Mrs. Wee Lee Ting for her administrative support, blinding and randomization for the study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
SJC, KH, CC, EHL, VCKY, and PTHW designed research; SJC, HZ, KH, CC, CTT, JH, RT, GH, and TVA performed research. SJC and PTHW wrote the paper.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.