
Abstract
Administration of anesthetic agents fundamentally shifts the responsibility for maintenance of homeostasis from the patient and their intrinsic physiological regulatory mechanisms to the anesthesiologist. Continuous delivery of oxygen and nutrients to the brain is necessary to prevent irreversible injury and arises from a complex series of regulatory mechanisms that ensure uninterrupted cerebral blood flow. Our understanding of these regulatory mechanisms and the effects of anesthetics on them has been driven by the tireless work of pioneers in the field. It is of paramount importance that the anesthesiologist shares this understanding. Herein, we will review the physiological determinants of cerebral blood flow and how delivery of anesthesia impacts these processes.
Introduction
Utilizing 10-fold more oxygen than would be predicted based on its weight the human brain is one of the most energetically dense organs in the body. 1 This feature is unique to humans and, in part, is a consequence of the complexity of tasks, such as language, tool use, and social relationships, that the organ performs.2,3 As the brain has relatively little capacity for energy storage, a continuous supply of oxygen and nutrients must be delivered by uninterrupted cerebral blood flow (CBF). 4 Multifaceted physiological regulatory mechanisms of CBF exist to ensure that this process occurs throughout the life of the organism. Disruption of CBF by disease, trauma, or iatrogenic causes can give rise to profound irreversible injury in the form of stroke.
Surgery is a form of controlled trauma that occurs concurrently with the establishment of some type of anesthesia. Neurosurgical interventions directly traumatize vulnerable tissues. Anesthetic agents either directly impact the primary functions of the brain and CBF secondarily and/or indirectly alter the hemodynamic factors that contribute to CBF. It is paramount that the anesthesiologist be cognizant of these effects to facilitate surgical procedures and to avoid undue iatrogenic injury during the perioperative period.
In this review, we will discuss the physiological mechanisms responsible for maintaining CBF homeostasis and their interplay with anesthetic agents. Special attention will be given to the contributions of Dr. Richard J Traystman, former editor-in-chief of the Journal of Cerebral Blood Flow and Metabolism, to our understanding of this field.
Physiological determinants of CBF
Prior to an examination of the effects of anesthetics on CBF, we will review the physiological mechanisms that define flow. Under conditions of normothermia and normoxia, CBF must remain at 50–60 ml/100 g/min to meet the metabolic demands of the functioning brain, with women having slightly higher flow rates.5,6 Reserve blood flow exists to a point, but ischemic injury generally occurs once CBF drops below 22 ml/100 g/min, although concurrent pathology such as traumatic brain injury (TBI) or hypothermia can change this threshold. 7 Regulation of CBF is the result of a host of complex and incompletely described intra- and inter-cellular signaling events. The regulatory mechanisms that preserve CBF can broadly be categorized into cerebral autoregulation (CA), neurovascular coupling (NVC), and vasomotor reactivity (VMR). 8 These mechanisms will be reviewed here in brief; for additional details the reader is directed to dedicated reviews of the subject.9–12 While categorization of these regulatory mechanisms as discrete entities provides a useful conceptual framework for this discussion, it should be recognized that significant overlap exists between these mechanisms.
Cerebral autoregulation
In response to any number of extrinsic and intrinsic stimuli, systemic blood pressure can rise or fall dramatically on a timeframe of seconds to minutes to hours, days, and years. Autoregulation of blood flow exists to ensure that perfusion of vital organs remains intact and stable across the dynamic range of pressures the vascular bed is exposed to. CA can broadly be defined as a proportional change in cerebral vascular resistance in response to changes in perfusion pressure to maintain a constant blood flow (Figure 1, blue inlay).
13
Although the result of the processes that give rise to CA is measurable in clinical and laboratory settings, a full understanding of the processes themselves has remained elusive.
Summary of the effects of anesthetic agents on global oxidative-metabolism (GOM) and cerebral blood flow (CBF) as well as the endogenous regulatory mechanisms such as cerebral autoregulation (CA), vasomotor reactivity (VMR) and neurovascular coupling (NVC). See text for additional details.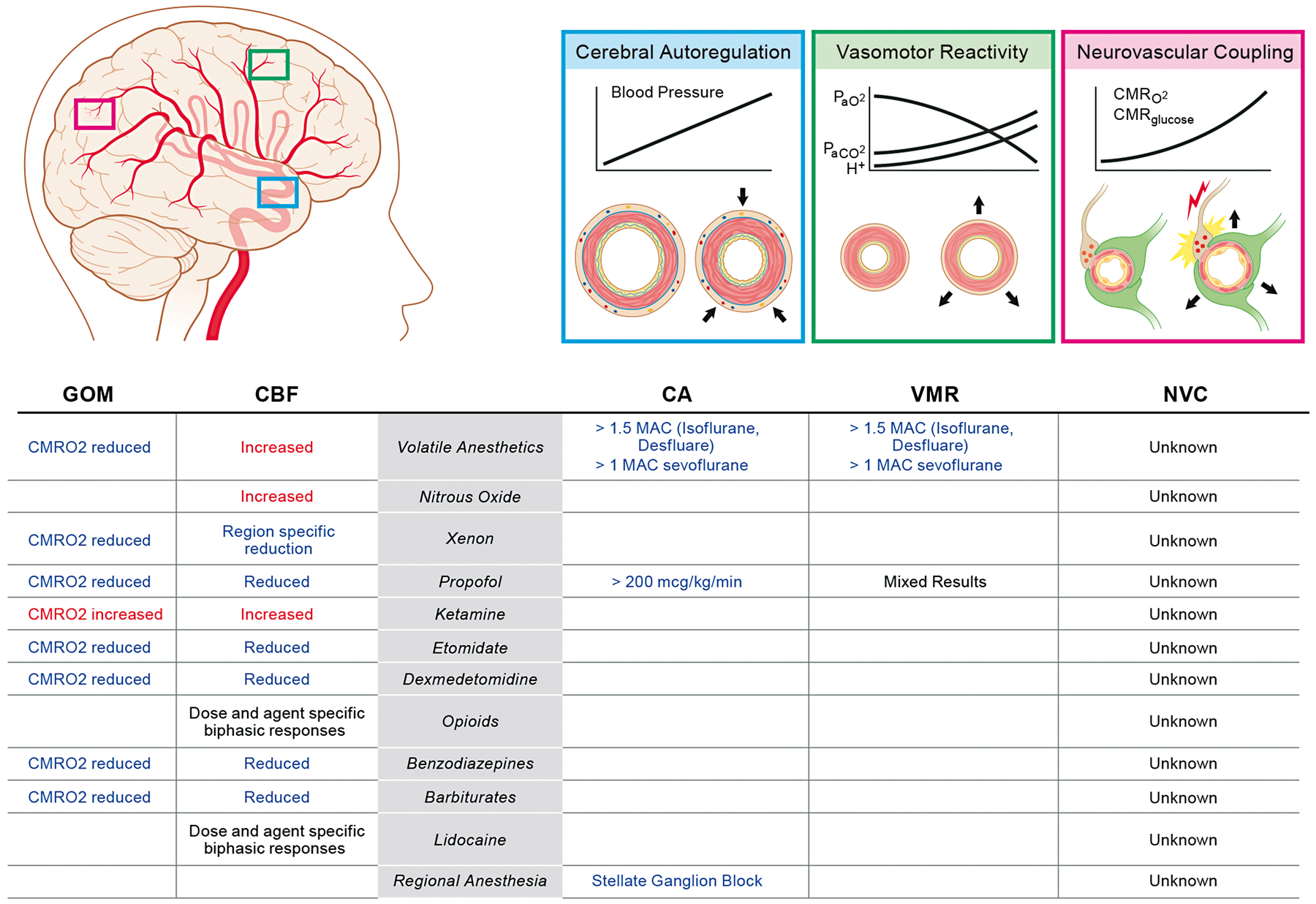
The range of mean arterial pressure (MAP) the CA mechanism can respond to was first described in humans by Lassen in 1959. In his seminal work, Lassen reviewed previous reports of CBF measured in 376 different individuals at 11 different MAP states which were established by normal physiology, administration of vasoactive agents, or systemic disease.
14
From this work, the “autoregulation curve” was generated, which depicts a plateau region wherein CBF is stable across an MAP range of ∼50–160 mmHg (Figure 2(a)). Since its first inception, the nature of the autoregulation curve has been used to guide intra-individual clinical management of patients. This practice assumes that the static inter-individual continuum reported by Lassen can be applied to the physiological experience of singular patients.
Evolving understanding of the relationship between MAP and CBF. (a) Inter-individual static cerebral autoregulatory curve (adapted from Lassen
14
). (b) Intra-individual static cerebral autoregulatory curve (adapted from Numan et al.
15
). (c) Dynamic cerebral autoregulatory curve (adapted from Tan
17
).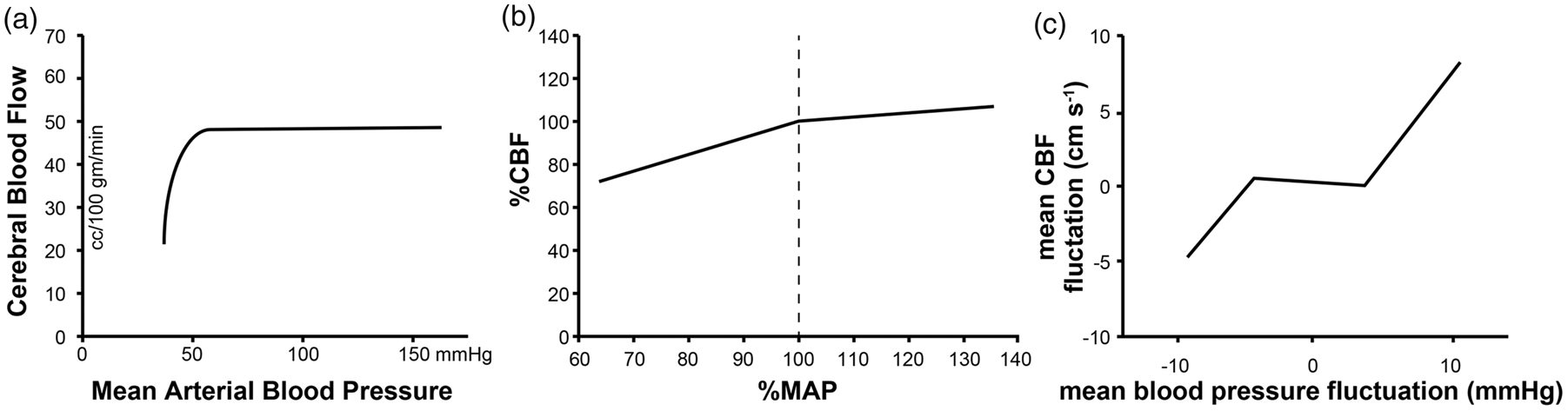
In the last three decades with the advent of non-invasive methodologies, such as transcranial doppler (TCD) ultrasonography which allows for the measurement of CBF velocity in real time, the validity of this assumption has been called into question. This technology has made it possible to evaluate the intra-individual relationship between CBF and MAP. Utilizing technology which allows for real-time evaluation of the CBF in singular individuals across a range of systemic blood pressures an autoregulatory curve quite unlike Lassen's has been described. Analysis of individual static CA relationships from multiple studies indicate that increases in cerebral vascular resistance provide effective CA when systemic blood pressure is increased in most individuals, whereas the decreases in cerebral vascular resistance during decreases in MAP are usually insufficient to maintain CBF perfectly constant (Figure 2(b)). 15
Evaluation of CBF in real time has also revealed that cerebrovasculature can respond rapidly to fluctuations in perfusion pressure and that these adaptations persist. This observation has led to the development of the concept of dynamic CA, i.e. changes in CBF occurring in response to MAP alteration over a timeframe of seconds to minutes. Dynamic CA is in contrast to static CA which arises over minutes to hours to days. 16 When a dynamic CA paradigm is used to generate an intra-individual autoregulatory curve, a frequency-dependent narrow plateau region is observed (Figure 2(c)). 17 The significance of this observation to the practice of anesthesiology is unknown, but with the availability of non-invasive technologies which allow for real-time monitoring of CBF surrogates, it is now possible to perform the clinical studies necessary to make this determination.
Application of CBF-surrogate monitoring in real time to evaluate CA may be of interest to anesthesiologists and intensivists during states of suspected neurological stress due to iatrogenesis or disease. During non-pulsatile cardiopulmonary bypass (CPB) wherein the time-domain fluctuation of MAP is reduced from seconds to minutes, it has been found that the intra-individual lower limit of the CA is incredibly variable and unpredictable. 18 Further, hemodynamic management during CPB guided by CBF-surrogate monitoring may result in a reduction in injury. 19 Additionally, application of the technology has revealed that CA is preserved during hypothermia. 20
Acute regulation of vascular tone generally is mediated by the autonomic nervous system (ANS); however, the contribution of the ANS to CBF and CA is obscure. Cerebral vasculature is known to be highly innervated by the ANS, and postsynaptic adrenergic receptors are present on vascular smooth muscle. 21 While the presence of these elements would suggest that the ANS regulates CBF, early work by Traystman and Rapela demonstrated that direct stimulation of the stellate ganglion produces minimal reduction of CBF. 22 Similarly, denervation does not alter resting CBF and only modestly reduces the upper limit of the autoregulatory curve. 23 Further, both vasoconstriction and vasodilation have been observed following direct application of catecholamines to isolated cerebral artery preparations.24–26 While controversial, an emerging consensus is that the contribution of the ANS to CA is minimal in comparison to the intrinsic mechanisms which establish cerebral vascular tone.27–31
The intrinsic mechanisms of the cerebral vasculature to set its myogenic tone have been intensely studied. The vascular smooth muscle, endothelium, and neighboring neurons and astrocytes, collectively referred to as the neurovascular unit, play direct and modulatory roles in establishing myogenic tone. 32 Smooth muscle of the cerebral vasculature is able to respond to transmural pressure gradients to establish a resting tone matched to the luminal pressure. Mechano-sensitive transient receptor potential (TRP) channel family members present throughout the cerebral vasculature have recently been identified as a component of the pressure detection mechanism of the neurovascular unit. 33 These channels respond to stretch and/or shear forces resulting in cation conductance ultimately leading to vasoconstriction.34,35 The remaining components of the neurovascular unit modulate the myogenic tone established by the vascular smooth muscle.
In addition to mediating vasoconstriction by vascular smooth muscle, TRP channel activity on vascular endothelium may lead to smooth muscle relaxation. In isolated cerebral vascular endothelium, activation of TRP channels results in the generation of nitric oxide. 36 Nitric oxide has long been recognized as one of the endothelium-derived relaxation factors in the cerebral vasculature. Following up on work performed in small mammals, Traystman and his colleagues demonstrated that inhibition of nitric oxide synthesis results in increased cerebral vascular resistance and decreased CBF in piglets and non-human primates.37–39 Later work demonstrated similar activity in humans.40,41 The endothelium is also a source of vasodilatory arachidonic acid-derived prostanoids. 42 Inhibition of prostanoid production reduces regional CBF and is synergistic with inhibition of nitric oxide production. 43 These finding have led to the appreciation for the role of endothelium-derived nitric oxide and prostanoids in establishing the basal tone of the cerebral vasculature.
Astrocytes and neurons also modulate the tone of surrounding vasculature. Astrocytes are a source of arachidonic acid-derived epoxyeicosatrienoic acids which when released diffuse to vascular smooth muscle and mediate vasodilation through membrane hyperpolarization. 44 Neurons also produce and release vasodilatory prostaglandins and nitric oxide. 45 It should be recognized though that additional, as yet incompletely described, mechanisms must also contribute to CA. Systemic application of calcium channel antagonist which negates ion channel-dependent regulation of myogenic tone does not ablate compensation for orthostatic perturbations of blood pressure. 46 Also, disruption of the endothelium secondary to chronic disease which would inhibit mechanical signal transduction does not impair pressure-dependent autoregulation. 47
Neurovascular coupling
NVC describes the modulation of regional CBF to meet the metabolic demands of neural activity (Figure 1, pink inlay). This process occurs at the level of the cerebral microvasculature of plial arterioles outside the pia mater and penetrating parenchymal arterioles. The mechanism by which the tone of these vessels is matched to the needs of the surrounding neurons is complex and involves all members of the neurovascular unit. In response to excitatory glutamatergic inputs, active neurons release nitric oxide which diffuses to nearby vascular smooth muscle and promotes dilation. 48 Adenosine produced from extracellular conversion of neuron- and astrocyte-derived ATP diffuses to vascular smooth muscle and induces changes in vascular tone.49–52 Neuron-derived prostaglandins also influence vascular tone.53,54 Prostaglandin E2 is produced by active pyramidal neurons and acts on EP2 and EP4 receptors present on cerebral vascular smooth muscle resulting in vasodilation. 55 Additionally, GABAergic interneurons release numerous vasoactive mediators directly onto vascular smooth muscle. 56 Metabolically active astrocytes which envelope the parenchymal arterioles also mediate the tone of the vascular smooth muscle. 55 Astrocytes are a source of vasodilatory epoxyeicosatrienoic acids released during cortical activation.57,58 Astrocytes also modulate vascular tone through release of vasodilatory K+ and Ca2+ to the perivascular space.59,60 Finally, in response to neuronal activity, the endothelium itself propagates vasodilatory signals throughout the vasculature of active cortical regions. 61 Collectively, these mechanisms allow for modulation of CBF that is temporally and spatially correlated with neuronal activity. Vessels surrounding active neurons to a distance of 0.5–1 mm respond within ∼ 1 s to the onset of neuronal activity to modulate CBF. 62
Vasomotor reactivity
The cerebral vasculature tone is influenced by changes in arterial blood CO2 and, to a lesser extent, O2 tension through a process referred to here as VMR (Figure 1, green inlay). 63 VMR to CO2 is stronger in the brain compared to other organs. 64 Both plial arterioles and large caliber cerebral vessels respond. 65 Alteration of cerebral vascular tone in response to changes in PaCO2 is an intrinsic property of the vasculature and occurs independent of adrenergic activity despite extensive innervation by the sympathetic nervous system. 66 The mechanism responsible for cerebral vasodilation and vasoconstriction in response rising and falling PaCO2, respectively, is incompletely understood. Nitric oxide production is dispensable for VMR. 67 Early studies indicated that alteration of extracellular pH due to diffusion of CO2 out of or into the vasculature is the dominant mechanism of VMR to CO2.68,69 However, more recent work suggests that transient changes in vascular smooth muscle intracellular pH also can modulate the response. 70 This mechanism establishes a roughly linear relationship between CBF and acute PaCO2 changes with a 1–6% change in CBF for every 1 mmHg across a range of 20–80 mmHg. 71 Adaptation to chronic alteration of PaCO2 occurs and CBF will tend to normalize over a 6–8-h period. 72 Acute changes in CO2 tension impact the CA mechanism with loss of eucapnia, leading to a narrowing of the CA plateau. 73 While impaired VMR to changes in PaCO2 is not predictive of stroke risk, it does correlate with increased mortality.74,75 This may indicate that impaired VMR is a symptom of systemic vascular disease which itself is the most detrimental pathology.
To guard against injury due to hypoxia or hyperoxia CBF is modulated such that oxygen delivery expressed by the product of CBF and CaO2 is roughly constant over PaO2 range of 30–430 mmHg. 76 Vasodilation occurs independent of peripheral chemoreceptors and may be accompanied by a small increase in CMRO2, although not all studies detect this increase.76,77 Under hypoxic conditions, the hypocapnic cerebral vasoconstriction activity is attenuated. 78 This prevents ischemic injury from arising due to hyperventilation-induced hypocapnia associated with hypoxic ventilatory drive.
Anesthetic agents and CBF
Anesthetic agents affect the dynamics of the cerebral vasculature through direct effects on the vessels as well as modulation of the endogenous regulatory mechanisms (Figure 1). In addition to impacting the delivery of oxygen and nutrients to metabolically active neurons, alteration of cerebral vessel dynamics by anesthetic agents can influence the tissue composition encountered during neurosurgical intervention. For these reasons, the anesthesiologist must have a full understanding of the effect of their interventions on the cerebral vasculature. Additionally, intrinsic neuroprotective properties have been attributed to anesthetic agents. The concept of neuroprotective intervention encompasses therapies which favorably shift the balance of cerebral oxygen supply and utilization as well as those that prolong survival in ischemic states. Where directly relevant neuroprotection will be discussed, in addition, the interested reader is directed to other recent reviews of this subject.79–82 Herein, emphasis is placed on the inclusion of studies conducted using human subjects as much as is made possible by the availability of data.
Volatile anesthetics
Modern halogenated volatile anesthetics, such as isoflurane, sevoflurane and desflurane, are thought to uncouple flow-metabolism matching. Suppression of CMRO2 is consistently observed in human and animal models exposed to volatile anesthetics using a host of assessment techniques. Assessment of CBF in humans exposed to volatile anesthetics using TCD and gas diffusion techniques generally report dose-dependent increases.83–86 However, magnetic resonance imaging (MRI)-based assessments reveal minimal changes in global CBF in humans, whereas non-human primates display dose-dependent increases.87–89 Further, conflicting data have been generated regarding sevoflurane with no change, increases and decreases in CBF being observed.88,90–96 The mismatch between CBF and oxidative metabolism likely arises from the direct effects of volatile anesthetics on cerebral vascular resistance.
Vasodilation mediated by volatile anesthetics occurs independent of the anesthetic depth and is the result of a direct effect on the cerebral vasculature. 97 Compared to isoflurane, sevoflurane is a less potent cerebrovascular vasodilator when administered at MAC equivalent doses. 98 A full understanding of the mechanism by which vasodilation occurs is being derived. ATP-sensitive K+ channels as well as increased production of endothelium-derived nitric oxide and prostanoids have been identified as being potentially modulated by volatile anesthetics.43,99,100
CA remains intact during isoflurane and desflurane administration at 1 MAC, but above 1.5 MAC CBF is pressure-passive with respect to MAP.101,102 However, concurrent hypercapnia does induce blockade of CA during administration of therapeutic doses of volatile anesthetic. 103 Conversely, hypocapnia restores CA during supratherapeutic isoflurane administration. 104 Sevoflurane at therapeutic doses has been reported to narrow the plateau region of the autoregulatory curve. 105 VMR is preserved during isoflurane administration unless supratherapeutic doses are administered. 106 Conversely, vasodilation due to hypercapnia as well as vasoconstriction due to hypocapnia are blunted by sevoflurane. 107
Volatile anesthetics have long been postulated to act as neuroprotective agents in the setting of cerebral hypoperfusion. Investigations in small animals and non-human primates suggest that use of volatile anesthetics could limit injury following experimentally induced ischemia. 79 Retrospective clinical studies have produced data to suggest the same. Isoflurane use during carotid endarterectomy appears to decrease the critical CBF below which electroencephalogram (EEG) evidence of injury arises. 108 Similarly, desflurane administration preserves cerebral oxygenation during temporary cerebral artery clipping. 109 However, in specific circumstances such as the neonatal period, toxicity attributable to volatile anesthetics has been described in preclinical trials.110,111 In the absence of results from prospective randomized controlled trials, it is premature to conclude that sufficient evidence exists to support the notion that volatile anesthetics are neuroprotective or neurotoxic agents in humans.
Nitrous oxide
Nitrous oxide is the oldest anesthetic gas still in regular use. When inhaled, it produces dose-dependent analgesia, dissociative amnesia, anxiolysis, and somnolence. Due to its low potency at atmospheric pressures, a state general anesthesia cannot be induced by nitrous oxide alone. Under hyperbaric conditions, general anesthesia can be achieved by nitrous oxide alone; however, this is associated with significant hemodynamic derangement. 112 Numerous biological targets for nitrous oxide have been described with antagonism of the NMDA receptor thought to be responsible for its anesthetic properties. 113 The efficacy of nitrous oxide as part of the anesthesia provided to neurologically vulnerable patients has been contested.
Rapid agent elimination following surgical intervention is desirable to allow for early clinical assessment in the postoperative period, but this feature of nitrous oxide is not widely accepted to outweigh the potential risks associated with its use. 114 Classical teaching is that nitrous oxide increases CBF, CMRO2, intracranial pressure (ICP), and moves into the potential space created by surgically induced pneumocephalus or could worsen venous air embolism and should therefore be avoided. Data supporting this teaching come from studies to model the use of nitrous oxide as a MAC sparing agent in general anesthetic states established by other agents. The varying nature of these models may give rise to misunderstanding of the intrinsic activity of nitrous oxide. When administered as a single agent, nitrous oxide results in increased CBF and no change or decreased CMRO2.115–119 However, when used as an adjunct to other anesthetics, the addition of nitrous oxide has been reported to enhance, blunt, or have no effect on CBF and CMRO2.120–127 Further complicating this picture is the observation that the temporal relationship between agent exposure and outcome is inconsistent. 128 VMR remains intact when nitrous oxide is administered alone or in combination with other anesthetic agents. 129
Data on outcomes associated with administration of nitrous oxide to neurologically vulnerable patients come from retrospective analysis of the IHAST trial. 130 When nitrous oxide was used as part of general anesthesia provided to patients undergoing surgical repair of ruptured subarachnoid hemorrhage, there were no long-term neurological deficits attributable to the drug. 131 Subgroup analysis of patients who underwent temporary occlusion of major cerebral vessels as part of the surgical intervention did reveal an increase in the occurrence of neurological deficits in the immediate perioperative period associated with nitrous oxide exposure; however, this association was no longer present three months following surgery. 132 These results have led to a renewed interest in the use of nitrous oxide as part of the anesthesia provided during neurosurgical interventions. 129
Xenon
A noble gas naturally occurring at low concentration in Earth's atmosphere, 0.0875 ppm, xenon been recognized as an agent able to achieve general anesthesia since the 1940s.133,134 The mechanism by which xenon induces a general anesthetic state is believed to arise from inhibition of NMDA receptors by tight association with the glycine-binding site.135,136 Xenon has features of an ideal anesthetic including rapid induction of and emergence from a general anesthetic state following initiation and discontinuation of administration, hemodynamic neutrality, and neuroprotective properties.137,138 Scarcity, cost of production, and competition with use in industrial applications all limit clinical use of xenon.
Studies conducted to examine the effect of xenon on CBF and CMRO2 using rodent, porcine, and non-human primate systems have produced conflicting results. In an elegant study by Laitio et al., regional CBF was measured in humans under general anesthesia induced and maintained by 1 MAC of xenon alone. In this study, adjunct remifentanil was used at the time of intubation in response to signs of responsiveness to laryngoscopy; however, a period of 10 remifentanil half-lives was allowed to pass prior to measurement of CBF by 15O-H2O positron emission tomography (PET) Laitio et al. found that areas of dense concentrations of soma, the cortex, cerebellum, and thalamus experience an overall reduction in blood flow, whereas structures arising from axon projections, white matter tracts, had an increase in blood flow during xenon administration. 139 In a follow-up study, this group was able to confirm that reduction of CBF correlated with reduction of CMRO2. 140 These findings are largely in concordance with those reported by Rex et al.141,142 The matched reduction in CBF and CMRO2 induced by xenon is in stark contrast to other NMDA receptor antagonists, such as nitrous oxide and ketamine which, as discussed above and below, increase CBF.
Propofol
Propofol is an intravenous anesthetic that achieves dose-dependent sedation or general anesthesia. The mechanism by which reduced consciousness occurs is by a direct interaction between propofol and the GABAA receptor leading to potentiation of the receptor activity.143,144 Compared to volatile anesthetics delivered at Bispectral Index (BIS)-equivalent equipotent doses, propofol causes significant reduction of CBF and similar decreases in CMRO2.122,145 The mechanism by which CBF reduction occurs is thought to arise from intact flow-oxidative metabolism coupling as vasodilation is observed when propofol is applied directly to vessels in in vitro preparations. 146 CA remains intact during propofol administration up to doses of 200 µg/kg/min. 102 An early study using equipotent doses of propofol and sevoflurane titrated to BIS suggested that propofol blunts reactivity VMR. 147 However, recent studies using NIRS spectroscopy have demonstrated that reactivity to hypocapnia secondary to hyperventilation is similar when either propofol or sevoflurane is used to maintain general anesthesia.148–150
Evidence for neuroprotection that may be attributable to propofol comes from the prospective SIESTA and Goliath trials which suggest an association between general anesthesia maintained with propofol and improved neurological outcome following endovascular clot retrieval for stroke.151,152 However, neither trial was structured to investigate an agent-specific effect, rather a state-specific effect, sedation versus general anesthesia, was the primary comparison of interest for these studies. A single agent-specific study exists wherein propofol administration was titrated to burst suppression on EEG during cardiac valve surgery; this study failed to demonstrate improved neurological outcomes following propofol administration. 153 Interpretation of the significance of this study is difficult, as burst suppression is itself thought to give rise to adverse neurological outcomes. 154 Additional prospective studies examining the administration of therapeutic doses of propofol are likely indicated at this time.
Ketamine
Ketamine is a phenylcyclidine derivative that when administered intravenously or intramuscular induces a state of general anesthesia with preservation of respiratory drive. In the catecholamine-replete patient, ketamine induces a weak and transient release of endogenous stores which counteracts the direct myocardial depressant effect of the drug. While many molecular targets for ketamine have been described, inhibition of the NMDA receptor is thought to give rise to its anesthetic properties. Enthusiasm for use of ketamine in the neurologically compromised patient was tempered by early preclinical reports of increased CMRO2 and small case series documenting elevated ICP associated with its use.155–159 However, recent studies conducted in human reveal that when used in conjunction with usual modern anesthetic practices, ketamine administration does not result in increased ICP. 160 Alteration of CMRO2 in humans is minimal and is likely a consequence of increased metabolite supply rather than simply increased utilization.161,162 In addition, a recent meta-analysis of available human studies has demonstrated that ketamine administration increases CBF. 163 Ketamine-mediated increase of CBF appears to arise from direct vasodilation of medium cerebral vessels. 164 There is renewed interest in ketamine use due to the neuroprotective properties attributed to the drug. A full discussion of these properties is beyond the scope of this review; for additional information the reader is directed to an excellent recent review article on the subject. 165
Etomidate
Etomidate is an imidazole derivative synthesized in the early 1970s and originally intended for use as an antifungal agent prior to appreciation of its sedative-hypnotic effects. When administered as a single bolus or continuous infusion, etomidate achieves sedation through potentiation and direct activation of synaptic and extrasynaptic GABA receptors. 166 Unique among anesthetic agents, administration of etomidate is not associated with hemodynamic depression. Using animal models of focal and global ischemia, reports have described etomidate as having neuroprotective properties.167,168 Etomidate administration to humans has been shown to result in reductions of ICP, CBF, and CMRO2.169–172 Reduction of CBF is speculated to arise secondarily from reduction of CMRO2 as well as direct vasoconstrictive properties of the drug itself possibly through inhibition of nitric oxide signaling.170,173,174
Due to its unique hemodynamic neutrality, an argument has been advanced for use of etomidate in the context of vulnerable cerebral perfusion in place of barbiturates. 175 However, administration during cerebral aneurysm clipping procedures and temporary vessel occlusion has revealed that tissue hypoxia attributable to etomidate occurs.109,176 Further, retrospective analysis of the data from the IHAST trial did not demonstrate an association between etomidate administration and neurological outcome following temporary vessel clipping. 177 Finally, reduced cerebral oxygenation has been observed following administration of induction doses of etomidate administered to patients with intact cerebral vasculature. 178 These results suggest that cerebral vasoconstriction and CBF reduction mediated by etomidate are not matched by reduction of CMRO2 and the presumption that neuroprotection arises in ischemic states is questionable.
Dexmedetomidine
Dexmedetomidine (Dex) is a centrally acting α2-adrenergic agonist that achieves sedation, anxiolysis, and analgesia without concurrent respiratory depression.179,180 Dex administered by continuous infusion is used as a primary sedative in the ICU setting or for procedural sedation and has been found to have opioid sparing effects when used as an adjunct for intracranial procedures.181–184 Dex reduces CBF possibly through direct activation of postsynaptic α2-adrenergic receptors located on cortical vasculature resulting in vasoconstriction.185–187 Clonidine, another α2-adrenergic agonist, also reduces CBF. 188 In addition, Dex administration results in a reduction in CMRO2. 189 In humans, the reduction in CBF induced by Dex occurs without producing significant alteration of cerebral oxygenation.186,189–191 However, concurrent hypoxia or systemic hypotension may result in impaired cerebral oxygenation during Dex administration.185,192 These results raise cause for caution with the use of Dex as part of an anesthetic for the patient with hemodynamic instability.
Opioids
As a class, opioids have generally been considered to be neutral in regards to alteration of CBF and CMRO2. Studies supporting this belief were generated using low-resolution detection techniques, such as TCD wherein flow velocity through the MCA is used as a surrogate for global CBF without consideration for regional differences. High-resolution studies using MRI and PET-based detection techniques have revealed that regional CBF is affected by opioids and use of class-wide generalities may incorrect. Fentanyl administration to opioid-naive humans has been found to increase regional CBF in areas of the prefrontal cortex and caudate.193,194 Morphine and hydromorphone have been found to have similar effects.195,196 Conversely, sufentanil and remifentanil administration have a biphasic effect on regional CBF with initial increases at low to moderate doses (<0.15 µg/kg/min remifentanil) followed by dose-dependent decreases in CBF and CMRO2 at supratherapeutic doses (>2 µg/kg/min remifentanil) under normocapnic conditions in brain regions known to be involved in pain processing.197–200 VMR appears to be intact during remifentanil infusion.201,202 Alteration of regional CBF has been speculated to be reflective of the mechanisms giving rise to the analgesic and euphoric properties of the drugs.195,203
Outcome studies comparing the use of different opioids in conjunction with propofol-based anesthetics have not revealed a difference in dreaded perioperative complications attributable to opioid choice. However, these studies do suggest a more rapid recovery following discontinuation of anesthesia when remifentanil is used, although this effect is likely attributable to remifentanil's unique pharmacokinetic profile rather than arising from differences in intraoperative cerebrovasculature dynamics.204–206 The lack of dreaded complications observed suggests that the clinical significance of the alteration of regional CBF induced by different opioids is small or non-existent. Additionally, neuroprotective properties have been attributed to opioid receptor subtype-specific agnonists.207,208 It is possible that this activity arises from preservation of CA and VMR in the face of ischemic injury.209,210 In addition, preclinical data suggest that endogenous opioids may play a protective role following TBI. 211 However, at present, no opioid receptor-specific agonist agent has been evaluated in clinical trials for therapeutic efficacy in ischemic states or TBI.
An additional point of note is the finding that administration of the opioid receptor antagonist, naloxone, has been found to result in decreased CBF. 212 Naloxone and related opioid receptor antagonists have been evaluated in the context of stroke injury, but efficacy has not been demonstrated.213–215 Neuroprotective properties have been attributed to naloxone in the context of incomplete spinal cord injury. 216 This effect may arise from preservation of blood flow to the spinal cord through alteration of the expression of mediators of vasogenic tone.217,218 It is possible that the CBF alteration and/or induction of a neuro-protected state that occurs following administration of exogenous opioids or opioid receptor antagonists arises from competition with endogenous opioids or alteration of opioid receptor signaling in a subtype and location-specific fashion.
Benzodiazepines
Benzodiazepines are commonly used to achieve anxiolysis, amnesia, and sedation during conscious sedation and, when administered at high doses, induce general anesthesia through potentiation of GABA receptors. Acute administration of benzodiazepines results in dose-dependent matched reductions in CBF and CMRO2.219–222 Enhancement of GABAergic activity seems to generally cause a reduction in CBF as, like benzodiazepine, the non-benzodiazepine GABA receptor modulator zolpidem has similar effects. 223 Studies assessing the cerebrovascular response to benzodiazepine administration have generally been performed in healthy volunteers naive to the drug following acute administration. A single study has investigated the relationship between chronic benzodiazepine use and CBF and revealed development of tolerance to the CBF reduction effects. 224 Regional CBF alteration has been observed with the largest declines in areas of the brain involved in memory formation, attention, and arousal; while the location of these changes correlates with the areas of the brains associated with the clinical effects observed following administration of benzodiazepines, a causal relationship has not yet been established.225,226 Interestingly, unlike the sedative effects, the reduction of CBF following midazolam administration is not reversed by doses of flumazenil, an observation that may be explained by direct vasoconstriction mediated by flumazenil.227,228 This effect though is controversial as some reports describe a recovery of CBF following administration of flumazenil following midazolam.222,229
Barbiturates
While short-acting barbiturates (e.g. thiopental) are no longer available for clinical use in the United States, a discussion on barbiturates is included here for completeness sake. With the exception of methohexital use in electroconvulsive therapy, thiopental was the only barbiturate used to induce and maintain general anesthesia in modern anesthetic practice. Unconsciousness occurs secondary to direct activation and potentiation of the GABAA receptor by barbiturate binding. 230 Barbiturate administration results in reduced CBF and CMRO2 with preservation of CA and blunting of VMR.231–234 Many studies using small and large animal models of cerebral ischemia have demonstrated neuroprotection attributable to barbiturate administration. 235 Thiopental has been subject to many clinical trials aimed at determining if its neuroprotective properties are clinically accessible. The results of these studies have been mixed and if beneficial affects are present, the timing of thiopental administration surrounding the onset of ischemia may be crucial. 236 Additionally, retrospective analysis of data generated in the IHAST study demonstrated no association between thiopental administration and neurological outcome. 177
Lidocaine
While not a general anesthetic agent itself, lidocaine is often administered during induction of general anesthesia at doses known to effect CBF. Data supporting this practice come from the observations that lidocaine administration blunts the pain associated with propofol injection and limits hemodynamic alteration during laryngoscopy.237,238 In humans, a moderate bolus (0.5 mg/kg) of lidocaine increases regional CBF whereas a large dose (5 mg/mg) results in modest reduction of CBF and CMRO2.239–241 In addition, bolus administration is associated with reduced ICP. 242 No studies exist which have examined the effects of lidocaine on CBF when administered with other agents during induction of anesthesia. Studies conducted in small mammals suggest that lidocaine may have neuroprotective properties during threatened CBF.243–245 Lidocaine has been examined in humans in the context of post-operative cognitive dysfunction occurrence; these studies have produced conflicting results, and at this time, no uniform body of evidence exists to define the clinical utility of the drug in this context.246–249
Regional anesthesia
As certain blocks alter intracranial sympathetic inputs, the possibility exists for disruption of CBF secondary to alteration of incoming supply. While contribution of the ANS to CBF is controversial, it has been found that direct stellate ganglion blockade results in a modest increase in CBF prominent on the side ipsilateral to the block. 28 Interscalene block has been found not to result in significant change to CBF. 250 Interestingly, afferent inputs during spontaneous movement increase regional CBF and peripheral regional blocks blunt this response.251–253
Conclusions
Regulation of CBF to ensure uninterrupted delivery of oxygen and nutrients is necessary for the prevention of irreversible ischemic injury to the brain. Through the work of pioneers in the field like Dr. Richard J Traystman, we are beginning to understand not just how the CBF regulatory mechanisms function but also how therapeutics interact with these processes. Clinical studies informed by this understanding are ongoing. While no single agent has yet been identified as being the “silver bullet” for neuroprotection, there is cause for hope. Reason for this hope was best expressed by Dr. Traystman himself when he wrote, (a)nesthetics have shown neuroprotective potential, but thus far, no single leader has come forward despite much research in this area. While it would be easy to give up looking for an anesthetic neuroprotective agent considering this poor track record, the lure of the benefit to patients of actually finding a neuroprotective agent is great. I say, we keep looking and be optimistic. It is just a matter of time!
235
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.