
Abstract
Ischemic stroke is caused by a regional interruption of cerebral blood flow to the brain. Rigorous pre-clinical and clinical research has made landmark progress in stroke treatment using thrombolytics and endovascular thrombectomy. Although numerous successful neuroprotective therapeutic agents for ischemic stroke have been reported in pre-clinical studies, most of them failed in clinical testing. Persistent pre-clinical research has demonstrated that the ischemic brain is not only passively dying but is also actively recovering. Within the neurovascular niche in the peri-infarct tissue, repair mechanisms thrive on the interactions between the neural and vascular compartments. In this review, we discuss exogenous therapy using mesenchymal stromal cell-derived exosomes to amplify endogenous brain repair mechanisms and to induce neurorestorative effects after stroke. Emerging evidence indicates that multiple communication axes between the various organs such as the brain, heart, kidney and gut, and whole body immune response mediated by the spleen can also affect stroke outcome. Therefore, in this review, we summarize this evidence and initiate a discussion on the potential to improve stroke outcome by amplifying multiple brain repair mechanisms after stroke, and by targeting peripheral organs and downstream events to enhance recovery in the injured brain and promote over all well being.
Introduction
Despite a declining trend in stroke-related deaths, stroke remains the second leading cause of death behind heart disease, and is a major cause of long-term disability worldwide. 1 Our understanding of the pathophysiological cascade following ischemic injury to the brain has greatly improved over the past few decades. Ischemic brain damage can be divided into two primary zones – the ischemic core and the ischemic penumbra. Failure to meet energy demands is the primary cause of cell death within the ischemic core. Ischemia decreases cerebral blood flow (CBF), impairs energy synthesis, increases extracellular adenosine triphosphate (ATP) as a result of neuronal and glial depolarization, which leads to metabolic and ionic imbalance, and excitotoxicity.2–4 Therefore, neuronal loss of transmembrane ionic gradients, neuronal depolarization, excessive release of excitatory neurotransmitters and pro-inflammatory molecules, and intracellular ionic imbalance evoke primary neuronal loss via necrosis and apoptosis. The ischemic penumbra is sandwiched between the poorly perfused ischemic core and normally perfused healthy tissue. The penumbra remains viable for hours due to collateral blood flow, presenting a window of opportunity to reperfuse and salvage this region. Reperfusion is a primary goal for thrombolytic and neuroprotective therapies, and is a predictor of improved functional outcome. 5 In addition to cell death mechanisms which dominate the ischemic core, stroke also stimulates survival and repair mechanisms especially in the penumbra. Activation of these endogenous brain repair mechanisms stimulates spontaneous functional improvement after stroke. Several clinical studies have shown that approximately 50% functional recovery occurs within the first two to six months after stroke onset.6–8 Initial stroke severity, age of patient, co-morbidities and other factors influence the extent and rate of such spontaneous recovery from stroke.6–8 Hypoxia and inflammation stimulate tissue repair mechanisms such as angiogenesis, neurogenesis, immunomodulation, neuronal functional reorganization, and rewiring of neuronal networks in the ipsilateral and contralateral brain hemispheres. 9 The primary aim of neurorestorative therapies after stroke is to enhance these endogenous restorative mechanisms and improve long-term neurological functional outcome.9,10
The knowledge that the ischemic penumbra is not only passively dying but also is actively recovering, has spurred research to amplify endogenous repair mechanisms to improve recovery after stroke. Following ischemic stroke, there is a graded transition from damage to endogenous repair mechanisms. These repair mechanisms and their signaling pathways are coupled and interdependent, particularly within the neurovascular niche.10,11 Treatments that can amplify multiple restorative and repair processes are most likely to have maximum therapeutic efficacy. In this review, we will focus our discussion of restorative and repair therapeutics of stroke on the therapeutic effects of mesenchymal stromal cell-derived exosomes (MSC-exosomes).
Remarkable progress has been made in pre-clinical research in elucidating various signaling pathways, and numerous novel therapeutics for stroke prevention, pre-treatment, thrombolytics, neuroprotection, neurorestoration and rehabilitation have emerged.10,12,13 Unfortunately, clinical translation of investigational drugs remains problematic. Various factors have been implicated in hindering bench to bedside translation of stroke treatments including age and gender bias resulting in widespread use of young male animals that are typically healthy without co-morbidities such as diabetes and hypertension, poor experimental design with insufficient sample size, and reporting and publishing bias.12,14,15 In this review article, we draw attention to another confounding factor based on emerging preclinical and clinical evidence that indicate multiple communication axes between various organs such as the brain, heart, kidney and gut, and whole body immune response mediated by the spleen that may also contribute to poor outcome after stroke. This new understanding calls for novel treatment approaches that target these peripheral organs to enhance recovery in the injured brain and improve neurological function after stroke.
Endogenous brain repair after stroke
Neurovascular coupling
Coupling of angiogenesis, arteriogenesis and neurogenesis enhances brain plasticity and improves neurological recovery after stroke. 10 The intrinsic ability of the central nervous system to undergo functional reorganization in response to ischemia involves rewiring of brain and spinal cord circuitry, including synaptogenesis to compensate for lost connections and function due to ischemic lesion.9,10,16,17 Angiogenic signaling is activated as early as an hour after stroke, and persists for several days to weeks. 18 Angiogenesis is a therapeutic target for post stroke recovery; however, there is an associated risk of vascular endothelial growth factor (VEGF) induction of immature and leaky blood vessels that can evoke adverse effects such as edema and vascular regression over time, particularly in diabetic stroke which is associated with extensive vascular damage and poor outcome.19,20 In addition to angiogenesis, promoting arteriogenesis to form new capillaries and remodel pre-existing collateral blood vessels also improves functional outcome after stroke. 21 Angiogenesis enhances post stroke synaptogenesis. 17 Increasing synaptophysin, a pre-synaptic vesicle protein, using cell therapy has been correlated with post stroke functional improvement in rodent models of stroke.22,23 Stroke induces white matter damage with loss of axons, myelin and oligodendrocytes. 10 Regeneration and rewiring of lost neuronal circuitry via axonal outgrowth, dendritic sprouting and remyelination are important for long-term functional recovery. 10 In the weeks following stroke, there is gradual and substantial increase in numbers of myelinated axons and apical dendrite spine density in the peri-infarct brain tissue of rodents. 24 Stroke induces differentiation of non-myelinating oligodendrocyte progenitor cells into mature and myelinating oligodendrocytes. 24 Post ischemic oligodendrogenesis is required to form myelin sheaths on the newly sprouting axons because mature oligodendrocytes are unable to proliferate and injured oligodendrocytes are unable to form myelin. Magnetic resonance imaging (MRI) of ischemic rodent brain demonstrated that stroke induces increased axon density with axonal projections oriented parallel to the rim of the ischemic boundary zone. 25 The temporal and spatial profiles of angiogenesis and axonal remodeling along the ischemic boundary zone coincide after stroke. 25 Interestingly, the intact cortical neurons of the contralateral hemisphere also undergo remodeling in response to ischemic injury to form axon collaterals and synaptic-like structures that extend to the denervated side of the spinal cord and improve sensorimotor function in experimental stroke.9,16 Such intercortical and intracortical axonal connections facilitated by corticospinal neuronal sprouting from the intact cortex to peri-infarct regions can be stimulated and amplified by cell therapies and pharmacological agents.16,26
Remodeling within the neurovascular niche
Post ischemic angiogenesis regulates several repair mechanisms, including secretion of growth factors that enhance cell survival in the ischemic penumbra, macrophage surge that facilitates debris removal, and scaffolding and guidance for neuroblast migration to the ischemic boundary region.27,28 Neuroblasts migrate from the subventricular zone in chains or individually to reach a neurovascular niche in the ischemic boundary zone.11,29–32 The neurovascular niche is a specialized microenvironment in the peri-infarct tissue, where interactions between neural progenitor cells (NPCs) and the vasculature promote adult neurogenesis. 11 When NPCs isolated from the subventricular zone of non-ischemic adult rats were co-cultured with cerebral endothelial cells isolated from the stroke boundary, there was a significant increase in NPC proliferation and differentiation into neurons. 33 Similarly, conditioned medium harvested from the SVZ of stroke rats promoted capillary tube formation of normal cerebral endothelial cells. 33 Inhibiting post stroke angiogenesis in the peri-infarct cortex decreased vascular density and resulted in a dramatic decrease in neuroblasts in the peri-infarct cortex indicating that angiogenesis and neurogenesis are causally linked at least within the neurovascular niche after stroke. 11 These studies demonstrate that post ischemic angiogenesis and neurogenesis are coupled. While post-stroke neurogenesis has been primarily studied using experimental stroke models in rodents, there is evidence of newborn neurons preferentially localized adjacent to blood vessels in the peri-infarct cortex of stroke patients. 34 Therefore, stroke initiates neuronal proliferation and expansion of neuroblasts in the subventricular zone which is followed by their migration to peri-infarct tissue where they differentiate into a region-appropriate phenotype reflecting phenotype of most degenerated neurons and replace neurons destroyed by ischemia.29,31,32 The innate ability of the brain to replace damaged neurons via adult neurogenesis can be amplified by trophic and growth factors 11 and presents a therapeutic target.
Mechanisms of neurovascular coupling
The neurovascular unit (NVU) comprises structural and functional interactions between neuronal, glial (astrocytes, microglia, and oligodendrocytes), and vascular (endothelial cells, pericytes, and smooth muscle cells) compartments, and the extracellular matrix of the brain. The molecular mechanisms underlying coupling of angiogenesis and neurogenesis after stroke remain unknown. Stroke increases ischemic brain expression of several growth and trophic factors and chemokines such as VEGF, insulin-like growth factor-1 (IGF-1), fibroblast growth factor-2 (FGF-2), brain-derived neurotrophic factor (BDNF), stromal cell-derived factor 1 (SDF1), monocyte chemoattractant protein-1 (MCP-1), endothelial nitric oxide synthase (eNOS), and angiopoietin-1/Tie2 (Ang1/Tie2) some of which are multi-functional and promote neurogenesis, angiogenesis, oligodendrogenesis, and may potentially mediate their coupling. 35 Neuroblast proliferation, migration, and differentiation are coupled to post ischemic oligodendrogenesis.11,24,36–38 In addition to the proliferation of oligodendrocyte progenitor cells in the corpus callosum, stroke stimulates NSCs in the SVZ to generate oligodendrocyte progenitor cells which then migrate to the white matter tracts of the corpus callosum and striatum as well as ischemic boundary zone and differentiate into mature myelin producing oligodendrocytes in the vicinity of sprouting axons. 24 In the neurovascular niche, secretion of SDF-1, VEGF and Ang1 by activated endothelial cells, angiogenic blood vessels, and astrocytes guide neuroblast and oligodendrocyte progenitor cell migration, and the inhibition of SDF-1 and Ang1 in remodeling cerebral vessels attenuates neuroblast migration toward the peri-infarct cortex.11,36–38 Exosomes derived from ischemic endothelial cells and NSCs can modulate neurogenesis and angiogenesis, respectively, via transfer of exosomal cargo microRNAs (miRs). 39 Ischemic endothelial cell-derived exosomes rich in cargo miR-146 a and miR-125 a were taken up by non-ischemic NSCs and promoted neuronal differentiation. 39 Similarly, ischemic NSC-derived exosomes exhibiting increased miR-106 b and miR-125 b were taken up by non-ischemic endothelial cells and significantly increased their capillary tube formation. 39 In-vitro, ischemia-activated NPCs were found to stimulate angiogenesis in cultured endothelial cells via secretion of VEGF. 33 MiR-9 has been implicated to link developmental neurogenesis and angiogenesis through the formation of neurons expressing VEGF-A. 40 Ischemia increased expression of serum response factor in oligodendrocyte progenitor cells and oligodendrocytes which coincided with decreased expression of miR-9 and miR-200 family in the injured white matter tracts of corpus callosum and striatum. 41 Additionally, inhibition of serum response factor or over expression of miR-9 and miR-200 in oligodendrocyte progenitor cells was found to inhibit oligodendrocyte progenitor cells differentiation into oligodendrocytes. 41 Therefore, exosomes and their cargo miRs may play an important role in facilitating neurovascular coupling. Treatments that regulate multiple brain repair mechanisms may improve long-term functional recovery after stroke. 10
Inflammatory responses after stroke
Disruptions of the NVU and blood–brain barrier (BBB) are key pathological steps that initiate secondary tissue damage after stroke. Platelets and endothelial cells are activated immediately after vascular occlusion, and the adhesion molecule P-selectin translocates to the cell surface membrane initiating leukocyte recruitment to the sites of brain injury. 42 BBB disruption facilitates the infiltration of peripheral/circulating neutrophils, macrophages, monocytes, and lymphocytes from the blood into ischemic brain.43,44 Inflammatory responses begin within a few hours after stroke and evolve over the following several weeks contributing to secondary brain damage, infarct expansion and secondary organ dysfunction after stroke. Neuronal, glial and transmembrane depolarization leads to an increase in extracellular ATP which can activate resident microglia. 4 Stroke-induced rapid local inflammatory responses in the brain parenchyma include microgliosis, astrogliosis, and cytokine and chemokine secretion. Exosomes secreted by microglia and astrocytes can store and release inflammatory cytokines such as IL-1β.45,46 Microglia-derived exosomes can also stimulate neuronal activity and enhance propagation of inflammatory signals. 47 Exosomes secreted by injured brain cells in response to ischemic stimuli can influence bystander cells by the transfer of dysregulated miRs that suppress the expression of essential genes in the recipient cells. 48 In their resting state, resident microglia continuously monitor the microenvironment using their motile processes and protrusions and aid in metabolic byproduct and debris removal. 49 Upon ischemic injury to the brain, microglia are rapidly activated, undergo structural and functional alterations to a phagocytic phenotype, accumulate near the injury site and secrete inflammatory mediators. 49 Following neutrophil invasion and microglial activation, macrophages derived from local microglia and hematogenous macrophages invade the ischemic tissue. 42 In their degenerative M1 phenotype, microglia and macrophages promote inflammation by increasing proinflammatory cytokines (interleukins (IL-6, IL-1β), tumor necrosis factor-α (TNF-α) etc.), integrins, adhesion molecules, chemokines and their receptor molecules and reactive oxygen species (ROS).4,50,51 During this sub-acute phase of stroke, the various pro-inflammatory cytokines, chemokines, ROS, and activated matrix metalloproteinases (MMPs) increase glial activation and peripheral immune cell infiltration into the brain which worsen BBB leakage, brain edema, neuronal death, and hemorrhagic transformation.51,52 During the recovery phase of stroke, microglia and macrophages assume an anti-inflammatory M2 phenotype and facilitate debris removal from the ischemic brain. 4 M2 phenotype microglia and macrophages decrease the expression of inflammatory factors, protect neurons, and improve cell survival in the ischemic environment. 53 Therefore, extending the M2 phase of these macrophages and microglia and inducing macrophage polarization from M1 to M2 phase are desirable neurorestorative effects. However, the distinction of M1 and M2 phenotypes is not clearly defined and the simple M1 and M2 classifications are likely inadequate to explain the complex role of macrophage polarization in mediating inflammatory responses. 54
Exosome therapy to amplify brain repair
Exosomes are nanosized vesicles (∼30–100 nm in diameter) that are generated by nearly all cells, and constitute major vehicles for intercellular communication. 55 Exosomes have multiple roles in regulating physiological and pathological processes. 55 Based on their parent cell and functional cargo, exosomes derived from various cell types have been reported to induce neuroprotective and neurorestorative effects by modulating gene, protein and miR expression in their target cells and tissues.55,56 MiRs are small non-coding RNA sequences approximately 22 nucleotides in length that can regulate multiple genes, pathways, and complicated cellular networks by acting either alone or in concert with other miRs. 10 MiRs regulate post-transcriptional gene regulation by binding to complementary sites in the 3′-untranslated region of messenger RNAs. 57 The miR signature in ischemic stroke is being unraveled and has potential use as diagnostic and prognostic markers as well as novel therapeutic targets. 58 Exosomes are transported by biological fluids and provide an effective means of communication between neighboring as well as remote cells. 59 Exosomes contain and deliver proteins, lipids, and nucleic acids that are characteristic of its parent cell to recipient cells. 59 Exosomes enable a graft-free delivery of therapeutic molecules and offer several advantages over cell-therapy. For instance, exosomes have low immunogenicity, low risk of microvascular thrombosis and can easily cross the BBB and be internalized by endogenous brain cells.56,60–62 Exosome content can be readily modified by manipulating contents of the parent cell and miR expression. Therefore, exosomes can be programmed to maximize therapeutic efficacy by targeting specific neuroprotective and neurorestorative pathways, thereby enabling the development of personalized targeted drug delivery vehicles to deliver designer genetic instructions as well as the active components of cell-based therapy to treat ischemic stroke.63–65 Exosomes derived from a variety of cell types such as embryonic stem cells, induced pluripotent stem cells (iPSCs), and adult stem cells such as mesenchymal stromal cells (MSCs), human umbilical cord blood cells (HUCBCs), and neural stem cells (NSCs) have been employed to treat ischemic stroke and other cerebrovascular diseases in pre-clinical studies.66,67 In this review, we will discuss the therapeutic effects of a multipotent restorative therapy for stroke, MSC-exosomes.
Stroke treatment using MSC-exosomes
MSCs are a prolific source of exosomes
68
and MSCs as well as MSC-derived exosomes have been widely studied for the treatment of stroke and other neurological diseases.
69
Treatment of stroke with MSCs was found to improve neurological recovery by secreting a plethora of growth and trophic factors and promoting endogenous brain repair mechanisms, primarily via the secretion of exosomes.63,70–76 The secreted exosomes were found to carry and transfer their cargo miRs (such as miR-133 b) to astrocytes and neurons in the brain and regulate target genes which stimulate restorative mechanisms such as neurite outgrowth and improve neurological functional outcome.
75
Pre-clinical studies using rodent models of ischemic stroke have demonstrated therapeutic efficacy, ability to promote endogenous brain repair mechanisms, and improvement of long-term neurological functional outcome after stroke when MSC-exosomes were administered intravenously at 24 h after stroke onset.63,73–76 When mice subjected to stroke were intravenously treated with MSCs at 24 h after stroke or with MSC-derived exosomes at one, three, five days after stroke, similar functional recovery was observed.
76
In addition to therapeutic efficacy which is at least equivalent to parent cell therapy,
76
treatment with exosomes has become popular due to advantages over cell therapy such as low risk of tumor formation and reduced immunogenicity, and their ability to be readily expanded in-vitro and stored until use. Figure 1 presents a simplified schematic summarizing MSC-exosome-derived therapeutic effect after stroke.
Summary of MSC-exosome-derived therapeutic effects after stroke. MSC-derived exosomes transfer microRNA to the various cells of the neurovascular unit as well as stimulate parenchymal brain cells to secrete growth and trophic factors which promote neurorestorative mechanisms such as angiogenesis, neurogenesis, synaptogenesis, oligodendrogenesis, and anti-inflammatory responses which contribute to neurological functional recovery after stroke.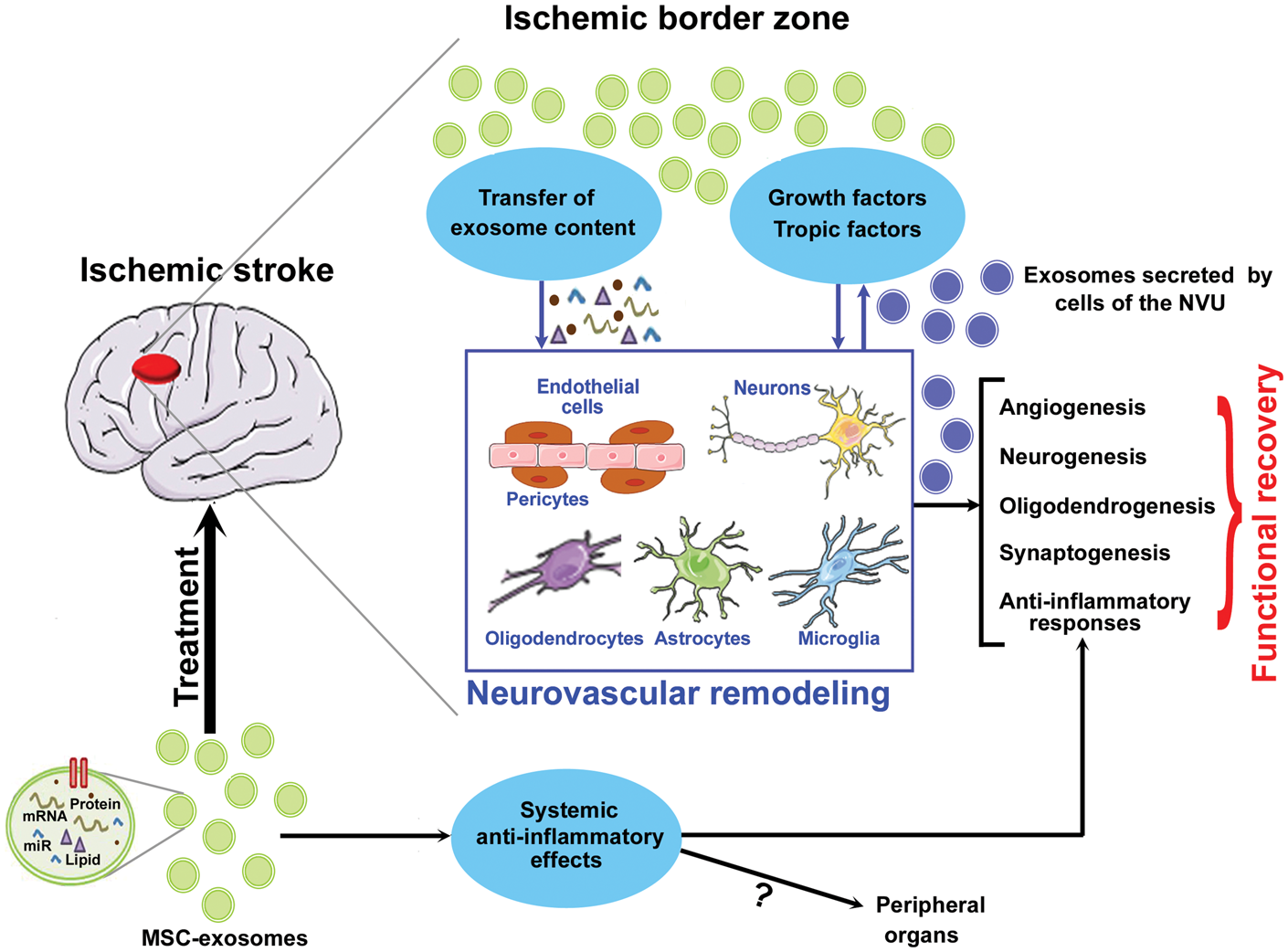
Apart from their therapeutic potential, plasma exosomal miRs can also serve as biomarkers for prognosis of stroke. 58 Compared to a healthy control patient population, serum exosomal content of brain-specific miRs (miR-9 and miR-124) was significantly increased in acute ischemic stroke patients, and the expression level correlated with stroke severity. 77 Another study compared plasma exosome levels in healthy and ischemic stroke patients and found that employing the combination of miR-422 a and miR-125 b-2-3 p may serve as blood-based biomarkers for monitoring and classifying stroke patients into acute (decreased expression) and sub-acute (increased expression) stages. 58
MSC-exosome induced neurovascular remodeling after stroke
MSC-exosome therapy favors neurovascular remodeling.63,76 Systemic administration of MSC-exosomes at 24 h after ischemic stroke was found to significantly increase axon density, myelin density, synaptic plasticity and angiogenesis in the ischemic border zone as well as promote neuroblast migration from the subventricular zone to ischemic border zone.63,76 In a model of sub-cortical stroke, intravenous administration of MSC-exosomes at 24 h after stroke was found to improve long-term functional outcome, axonal sprouting from the intact cortical tissue to injured striatum leading to increased axon density in the injured striatum, and increased oligodendrocyte progenitor cell number as well mature oligodendrocyte counts resulting in increased number of myelinated axons compared to control stroke animals. 78 The mechanisms underlying MSC-exosome-induced brain plasticity after stroke and other cerebrovascular diseases are under investigation.79,80 The characteristics of the parent cell and cargo of exosomes particularly miR content appear to influence therapeutic efficacy. MSC-exosomes are internalized by endothelial cells, neurons, oligodendrocytes and microglia in the ischemic brain. 78 MSC-exosomes are internalized by both neuronal cell bodies and axons resulting in increased axonal outgrowth in cultured primary cortical neurons. 81 The miR content of exosomes appears to mediate neuronal plasticity as inhibition of argonaute 2 protein (a primary miR machinery proteins) in MSC-exosomes blocked MSC-exosome effects on axonal outgrowth. 81 In addition, modified MSC-exosomes over expressing miR-17-92 cluster amplified axonal growth by activating the PTEN/mTOR signaling pathway in recipient neurons. 81 Therefore, MSC-exosomes can directly transfer their cargo miR to recipient neurons and thereby influence neuronal remodeling. Glial cells and glial cell-derived exosomes likely modulate neurovascular coupling and brain plasticity by mediating communication between neuronal, glial, and vascular compartments.82,83 Exosomes derived from miR-133 b-overexpressing MSCs were found to amplify therapeutic effects of MSC-exosome in rats subjected to stroke. 84 Interestingly, the amplification of neural plasticity following miR-133 b+MSC-exosomes was attributed to a stimulated secondary release of neurite-promoting exosomes from astrocytes. 84 Therefore, it is likely that MSC-exosomes exert their therapeutic potential by acting directly on parenchymal and vascular cells and also via secondary secretion of exosomes by astrocytes and other parenchymal cells and promote remodeling of the ischemic brain.
Effect of MSC-exosome therapy on inflammatory and immune response
Intravenous transplantation of MSCs at a chronic time point of 60 days after stroke in rats showed that MSCs preferentially home to the spleen over the ischemic brain, regulate inflammatory responses and decrease ischemic lesion volume compared to control rats. 85 MSC-exosomes are less immunogenic compared to MSCs due to the reduction in membrane-bound protein content. 86 MSC-exosomes are immunologically active and promote anti-inflammatory effects.87,88 Exosomes derived from normal MSCs or hypoxia-preconditioned MSCs were found to exhibit anti-inflammatory properties and induce M2 polarization of bone-marrow-derived macrophages in-vitro and in-vivo in a model of skeletal muscle injury. 88 MSC-exosome treatment in stroke mice did not alter the infiltration of immune cells such as neutrophils, dendritic cells and macrophages. 76 However, at six days after stroke, post ischemic immunosuppression characterized by substantial reduction of total white blood cells counts (∼50%), monocyte (∼75%) and lymphocyte subsets (∼60%) in the peripheral blood of saline-treated stroke mice was significantly attenuated in MSC-exosome-treated stroke mice which had significantly increased numbers of B cells, NK cells, and T cells. 76 Therefore, MSC-exosomes can potentially attenuate post stroke immunosuppression which in addition to extending protection against infection also creates a hospitable environment favoring brain remodeling.
Peripheral organ dysfunction after stroke
Secondary organ dysfunction is present after stroke and treatment strategies should expand beyond treating the injured brain to also target secondary organ recovery.89–95 In recent years, there has been a focus on elucidating the mechanisms underlying peripheral organ dysfunction after stroke and the signaling pathways that may mediate inter organ communication, that are discussed in this section. Almost all the cell types constituting the NVU can produce extracellular vesicles such as exosomes and microvesicles which may mediate intercellular communication within the NVU as well as with distant target cells. Extracellular vesicles are emerging as intrinsic communication mediators between the brain and immune system.
96
Stroke-induced BBB disruption facilitates brain-derived antigens, and extracellular vesicles derived from injured brain cells to enter the blood stream alter peripheral immune response and potentially induce dysfunction of peripheral organs.
96
For instance, activated endothelial cells and astrocytes at the site of inflammation secrete extracellular vesicles that can cross the BBB, enter the peripheral circulation, and regulate acute cytokine response and mediate trafficking of peripheral immune cells to injured brain tissue.
96
On the other hand, systemic inflammation and circulating exosomes in response to infectious stimuli are potential neuroinflammatory mediators that can stimulate microglial and astrocytic activation and increase the expression of inflammatory cytokines in the brain.
97
High doses of endotoxins and severe peripheral inflammation can potentially damage the BBB and aggravate injury to the brain.
97
Therefore, it is important to understand the various communication axes between the brain and other organs and the role played by systemic inflammatory and immune responses in mediating secondary organ dysfunction after stroke. Figure 2 represents simplified schematic summarizing peripheral organ injury after stroke.
Summary of stroke-induced peripheral organ injury. Stroke induces systemic inflammation and activates peripheral immune response which aggaravates ischemic brain injury as well as contributes to stroke-induced peripheral organ injury such as gut dysbiosis, cardiac dysfunction and renal disease.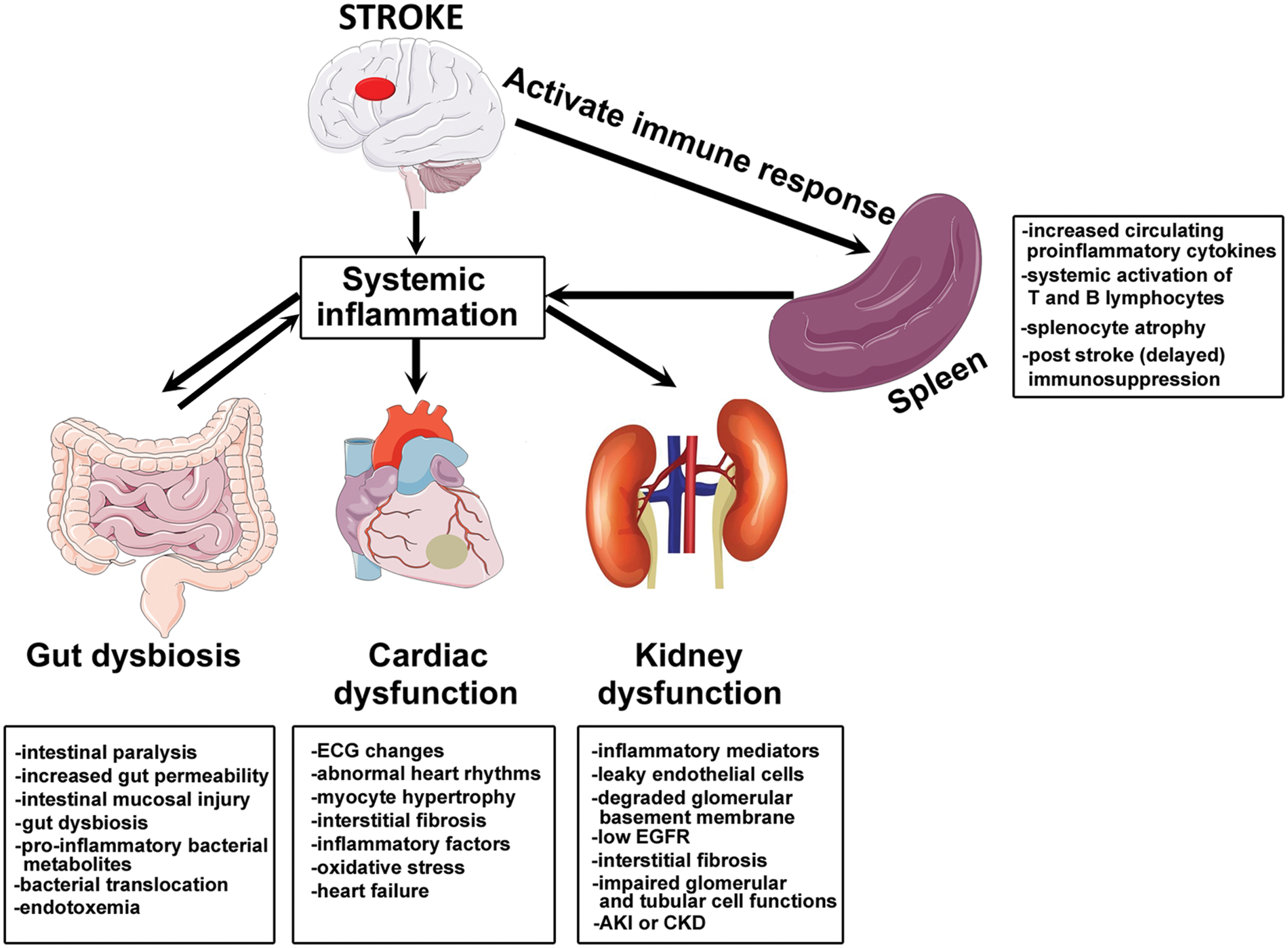
Brain–immune axis
Systemic inflammatory responses after stroke are mainly driven by cytokines, chemokines and stress hormones. 98 Exosomes can cross the BBB and potentially transfer brain-derived antigens to the periphery and activate/aggravate peripheral immune response after stroke.99,100 Exosomes can also transfer pro-inflammatory messages from the periphery to recipient brain cells. 101 The spleen is a major regulator of peripheral immune response after ischemic stroke.98,102–104 Large artery occlusion resulting in large lesions affecting cortical and sub-cortical areas stimulates systemic inflammatory responses irrespective of left- or right-sided stroke. 102 Rats that underwent splenectomy two weeks prior to stroke induction were found to have significantly lower lesion volume and decreased local inflammation in the ischemic brain tissue characterized by reduction in number of activated microglia, macrophages, and neutrophils, indicating that peripheral inflammation mediated by the spleen plays an important role in neurodegeneration after stroke. 105 Stroke significantly alters spleen function, increases production of circulating proinflammatory cytokines, induces systemic activation of T and B lymphocytes, aids invasion of peripheral immune cells and inflammatory mediators to the brain, and exacerbates local inflammatory responses.98,102 Lymphocyte infiltration in the ischemic brain begins about 48 h after acute ischemic stroke, and the invading T lymphocytes promote detrimental inflammatory cascades and contribute to secondary brain injury.43,50 Cytotoxic CD8+ T-cells and CD4+ T-helper 1 (TH1) cells aggravate ischemic injury via secretion of proinflammatory cytokines such as TNF-α, Interferon-γ, interleukins (IL-2, IL-12), while CD4+TH2 cells secrete anti-inflammatory cytokines such as IL-4, IL-5, IL-10, and IL-13 which may exert neuroprotection. 106 In pre-clinical studies using rodent models of ischemic stroke, early after stroke (6–24 h after stroke) activated spleen cells were found to increase proinflammatory cytokines, while at later time points (96 h after stroke), thymus and splenic atrophy (gross spleen size reduction increased apoptosis and decreased splenocyte numbers) decreased CD8+T-cell counts, reduced inflammatory cytokine secretion and increased Tregs, resulting in a state of profound immunosuppression.98,103,104 Post stroke immunosuppression increases risk of infection and contributes to post-stroke mortality. 107 Infection complicates acute stroke outcome in approximately 30% of patients. 108 Advancing age, history of alcohol abuse, hyperglycemia, hypertension, and cardiac abnormalities such as atrial fibrillation increased susceptibility to post stroke pneumonia. 109 Stroke survivors often encounter respiratory complications and abnormal breathing patterns. 110 Stroke-induced immune suppression, altered stress responses and environmental factors/hygiene status (urinary catheterization, feeding tubes) are some of the factors that have been implicated in post stroke lung infection. 109 Immunomodulation after stroke has been found to improve neurological outcome after stroke and is a promising therapeutic strategy.111,112
Brain–heart axis
Cardiac abnormalities are common in patients with neurological injuries, even in the absence of primary cardiac disease.89,90 Stroke patients often present with symptoms of myocardial injury, ischemia-like electrocardiographic (ECG) changes and arrhythmias.89,90,113 Cardiac complications such as heart attack, congestive heart failure, cardiac arrest and abnormal heart rhythms-like atrial fibrillation can contribute towards mortality and morbidity in stroke patients.114,115 The risk of cardiac complications after stroke increases with stroke severity, neurological deficits and co-morbidities such as hypertension, diabetes, high cholesterol, and age116,117 which are also predictors of worse functional outcome and secondary complications such as vasospasm, delayed cerebral ischemia and pulmonary edema. 118 Ischemic stroke affecting the right insular cortex decreases sympathetic tone and causes parasympathetic over activity. 119 Compared to left hemispheric stroke, right insular lesions are associated with higher mortality and cardiac dysfunction.120–122 Inflammation and immune response may play an important role in mediating brain–heart interaction in the context of stroke. 123 Mice subjected to stroke exhibit cardiac dysfunction with evidence of cardiomyocyte hypertrophy, interstitial fibrosis and increased inflammatory factors, macrophages and oxidative stress in the heart compared to non-stroke control mice. 124 Concomitant with cell death in the ischemic brain, cardiomyocyte death in the heart tissue was also observed in rats subject to ischemic stroke. 125 Ischemia-damaged brain cells can secrete soluble factors that can transmit cell death signals to cardiomyocytes and induce cardiac dysfunction after stroke.125,126 Proinflammatory cytokines secreted by damaged neuronal and glial cells can stimulate the posterior hypothalamus to increase sympathetic output and circulating catecholamine level.127–129 Catecholamine surge is known to cause cardiac hypertrophy or myocardial ischemia.130,131 Preclinical studies have demonstrated that elevated plasma catecholamine levels following ischemic stroke are directly proportional to the incidence of myocardial lesions and cardiac damage. 132 Detailed mechanisms of brain–heart interaction after stroke have been discussed elsewhere. 123
The role of exosomes, miRs and immune response in mediating cardiac dysfunction after stroke and other cerebrovascular diseases is emerging.123,124,133 Since stroke alters miR profile in the brain as well as in the circulation and some of these miRs may play critical roles in regulating the functioning of other organs such as heart and kidney, it is likely that alteration of exosomes and miR content influences secondary organ damage. For example, miR-126 has been well documented to play critical roles in maintaining vascular integrity and regulating angiogenesis. 134 Stroke significantly decreases circulating miR-126 level and miR-126 deficiency has been associated with several cardiac abnormalities and diseases including heart failure, atrial fibrillation and coronary artery disease.124,134–136 Employing a rodent model of stroke, ischemic stroke in rodents significantly decreases serum and heart miR-126 expression, and induces post stroke cardiac dysfunction. 124 In addition, endothelial cell miR-126 knockout mice exhibited cardiac dysfunction and increased cardiomyocyte hypertrophy, fibrosis, and inflammatory factor expression after ischemic stroke compared to control stroke mice. 124 On one hand, employing miRs as biomarkers to predict and diagnose such cardiac dysfunction may be clinically useful, and on the other, targeting these miRs might be necessary and/or amplify the therapeutic benefits of exosome therapy. Thus, improving cardiac function after stroke may provide a means to improve stroke recovery.
Brain–kidney axis
Stroke patients often present with symptoms of kidney dysfunction depending on severity and stroke subtype.91,92 Patients with large artery atherosclerotic stroke often have low estimated glomerular filtration rate that is associated with increased stroke severity and six-month mortality rate, and worse neurological functional outcome. 137 Ischemic stroke patients can also develop acute kidney injury or chronic kidney disease.138–140 Post ischemic systemic inflammation likely plays an important role in mediating kidney dysfunction after stroke. Stroke-induced elevation of inflammatory mediators such as C-reactive protein, IL-6 and ROS can also affect endothelial cell permeability in the kidney and enable entry of high-molecular-weight compounds, inflammatory mediators, immune cells and immune complexes, into the endothelium of glomerular filtration membrane. 141 ROS can degrade the glomerular basement membrane, impair glomerular and tubular cell functions and promote renal fibrosis.141–143 Interestingly, systemic administration of microvesicles derived from MSCs has been employed successfully to treat acute kidney injury in rats subject to renal ischemic reperfusion injury as well as extended protection against subsequent chronic kidney disease. 144 Future studies are needed to understand brain–kidney axis and how injury to one organ affects the other.
Brain–gut axis
It is increasingly evident that there exists a gut–brain axis which affects normal brain functioning as well as influences disease outcome under pathological states. 145 Gut dysbiosis after stroke is proportional to stroke severity and is characterized by an increase in opportunistic bacterial count and diversity often with decline in beneficial bacterial diversity numbers, and can adversely affect stroke outcome via several intersecting mechanisms.93–95 Gut dysbiosis after stroke can affect neuroinflammatory and immune responses as the brain–gut axis consists of neural and humoral pathways that can regulate peripheral immune response and lymphocyte population and worsen neurological function.145–147 Bacterial metabolites can also alter immune responses in favor of proinflammatory signaling. 148 In rats subjected to stroke, significant decrease in gastrointestinal motility and intestinal mucosal injury including necrosis and shedding of the intestinal epithelium was observed at 6 h after stroke which worsened by 24 h after stroke.93,95 Intestinal lymphocytes infiltrate into the brain and aggravate ischemic injury. 146 Using a mouse stroke model, antibiotics were shown to induce alterations in the intestinal microbiota composition, and reduce ischemic lesion volume and neurological deficits. 146 Systemic inflammation can increase gut permeability. 149 In mice subjected to stroke, increased gut permeability facilitated bacterial translocation and leads to infection, sepsis and increase mortality. 150 While stroke increases gut permeability and bacterial translocation in both young and aged mice, aged animals were found to be particularly more susceptible to sepsis and immune dysfunction. 150 Bacterial translocation from the gut to mesenteric lymph nodes and peripheral organs such as spleen, liver and lungs triggers adaptive and innate immune responses. 150 Gut dysbiosis-mediated inflammatory responses can also affect cardiac and kidney function after stroke.123,151,152 Interestingly, MSCs are known to have anti-microbial effects and can attenuate bacterial infection-induced sepsis, sepsis-induced inflammatory responses and sepsis-related mortality while improving bacterial clearance in sepsis rodent models.153,154 Thus, there is promising therapeutic potential in amplifying the anti-microbial effects of MSCs-exosomes via cell conditioning and/or drug loading. 155
Conclusions and future directions
The interaction between various cell types within the NVU and neurovascular niche dictates the injury and repair processes after ischemic stroke, respectively. Understanding the sequential course of pathological events and transition from injury to repair mechanisms within the brain will enable neuroprotective and restorative interventions to amplify endogenous brain repair mechanisms to reach therapeutic levels. A rigorous understanding of the interdependence and coupling between various restorative events after stroke await fruition and it is crucial to develop therapeutics that targets multiple signaling pathways to maximize therapeutic efficacy. MSC-derived exosomes offer a powerful therapeutic tool to promote endogenous brain repair mechanisms after stroke while overcoming limitations of cell-based therapy. Current and future research focusing on optimization of MSC isolation and culture conditions, collection and characterization of exosomes, and importantly technology for expansion of MSC-derived exosomes to obtain large quantity of clinically effective exosomes are warranted. Optimizations of MSC-exosomes including manipulation of key miRs that promote neurorestorative effects to amplify therapeutic effects of MSC-exosomes are needed. The effects of MSC-exosome therapy on systemic inflammatory and peripheral immune response are yet to be fully understood. Local and systemic inflammatory and immune responses mediate delayed brain injury after stroke and likely regulate peripheral organ dysfunction after stroke. Recent and emerging literature delineating the effect of stroke on other organs such as the heart, gut, spleen, lungs, and kidney stress the importance of treating secondary injury to peripheral organs after stroke to improve overall recovery. It is likely that there are several such communication axes between the various organs which dictate over all brain recovery after stroke. When employing systemic administration of MSC-exosomes to treat the ischemic brain, it is likely that there is some level of direct/indirect protection rendered to peripheral organs such as heart, kidney, spleen and gut, and further studies are warranted.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Institute of Neurological Disorders and Stroke R01NS083078 (JC), R01NS099030 (JC), R01NS097747 (JC) and R01NS088656 (MC), National Institutes of Health R01HL143432 (JC), and American Heart Association award 17POST33410580 (PV).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.