
Abstract
In order to understand the nature of the relationship between cerebral blood flow (CBF) and primary headaches, we have conducted a literature review with particular emphasis on the role of perivascular neurotransmitters. Primary headaches are in general considered complex polygenic disorders (genetic and environmental influence) with pathophysiological neurovascular alterations. Identified candidate headache genes are associated with neuro- and gliogenesis, vascular development and diseases, and regulation of vascular tone. These findings support a role for the vasculature in primary headache disorders. Moreover, neuronal hyperexcitability and other abnormalities have been observed in primary headaches and related to changes in hemodynamic factors. In particular, this relates to migraine aura and spreading depression. During headache attacks, ganglia such as trigeminal and sphenopalatine (located outside the blood-brain barrier) are variably activated and sensitized which gives rise to vasoactive neurotransmitter release. Sympathetic, parasympathetic and sensory nerves to the cerebral vasculature are activated. During migraine attacks, altered CBF has been observed in brain regions such as the somatosensory cortex, brainstem and thalamus. In regulation of CBF, the individual roles of neurotransmitters are partly known, but much needs to be unraveled with respect to headache disorders.
Keywords
Introduction
Migraine is a frequent primary headache disorder, with higher prevalence in females (15.5%) than males (4.5%), 1 and associated with clinical symptoms (typical headache, and phonophobia/photophobia/nausea/vomiting that occur over 4–72 h). 2 On the contrary, cluster headache is more prevalent in males than females for most age groups. 3 The origin of headache attacks is still under discussion, with particular focus on whether they represent a vascular or neurogenic disorder.4,5 During the different phases of the migraine attack (premonitory and during attacks), there are variations in cerebral blood flow (CBF), coinciding with changes in brain activity, observed in the somatosensory cortex, brainstem and thalamus.6,7 In headache-free periods (inter-ictal), elevated CBF in the primary somatosensory cortex has been reported for migraineurs. This effect was positively correlated to headache attack frequency, but also to the number of cutaneous allodynia symptoms. 6 During headache attacks, autonomic and sensory ganglia are known to be variably activated and sensitized,8,9 which gives rise to neurotransmitter release, believed to be important in the migraine pathophysiology.10,11 In particular, trigeminal ganglion (TG) stimulation in cats resulted in increased CBF and calcitonin gene-related peptide (CGRP) release. 12 Several signaling molecules are involved in both migraine and innervation of the cerebral vasculature.
The cerebral vasculature is known to be innervated by (i) sympathetic nerves storing adenosine triphosphate (ATP), noradrenaline and neuropeptide Y (NPY), (ii) parasympathetic nerves with acetylcholine, vasoactive intestinal peptide (VIP), peptide histidine methionine, pituitary adenylate cyclase-activating peptide (PACAP) and nitric oxide (NO), and (iii) sensory nerves containing CGRP, PACAP, NO, substance P (SP) and neurokinin A (NKA).13,14 The extrinsic innervation ends close to the entry of the vasculature into the brain parenchyma; a transition zone in the Virchow-Robin space. Moreover, the intraparenchymal nerve supply to arterioles and microvessels stems primarily from central nervous system (CNS) neurons. 14 Therefore, it is necessary to discuss migraine in the light of extrinsic and intrinsic neural inputs, and their role in regulation of CBF.
The link between headache, CNS symptoms, intracranial vessel diameter and regulation of blood flow has long been pursued. In general, primary headaches are considered complex polygenic disorders (genetic and environmental influence) with pathophysiological neurovascular alterations.15–17 Several primary headache theories have been proposed over time, with the purpose of elucidating the underlying cause of headache attacks and associated characteristics. Early studies on involvement of extracranial vasodilation and a link between intracranial vasospasm and cortical spreading depression (CSD) in migraine with aura led to the vascular theory of migraine, originally forwarded by Wolff in the early 1940s.18,19 Although a pure vascular theory has now lost its role as the leading hypothesis to explain migraine, involvement of extracranial and intracranial vasculature has persisted over time as an etiological or coexisting factor.20,21 Direct experimental evidence, contrary to the vascular theory, came from a magnetic resonance angiography study showing no/minor alterations in the diameter of meningeal and cerebral arteries in genuine migraine attacks. 22 Nevertheless, this study is not entirely conclusive, as the intracranial middle meningeal artery (MMA) was not measured due to technical limitations. 23
Today CSD remains one of the possible early links between vascular observations and the neurogenic theory. CSD is a self-propagating wave of neuronal and glial cortical depolarization resulting in biophysical and biochemical alterations such as changes in parenchymal blood flow and ionic concentrations. 24 The aura, a clinical symptom (mostly visual) seen in about 20–30% migraineurs, 25 is supposedly due to CSD.26,27 It has been suggested that CSD directly activates trigeminal sensory nerve terminals in dura mater/meninges followed by trigeminovascular system activation.24,28 For decades, activation and sensitization of the trigeminovascular system have been considered a pathophysiological mechanism/consequence behind primary headaches.29–31 Interestingly, CSD not only affects meningeal arteries, but also cortical cerebral arterioles and CBF. 32 An alternative hypothesis involves activation of CNS regions by CSD which in turn, via thalamus and brainstem, causes activation of the trigeminovascular system with dural inflammation being secondary to the CNS activation. Moreover, during the headache phase in genuine attacks, waves of flow reductions slowly spread over the cortex bilaterally. This pattern resembled CSD even though the subject did not have obvious aura symptoms. 33 Other studies also report the occurrence of local depression of CBF in migraine. 34 More details on this research topic has been discussed by Pietrobon and Moskowitz26,27 and by Ayata and Lauritzen. 35
Today, leading scientists advocate that already in the premonitory phase of an acute migraine attack, distinct blood flow alterations within, e.g. hypothalamus-thalamus and the adjacent brainstem (periaqueductal gray) can be observed.36,37 The proposal of a “migraine generator” originates from Weiller et al. 38 demonstrating activation of brain regions such as the periaqueductal gray, raphe nuclei, and locus coeruleus in the early phase of a migraine attack. Following injection of sumatriptan for acute treatment of migraine, pain subsided but the areas remained activated. After some hours, the pain returned, probably due to disappearance of the triptan from the involved receptors. 38 Thus, evidence suggests that migraine pathophysiology includes dysfunction of subcortical structures such as diencephalic and brainstem nuclei which can modulate the trigeminovascular system and perhaps also participate in CSD generation. 39
Complexity of neurovascular coupling and CBF
Brain homeostasis is known to be controlled by a tight regulation between neuronal function, local blood flow and maintenance of neurovascular coupling.40,41 More precisely, the CBF is controlled by (i) autoregulation, maintenance of adequate CBF regardless of blood pressure (BP) fluctuations (within some limits),42,43 (ii) chemical and metabolic factors, e.g. interstitial pH 44 and CO2, 45 and (iii) neurogenic factors such as perivascular innervation.46,47 Regulatory mechanisms of CBF depend on variations in the rate of local brain metabolism. 48
The changes of CBF in primary headaches are obviously not as drastic as observed in ischemic stroke. Weiller et al. 38 reported minor changes in cortical blood flow which was significant only if all migraine patients were normalized and grouped (about 5% of resting flow). In migraineurs with aura, a reduction in CBF was observed closely in time to the aura symptoms.49,50 In the interictal period, Loehrer et al. 51 reported a higher total and parenchymal CBF in migraineurs compared to controls, suggested to be driven by a higher basilar artery flow. In experimental animals, induction of CSD causes an abrupt rise in CBF within a minute followed by depression of function and flow. 52
Relevant for primary headaches, an association between neurovascular dysfunction and CSD has been proposed.52,53 It is important to clarify that the intraparenchymal neurovascular system is distinctly different from that of the large cerebral arteries and arterioles located around the brain and leading into the intraparenchymal system.14,41 The extraparenchymal vasculature (arteries belonging to the circle of Willis, and arteries located on the cortical surface) receives a dense supply of perivascular nerves that originate in cranial ganglia. 47 The intraparenchymal neurovascular system is thin and mainly contains pericytes and endothelial cells. Their connections with the local environment come from local neurons and astrocytes, inter alia. They are not supplied by innervation from the cranial ganglia, and 47 hence there are two different sites of neuronal regulation of the cerebral circulation.
Extrinsic and intrinsic cerebral vasculature: Neurogenic factors and neurotransmitter release
Extrinsic perivascular nerves in the cerebral circulation and dura mater
Three centuries ago, Thomas Willis noted nerve profiles on major cerebral arteries and speculated on a role in regulation of brain blood flow. 54 The role of perivascular nerves took a step forward in 1967 when Nielsen and Owman 55 demonstrated sympathetic noradrenaline-containing perivascular nerves. As a result, a new era began with analysis and demonstration of a multitude of associated receptors. 56 Originally, it was thought that they were an anomaly based on experiments showing that denervation had no effect on basic regulation of flow-metabolism coupling, chemical control or autoregulation.47,57 However, it is now known that the upper and lower limits of autoregulation are modified by sympathetic nerves arising from the superior cervical ganglia (SCG). 58 The parasympathetic fibers originate from the sphenopalatine (SPG) and otic ganglia (OTG), storing acetylcholine and numerous neuropeptides (the first to be found was VIP 59 ). Currently, these ganglia have been linked to facial symptoms in, e.g. cluster headache. 60 The third system innervating the cerebral circulation is the sensory system which originates in the TG. 13 For long, we were seeking a role of this system, and in 1986, we reported that it was a reflex designated to counterbalance any reductions in local CBF. 61 Recent work has focused on understanding of the intimate coupling process. CBF alterations, induced by neuronal responses, have been studied by functional magnetic resonance imaging (fMRI) as an indirect measure of neurovascular coupling. 62 Transient photophobia was investigated by fMRI in a healthy human subject and was found to be associated with trigeminal system activation. 63
Advances in our understanding of the neuronal innervation of the extraparenchymal cerebral vasculature
In the 1970s, characterization of neurotransmitters in perivascular nerve fibers was demonstrated in more detail due to the availability of novel histochemical methodologies, immunohistochemistry in particular. This increased the theoretical understanding of possible pathophysiological mechanisms involved in primary headaches (e.g. regulatory mechanisms of CBF). The origin of perivascular nerve endings on cerebral vasculature64,65 has been investigated by a nerve tracing and immunohistochemical localization study. Using True Blue applied on the vessel walls, we traced the dye to both uni- and bilateral ganglia, and showed co-localizations with NPY (sympathetic), VIP (parasympathetic) and SP and CGRP (sensory).66–68 The brainstem was subsequently analyzed using transganglionic tracers and the sensory system was found to localize in the trigeminal nucleus caudalis and brainstem C1-3, laminae I-II.69–71 Innervation studies of the trigeminal system showed that CGRP and SP originate in the TG. 72 Another study showed that PACAP-containing rat nerve fibers, innervating the extrinsic cerebral vasculature, primarily originate from neurons in the parasympathetic SPG and OTG and to a lesser extent from neurons in the sensory TG. 64 Most of PACAP and VIP were expressed in the SPG and OTG while PACAP only showed minor expression in TG, 73 also shown in man.74,75 The TG has, by far, retained most focus over time when investigating the pathophysiology of primary headaches. Neuronal sensory innervation of cerebral blood vessels, by vasoactive neuropeptides, has been linked to the TG.64,72 Electrical stimulation of TG resulted in increased CGRP, PACAP, SP and NKA levels,29,76–78 in agreement with immunohistochemical localization studies investigating TG and perivascular nerve fibers.64,79,80 PACAP immunoreactivity was also found in nerve fibers on cerebral arteries in cats. 81 Interestingly, PACAP-38 was increased in the plasma and trigeminal nucleus caudalis, but not in the TG itself or in the spinal cord 78 indicating stimulated signaling pathways upon TG activation.
Somatic afferents of the trigeminal nerve innervate considerable regions of the face, scalp, and meninges amongst others. 82 In coherence, increased ipsilateral facial skin blood flow is induced by experimental activation of TG in rats. 83 The trigeminal nerve is linked with the SPG,84,85 which has gained interest, particularly in cluster headache research. Repetitive transnasal blockade of SPG has shown effectiveness in treating both cluster headache 86 and chronic migraine.87,88 The structure is located in the pterygopalatine fossa below the maxillary nerve (trigeminal nerve branch, V2) where sympathetic and parasympathetic nerve fibers pass through. The superior salivary nucleus in the pons is connected to the SPG through nerve fibers 82 followed by nerve projections to the cranial vasculature from the parasympathetic ganglion. 39 Nerve projections originating from the SPG were traced to the internal ethmoidal, internal carotid and cerebral arteries by anterograde labelling of nerve fibers. 89 A study observed increased CBF in the ipsilateral and contralateral parietal cortex as a response to electrical SPG stimulation in rats 90 possibly as a response to neurotransmitter release. 91 Immunohistochemical localization studies revealed expression of PACAP, VIP and NO synthase (NOS) in rat and human SPG85,92 and expression of CGRP nerve fibers likely from the TG. 84 SPG extirpation, on the other hand, affects expression of VIP and acetylcholine in cerebrovascular structures. 93 Together these results indicate that there is a link between autonomic and sensory ganglia, CBF and migraine.
Nerve projections from the SCG, cervical C2 dorsal root ganglia (DRG) and OTG onto extrinsic cerebral arteries have been studied by retrograde tracing showing that the ganglia contain NPY, CGRP and SP, and VIP immunoreactive neurons, respectively.
66
The effects of these ganglia have been investigated less intensively compared to that of the TG and SPG with respect to primary headaches. In general, sympathetic nervous activity has been associated with cerebrovascular constriction and parasympathetic nervous activity with cerebrovascular dilation
41
of which parasympathetic activation is known to result in sympathetic hypoactivity and vice versa.
94
DRG has been associated with neuropathic pain as a response to pathological changes in the structure.
95
Since headache only seems to arise as a response to C1–C3 nerve innervations referred to as cervicogenic headache, the DRG of interest are the ones positioned at the C1–C3 level of the cervical spine.96,97 Autonomic and sensory ganglionic innervation of intracranial vessels by neuronal messenger molecules is illustrated in Figure 1.
Overview of innervation of intracranial vasculature. (a) Whole-mounted intracranial vessel immunohistochemically processed using anti-transmitter antibodies. Asterisk point at transmitter innervation of a nerve, and also at the region of perpendicular section shown in Figure 3(b). (b) An intracranial vessel (from the inside and out): endothelium (red), lamina elastic interna (green), unstained smooth muscle cells, transmitter immunoreactive nerves (red) and nuclei (blue). Transmitter innervation is displayed by schematic drawings of neurons. Abbreviations: Ach: acetylcholine; ATP: adenosine triphosphate; CGRP: calcitonin gene-related peptide; NA: noradrenaline; NKA: neurokinin A; NO: nitric oxide; NPY: neuropeptide Y; PACAP: pituitary adenylate cyclase-activating polypeptide; SP: substance P; VIP: vasoactive intestinal peptide.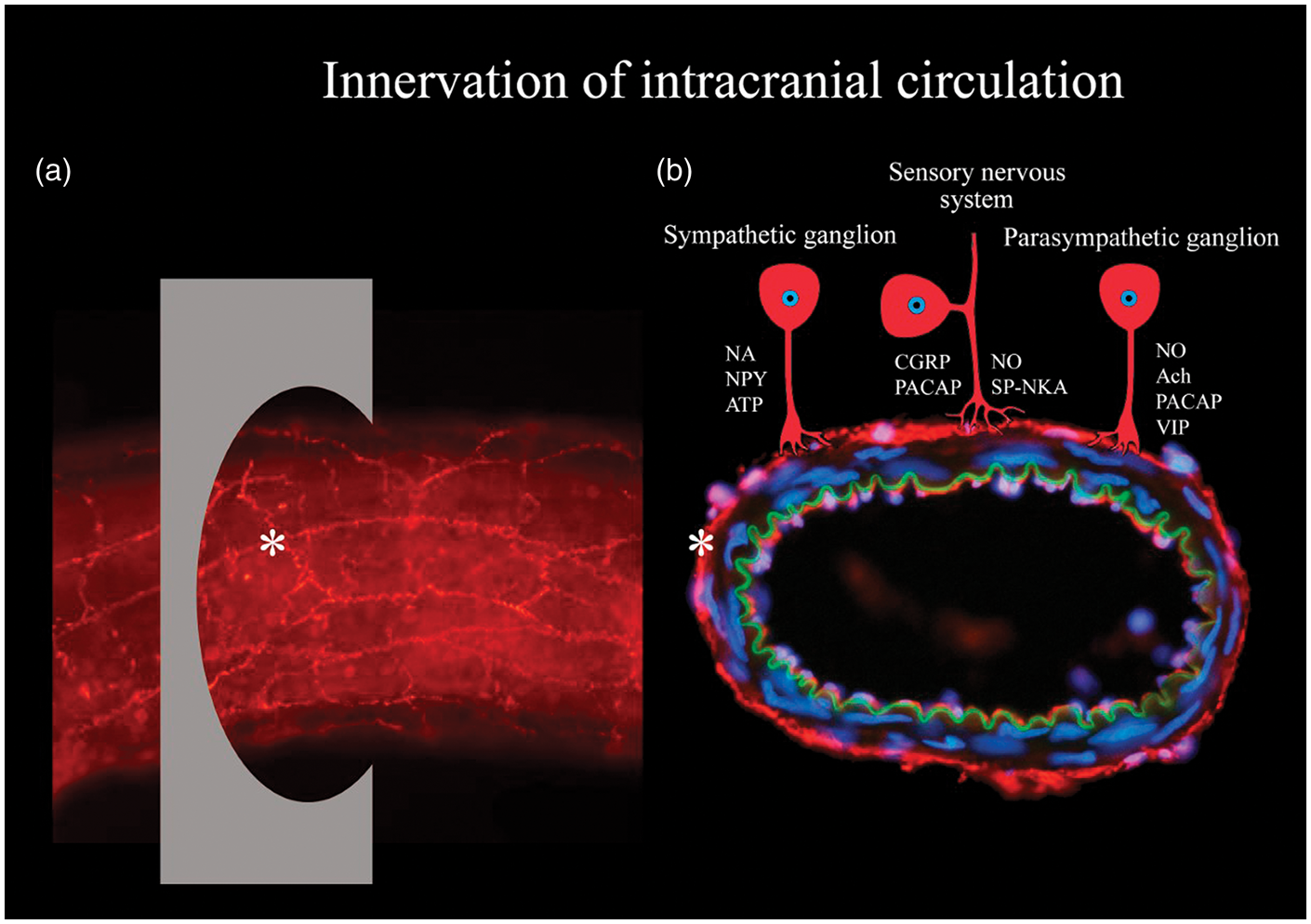
At this stage, it is pertinent to state that we, so far, have discussed innervation of the extraparenchymal system regulating the major cerebral and meningeal circulation. The current view on intraparenchymal regulation is still under analysis; so far, the prevailing view is a balance between local neuron and astrocyte activity, both in terms of their metabolism and direct activation of pericytes, smooth muscle cells and endothelium actions.98,99
Vasoconstrictive agents
In the relation to the development of a headache/migraine, the primary focus has been on vasodilating neurotransmitters. However, vasoconstrictive neurotransmitters are important in regulation of CBF and could also counteract a possible migraine-induced dilation. Therefore, studies have been performed on endothelin-1 (ET-1), 100 NPY 101 and 5-hydroxytryptamine (5-HT).102,103
High plasma levels of ET-1 have been recorded in the beginning of a migraine attack, which then decrease significantly over time. Increased levels of ET-1 have been proposed to account for part of local vasoconstriction observed early during migraine attacks. 104 It might be a counterbalance-mechanism to ganglionic release of vasodilatory neurotransmitters in order to maintain vascular tone, possibly until vasodilation overpowers the vasoconstrictive properties, e.g. of ET-1. There are currently two lines of research carried out regarding ET-1 and its receptors: (i) Uddman et al. 105 showed presence of ET receptors in TG neuronal cell bodies, and (ii) the drug industry developed and tested ET receptor blockade in migraine; however, the result was not positive. 106 NPY has been detected in nerve fibers on human cerebral arteries, in addition to CGRP, SP and VIP, reporting expressional variations between the neuropeptides (Figure 1). 80
Studies of 5-HT (or serotonin) indicate that migraine with aura leads to lower interictal 5-hydroxytryptophan (5-HTP) plasma levels, a serotonin precursor, compared to controls and migraineurs without aura. 107 Multiple studies have shown low interictal 5-HT levels with elevations recorded during attacks, however, with conflicting results. 108 Intravenous 5-HT and 5-HTP injection lead to symptomatic pain relief in migraineurs, however, with substantially varying outcome, for example with different injection rates. 103 The triptans (5-HT analogues) were further developed, and have been a partial successful treatment in migraine. The 5-HT1B/1D agonists are still the preferred treatments for acute migraine headaches.
Vasodilating agents
Numerous studies have examined the effects of vasodilator agents in patients in order to find key candidates that cause specific migraine attacks. Over the years, glyceryl trinitrate (GTN) and other headache-inducing agents have attracted much interest. It has been known for centuries, that GTN causes headache, in particular noted with workers in the dynamite industry. It is well known by clinicians that GTN, used to treat angina pectoris, results in headache at an early point in time. However, in a study in healthy volunteers, Daugaard et al. 109 reported that after 20 min of intravenous infusion, the recording of GTN-induced headache and a subsequent reduction in headache, a late phase called migraine-like headache occurred after some hours. Subsequently, >10 other agents have been tested in human pharmacological models resulting, not only in extracranial vessel dilatation, but also in different types of headache. 110 Of particular interest, CGRP has attracted vast attention since the 1980s in the search for underlying mechanisms explaining part of primary headache etiologies. 111 In cerebral arteries and arterioles (more precisely the middle cerebral arteries (MCA) and pial vessels), studies demonstrate that vasodilation caused by CGRP was more prominent and potent compared to that of SP and VIP.61,112 CGRP was also more potent compared to NKA, NKB, PACAP-38, dynorphine, galanin, peptide histidine methionine/isoleucine and nociceptin. Over time, this trend was observed in multiple studies investigating the effect of CGRP in headache disorders. Lately, this resulted in tests of CGRP receptor antagonists for prophylaxis and acute treatment of migraine. 113 Following CGRP, PACAP was isolated from ovine hypothalamic tissues. 114 Originally, PACAP was shown to have its main expression in the SPG and OTG, in parallel with VIP. 74 While VIP is not present in the trigeminal system, a subpopulation of CGRP positive neurons expresses PACAP-38 in the TG. 79 Interestingly, PACAP immunoreactivity was also seen along with CGRP, SP and neuronal NOS in the trigeminal nucleus caudalis and C1-2 laminae I-II, which are the central projection areas from the TG. 115 Infusion of PACAP-38 demonstrated vasodilation of extracranial arteries (external carotid artery and superficial temporal artery), but not intracranial arteries (internal carotid artery, basilar artery and MCA) using magnetic resonance angiography.116,117 In humans without a history of headache/migraine, 89% and 100% of the subjects developed headache in the immediate and delayed phase, respectively. 116 In functional studies, it was demonstrated that PACAP-38 and PACAP-27 both possess vasodilatory effects on MCA. It was suggested that the neuropeptides were involved in neuronal regulation of CBF reporting increases in CBF, as a response to PACAP-38 injection. 81 In addition to direct vasoactive effects, the use of PACAP knockout (−/−) mice has provided insight into the role of PACAP in migraine. PACAP has an essential role in activation and sensitization of the trigeminovascular system but also in occurrence of photophobia and vasodilatory responses. 118 Both α-CGRP and PACAP knockout (−/−) mice are viable, which suggest the presence of compensatory mechanisms. This might explain why there are so few adverse effects of the drugs that target CGRP and its receptor in humans.
The effect of exogenous administration of vasoactive peptides
Intravenous CGRP/PACAP-38 administration triggers migraine-like attacks in migraine subjects.117,119 Furthermore, MMA vasodilation has been detected upon α-CGRP and PACAP-38 infusions.116,120,121 The MMA consists of several segments: An extracranial (branch from the external carotid artery), intraosseous and intracranial part 122 of which we mainly are interested in the intracranial part due to the focus on CBF in primary headaches. Moreover, the inability of infused neuropeptides to influence intracranial arteries in vivo is proposed to be due to an incapability to pass the blood–brain barrier (BBB) based on intravenous infusion and myograph studies.117,123 Tuor et al. 124 investigated intracarotid/intracerebral NPY injection in rats and detected a cortical CBF reduction, although emphasizing that the change is only observable if NPY is administrated on the abluminal side of the BBB. In 2011, a myograph study revisited vasodilation of cerebral arteries, and observed vasodilation induced by PACAP and VIP. 125 In vivo, vasodilatory effects of VIP are, however, not as pronounced as the ones for PACAP-38. 117 In contrast, PACAP-38 did not produce vasodilation in isolated human intracranial MMA. 126 It is worth pointing out that intravenous infusion of VIP (18%) did not result in generation of migraine-like attacks to the same extent as intravenous infusion of CGRP and PACAP-38 (63–73%) in migraineurs without aura, even though vasodilation was recorded.117,119,127
Plasma–protein extravasation
Neurotransmitter release has been associated with plasma–protein extravasation,120,128,129 which might cause local edema.130–132 Extravasation has been described as follows: “proteins accumulate in the extravascular space when the rate of delivery exceeds the rate of dispersion into the surrounding tissue.” 133 Electrical TG and SPG stimulation have been associated with plasma–protein extravasation predominantly around small blood vessels in dura mater (which lacks BBB), with SP and NKA believed to be the most influential molecules involved.134,135 In contrast, CGRP did not induce plasma–protein extravasation, 136 but only vasodilatation. Perivascular projections from the TG have been detected on meningeal blood vessels in cats by tracing horseradish peroxidase, 65 and in rats using True Blue. 66 Moreover, induction of CSD, believed to stimulate trigeminal axons, resulted in plasma–protein extravasation in dura mater in rats. 130 Intracranial plasma extravasation has also been detected during a migraine attack in a female patient with episodic migraine evaluated by single photon emission computerized tomography. 137 Perivascular neurotransmitter release might affect vascular permeability since intravenous SP injection enhanced dural vascular permeability in rats possibly as a response to alterations of endothelial cells. Widening of intercellular junctions, elevated amount of gaps and vesicles and structural modifications of endothelial cells might occur. 138 More than a decade ago, neurokinins were considered the key molecules, and neurogenic inflammation the mechanism and target for migraine therapy.139,140 However, after the industry produced a series of specific neurokinin blockers and tested them in migraine (without significant effect), this line of research was abandoned. 141 In fact, SP was at this time not considered a major peptide involved in pain. 142
Integration of vasoconstrictors and vasodilators
In headache-free periods, research on blood samples revealed a significantly higher neurotransmitter baseline level of CGRP, SP and VIP in migraineurs when compared to controls,143–146 and significantly lower baseline levels of ET-1. 147 In chronic migraine, interictal VIP levels were significantly increased, whereas interictal PACAP levels were not significantly different between groups (chronic migraine, episodic migraine and controls). 148
CGRP in particular, is released in excess during headache attacks.10,11,149,150 As a response to CGRP depletion in mice, elevated baseline systolic BP and mean arterial pressure have been reported thereby revealing general effects of CGRP on vascular function.
151
Even though CGRP has been reported to be the main peptide in primary headaches, PACAP is also considered an important player. A significant change in the neuropeptide plasma levels was seen when comparing the interictal and ictal phase in migraine and cluster headache.150,152,153 In clinical tests, intravenous PACAP-38 infusion resulted in decreased MCA blood flow velocity in the beginning of the ictal phase (experimental).
154
Relative release of CGRP, PACAP, SP and VIP in migraine is summarized in Figure 2.
Summary of neurotransmitter release into the bloodstream in episodic and chronic migraine. Neurotransmitter baselines of calcitonin gene-related peptide (CGRP),11,143,145,146,155,258 pituitary adenylate cyclase-activating polypeptide (PACAP),148,150,259 substance P (SP)11,146,258 and vasoactive intestinal peptide (VIP)143,148 in episodic migraine (a) and chronic migraine (b) comparing the interictal headache period with healthy controls (measured as ratio). Neurotransmitter release of CGRP,10,11,149,150 PACAP
150
and SP
11
in episodic migraine (c) comparing the ictal with the interictal headache period (measured as ratio).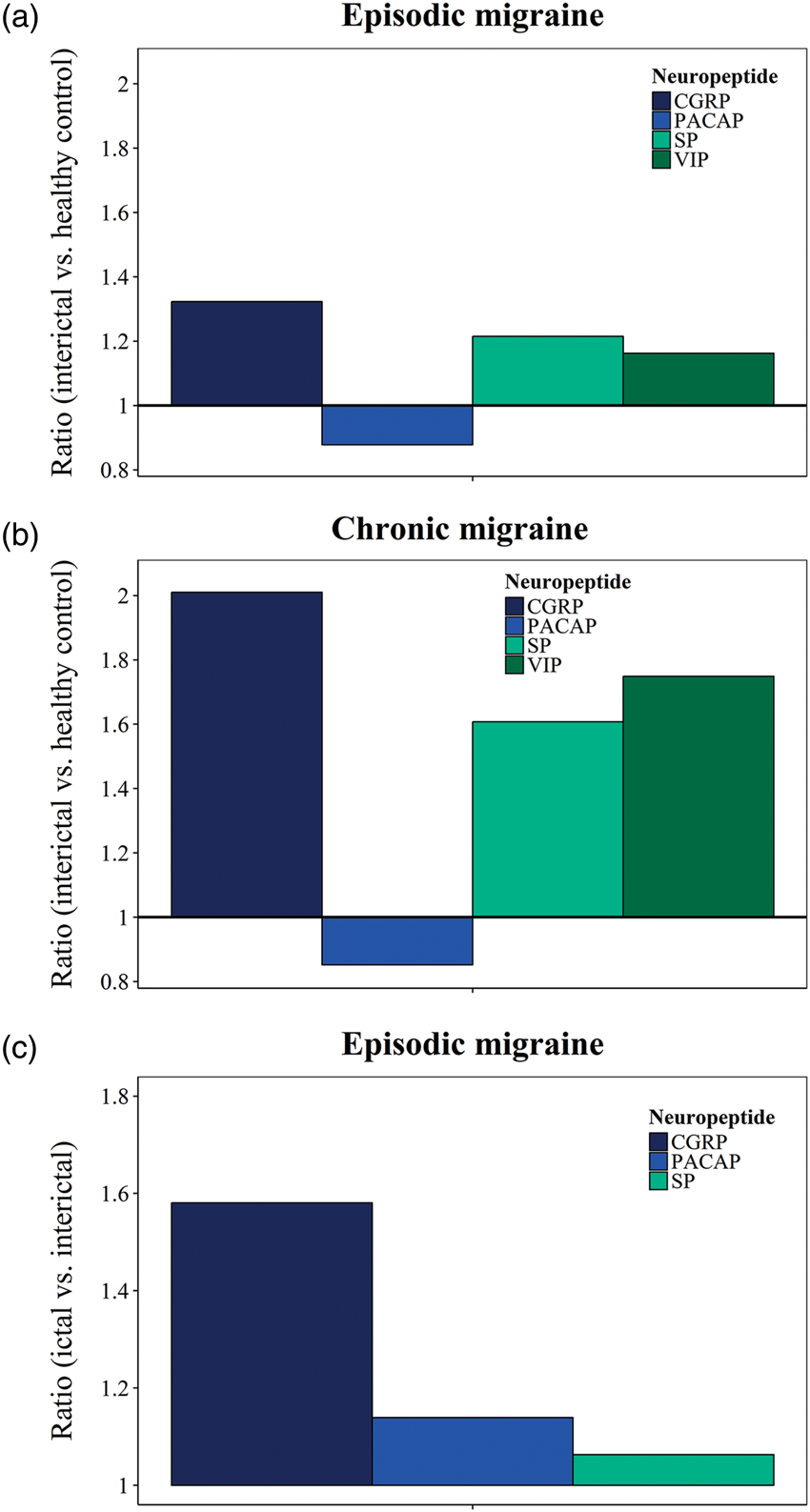
Even though migraine and cluster headache have been associated with autonomic and sensory neurotransmitter release during attacks,10–12,15,29,104,155,156 there still exists some degree of inconsistency between the findings. 157 Our view is mainly based on the very short half-life of released neuromodulators in plasma, high levels of peptidases in the blood and lack of proper site of sampling, important factors to consider for the detection of an effect and to obtain consistent results across studies.158,159 Moreover, because of the inability of peptides to pass the BBB, it is likely that neuropeptide release from cranial ganglia emanates from sites outside the BBB. In addition, Hoffmann et al. 160 investigated the effect of destroying primary trigeminal afferents on CGRP release into the jugular vein and cerebrospinal fluid, and suggested that stimulus-induced CGRP release mainly come from trigeminal afferents.
Multiple studies report an interconnection between neurotransmitter release in migraine, but also between neurotransmitter release and symptomatology/clinical phenotypes.146,161,162 To summarize, CGRP, PACAP, SP and VIP are considered cerebrovascular dilators, and ET-1, NPY and 5-HT cerebrovascular constrictors. However, potency and site of action vary between neurotransmitters. The observed neurotransmitter fluctuations have been proposed as a failing self-defense response 163 and being due to dysfunctional biosynthesis (e.g. de novo synthesis), release and uptake. 107
Possible importance of the endothelium
Endothelial dysfunction in migraine has been suggested by some authors. 164 Indeed, a considerable number of patients show signs of dysfunctional endothelial cells in cerebral arteries evaluated using a breath holding or reactive hyperaemia index.165,166 Increases in CBF cause flow-mediated dilation induced by production of NO in endothelial cells, as a response to shear forces, which depend on blood flow and velocity, and pressure on the vessel walls. If the regulatory mechanisms of CBF cannot keep up with changes in physiological conditions, cellular stress, morphological abnormalities and altered gene expression patterns might occur and give rise to inflammatory responses.167,168 Parameters such as intima-media thickness, flow-mediated dilation, intracellular adhesion molecules (ICAM) and vascular cell adhesion molecules (VCAM) have all been described as important factors in the evaluation of endothelial function. Elevated serum levels of VCAM and ICAM have been linked to migraine in children and young adults. 169 Cellular adhesion molecules are essential for immune cell transmigration across the BBB.170,171 Interestingly, there is a strong correlation between serum levels of ICAM-1 and interleukin 6 (IL-6) (proinflammatory cytokine) during migraine attacks; however, no correlation was observed in the headache-free period 172 suggesting shared regulatory ictal mechanisms. In addition, tumor necrosis factor α (TNF-α) (proinflammatory molecule) results in increased VCAM-1 expression in human cerebral endothelial cells. 170
Inflammation and BBB integrity and permeability
At present there is only limited evidence that points to an altered BBB permeability in migraineurs.173–175 BBB integrity has been investigated in animal models by means of Evans blue.
176
The marker revealed that cranial ganglia are not protected by the BBB including the TG,79,175 while the brain and cerebral vessels have a patent BBB. Figure 3 visualizes the spinal cord and selected ganglia from Sprague-Dawley male rats in relation to the BBB. It shows that TG, C1-C3 DRG, SPG, OTG and SCG are located outside the BBB (blue structures colored primarily by the Evans blue albumin complex, but also other protein complexes,
176
which indicates endothelial permeability). SPG stimulation is known to give rise to vasodilation and increased CBF, but these changes do not seem to affect the BBB permeability in rats.
91
Experimentally induced dural inflammation, and secondarily ganglionic inflammation, does not seem to alter BBB passage either,
175
even though inflammatory pain has been linked to BBB damage.177,178
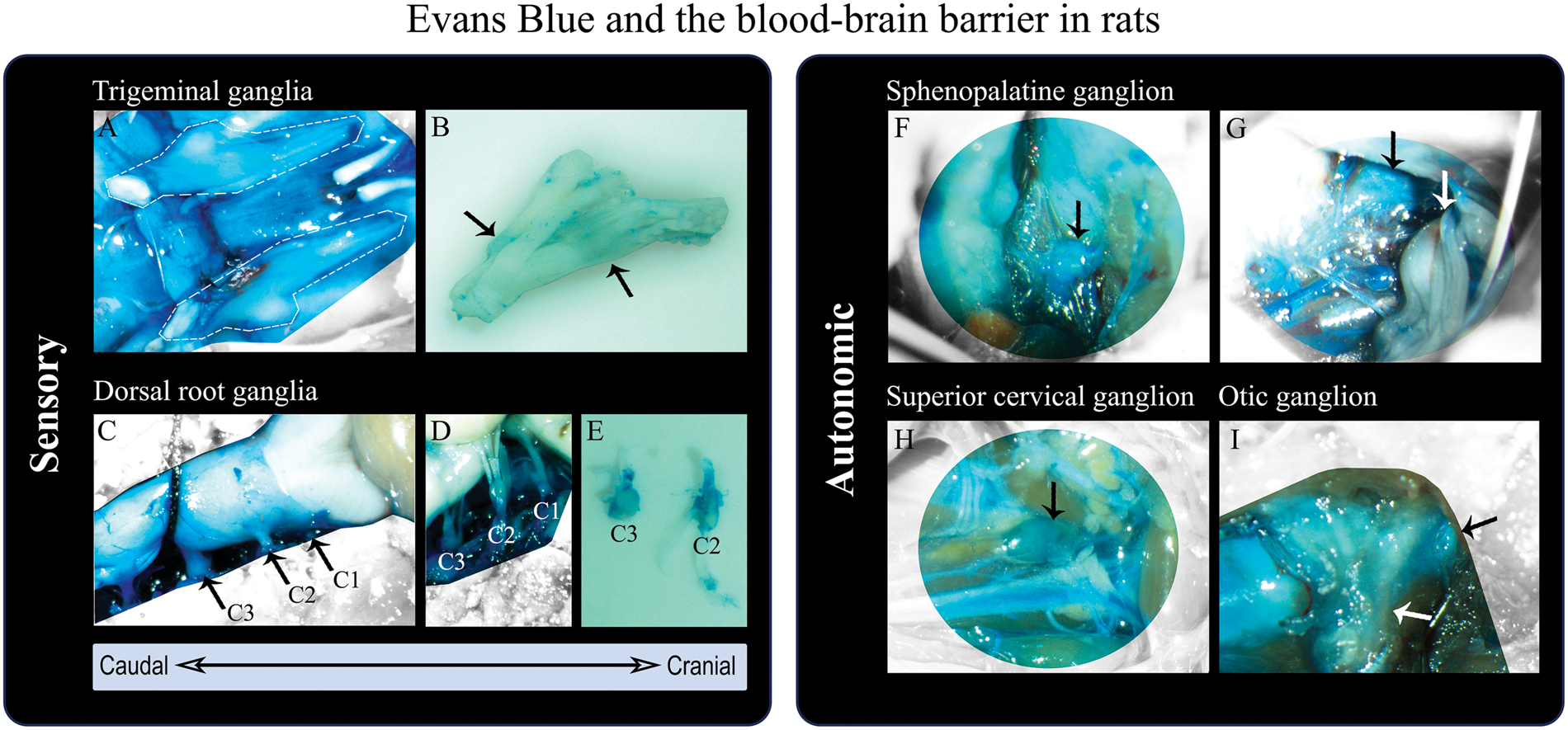
Sensory and autonomic ganglia in rats outside the blood–brain barrier (BBB) evaluated by means of in vivo staining with Evans Blue. (a) Trigeminal nerves, encircled by white dashed lines, after removal of the brain (dorsal view). The trigeminal nerves are covered by dura mater,
82
colored by Evans Blue and thus outside the BBB. (b) Trigeminal nerve (removal of dura mater) revealing Evans Blue leakage at sites of the trigeminal ganglion (black arrows). (c) Spinal cord, brainstem and cerebellum (dorsal view) protected by the BBB (not colored by Evans Blue). Dura mater, encasing the spinal cord, is stained by Evans Blue (outside the BBB). The cervical nerves C1–C3 are marked and the C3 dorsal root ganglion (DRG) visible. (d,e) C1–C3 DRG, not protected by the BBB (blue), after removal of dura mater. (f,g) Sphenopalatine ganglion (SPG) (black arrows) located below the maxillary nerve (white arrow).
82
The SPG is also outside the BBB (colored by Evans Blue). (h) Superior cervical ganglion (black arrow) not protected by the BBB (blue). (I) Otic ganglion (black arrow), outside the BBB (blue), positioned on the medial surface of the mandibular nerve (white arrow). The methodology is described by Eftekhari et al.
79
If BBB disruption occurs in an episodic disorder such as migraine, it would for example occur through endothelial tight junctions to result in increased permeability. 179 CGRP, SP and interleukin 1β (IL-1β), involved in primary headache pathophysiology, seem to have an effect on the brain endothelial permeability. 180
In an ischemic stroke model, results indicate that expression of tight junction proteins gets enhanced when inhibiting matrix metallopeptidase (MMP), which points to BBB restoration. 181 Focusing on primary headaches, specific MMP-2 and MMP-9 gene or haplotype polymorphisms have been associated with increased levels of MMP-2 1 82 and MMP-9 1 83 in the blood of migraineurs and are believed to be associated with alteration in BBB patency (however only studied in peripheral blood samples). Another study reported that CSD resulted in elevated MMP-9 in the ipsilateral cortex suggesting subsequent disruption of the BBB. Plasma protein leakage and edema were also observed, but not in MMP-9 null mice. 184 An ex vivo study revealed that cytokines (e.g. TNF and interleukin subtypes) also might contribute to disruption of the BBB. A NeuroVascular Unit microfluidic two chamber system (vascular and brain chamber) was used to investigate the effect of inflammation on the BBB. TNF and interleukin subtypes were released as a response to lipopolysaccharide exposure, but the release varied in regards to chamber and exposure time when comparing each cytokine separately. 185 Brown et al. 185 suggested that cytokine release patterns possibly are involved in alteration of membrane permeability over time. 185
In a BBB study involving gadolinium-enhanced MRI, cortical opening of the BBB and edema was identified in a familial hemiplegic migraine (FHM) type 2 patient where the attack was classified as severe. 132 However, there are no clinical studies to date that link primary headaches and opening of the BBB. This is reviewed by Edvinsson and Tfelt-Hansen. 174 In agreement, Schankin et al. 173 recently found no change in the BBB (interictally and ictally) in acute GTN-induced migraine attacks in humans. This field requires more studies in order to prove or disprove the effect of primary headaches on BBB integrity and permeability.
Internal and external factors and genetic predisposition in primary headaches
Internal and external factors and genetic predisposition are known to affect the susceptibility to develop primary headaches. Therefore, individual variation in regards to response to triggers, neuronal hyperexcitability and vascular regulation might exist. Figure 4 illustrates a proposed hypothesis of migraine development due to variable predispositions from genetic and environmental factors. It is evident when comparing subject B-D (migraineurs), that there can be individual differences in the factors leading to development of migraine, and hence they will probably respond differently to treatment strategies (e.g. medication). Therefore, creation of individual treatment strategies might be beneficial for treating primary headache disorders.
Genetic and environmental factors and migraine predisposition. Migraine conditions are primarily considered polygenic complex disorders of which genetic (inherited) and environmental risk factors, as well as interactions, constitute headache predisposition. (a) The bar plot illustrates a proposed hypothesis of individual migraine development. The migraine threshold, termed cutoff, shows the point at which subjects are diagnosed with migraine (ratio ≥ 1). Subject A is a non-migraineur and subject B-D migraineurs. (b) Subject B has developed migraine due to an equal predisposition from genetic (50%) and environmental factors (50%), subject C mainly due to environmental predisposition (92%) and subject D mainly due to genetic predisposition (75%).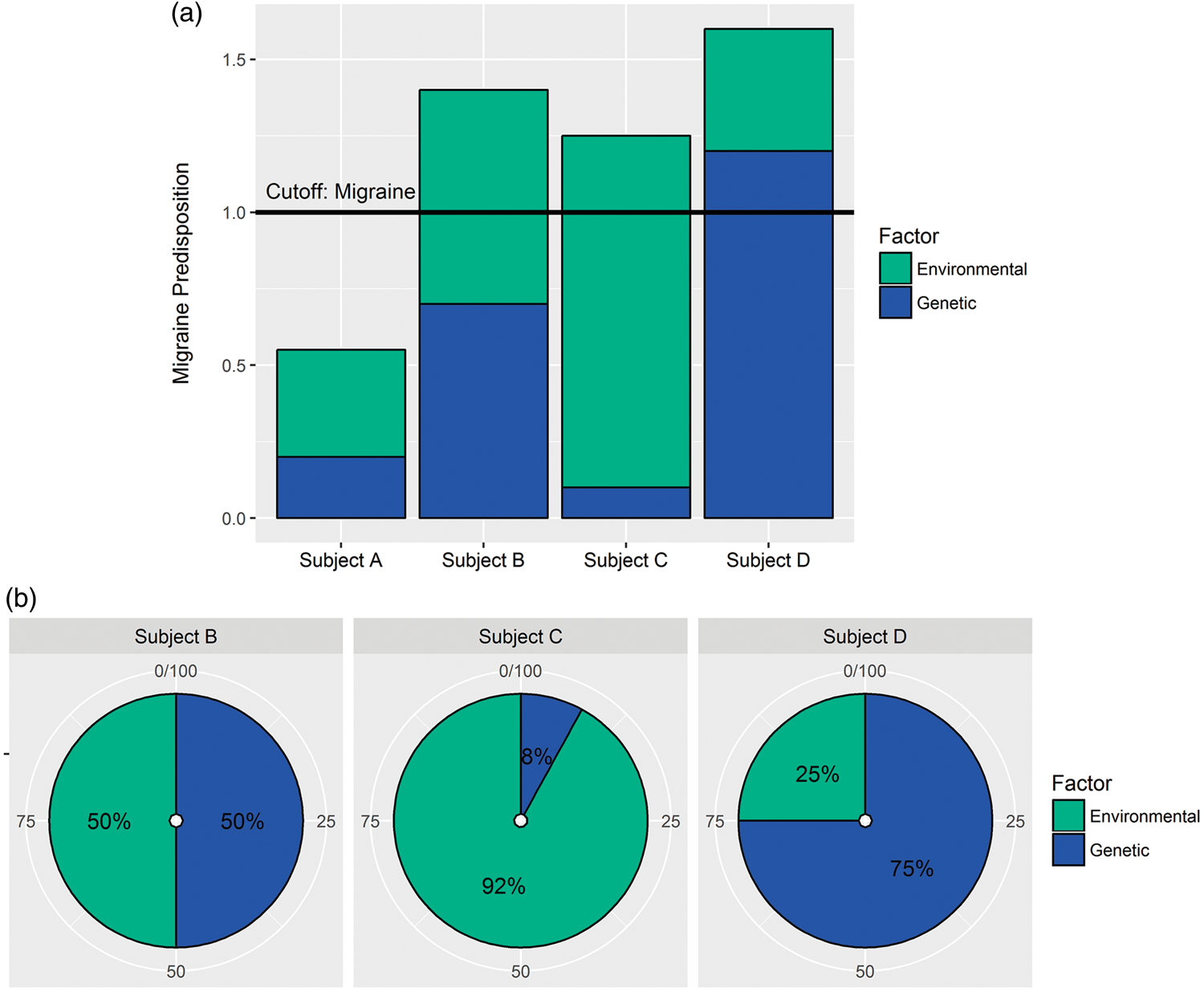
Neuronal hyperexcitability and over-activated neuronal circuits are considered functional abnormalities in migraine and believed to be important in triggering headache/migraine attacks. 186 More information on multiple internal and external headache triggers (e.g. sleep disturbance, stress, light, odors) can be found in thorough reviews by Ho et al. 15 and Borkum. 187 Migraine patients have subjectively reported a link between headache attacks and triggers (e.g. alcohol, food, visual triggers, stress/tiredness, environment), categorized by a principal component analysis. 188 Migraine triggers have been proposed to lead to fluctuations in neurotransmitter levels and hypersensitivity in specific regions of the brain.12,15 In humans, short exposure to noise resulted in sensitization in subjects without headache; however, prolonged exposure led to desensitization. For headache patients, the trend was not clear. 189 The effect of stress in rats included satellite glial cell (SGC) activation and elevated SP expression in the TG. IL-1β and interleukin 1 receptor type 1 (IL-1R1) expression in TG neurons were also elevated. 190
Development of migraine and cluster headache also seems to be age- and gender-dependent,3,191,192 e.g. with particular dependence on gender hormones (e.g. rate of estrogen withdrawal in females). 193 During the menstrual cycle, fluctuations in steroid hormones seem to be associated with changes in cerebrovascular reactivity of which vasodilation primarily is observed in the right hemisphere. 194 Regarding the effect of age and sex on cerebrovascular regulation, inconsistency exists between human studies. One study reported that these two factors did not seem to affect cerebrovascular regulation to any major extent, 195 while another study suggested an effect of age on BP and CBF. 196 Moreover, neuronal activity and CBF are coupled tightly to brain metabolism.197,198 Increased brain activity causes increased local energy expenditure and is thus correlated with increased local CBF. For long, it has been discussed which physiological parameter comes first, metabolism or flow? Recent studies suggest that they indeed may be closely coupled.62,199 Moreover, Gantenbein et al. 200 put forward a hypothesis stating that processing of sensory stimuli in migraine has a higher energy demand for neuronal function compared to that of healthy subjects, possibly linked to the theory of neuronal hyperexcitability.
In terms of genetic migraine predisposition, there are several lines of evidence including rare mutations in known proteins, single nucleotide polymorphisms and microsatellite markers. For the rare condition FHM, there are now at least three subtypes (FHM1-3) reporting gene mutations of calcium voltage-gated channel subunit alpha1 A (CACNA1A), ATPase Na+/K+ transporting subunit alpha 2 (ATP1A2) and sodium voltage-gated channel alpha subunit 1 (SCN1A). 201 The subtypes seem to share the potential to modify glutamate signaling in the CNS, making the neurons more susceptible to depolarize (cortical hyperexcitability). 202 The first mutation (FHM1) was detected in locus 19p13 203 which relates to the P/Q calcium channel in the CNS, and affects calcium-dependent facilitation and synaptic plasticity. 204 In a genetic family study, a considerable number of individuals (86%), carrying a mutation in ATP1A2, were diagnosed with FHM, 205 which implies a high effect size.
Selected candidate migraine genes detected by GWAS meta-analyses.

GWAS: genome-wide association studies.
Note: Results indicate involvement of the selected candidate genes in vascular disease/disorder, smooth muscle contractility or regulation of vascular tone amongst others (*Gormley et al. 208 ; **Anttila et al. 207 and Gormley et al. 208 )
Reported as
End-feet defined as “specialized foot-processes of perivascular astrocytes that are closely opposed to the outer surface of brain microvessels, and have specialized functions in inducing and regulating the BBB” by Abbott et al. 282
Neuronal hyperexcitability and vascular regulation
Neurophysiological alterations such as altered neural plasticity, hyperexcitability and lack of habituation to sensory stimuli have been associated with primary headaches.213–215 Recent research focuses on the epigenome, which is believed to affect the susceptibility to develop migraine, 216 with the field being at an emerging state. For example, a stressful environment during childhood has been linked to stress-induced alterations of homeostasis systems and development of migraine later in life 217 thereby including the psychological aspect of primary headaches. One postulation is that epigenetic changes, occurring as a response to chronic stress, results in hyperexcitable neurons, 218 thereby linking the epigenome to the CSD and trigeminovascular theory. However, methylation patterns have been found to vary considerably both between tissues and genes reported in a study investigating migraine-related genes. 219
Focusing on vascular diseases, hypoperfusion in the white matter has been observed in CADASIL patients, 220 who seem to have prolonged and more frequent CSD 221 possibly due to parenchymal hyperexcitability. 35 Interestingly, a study showed that migraine is present in 75% of CADASIL patients of which many reported aura. The migraine aura seemed to increase the odds of encephalopathy. 222 Moreover, a microvascular vasoconstrictor anomaly has been associated with CADASIL in noradrenaline and angiotensin pathways, but not with a vasodilator anomaly. 223 There are also some gender differences; brain vessels from males seem to be more sensitive to vasoconstrictors (angiotensin II and endothelin) than from females. 224 Overall, these parameters might predispose individuals to enhanced migraine susceptibility or vice versa.
As a response to neuronal hyperexcitability, one would expect increased neurotransmitter release from perivascular ganglionic nerve endings on the cerebral vasculature. Moreover, neurotransmitters might be released directly into the extracellular space surrounding the neuronal cell bodies in the ganglia, triggered by neuronal depolarization evoking SGC activation through neurotransmitter-receptor binding.225,226 CGRP evoked release of NO and inflammatory responses in these glial cells, which was blocked by a CGRP receptor antagonist.
226
The exact functional role of SGCs is still unknown; however, they are believed to be involved in control of the neuronal microenvironment.190,227 Neuronal excitability has been associated with SGC activation
227
and various pain syndromes seem to be related to diverse activation states of the glial cells.
228
The SGCs may thus be an important player in the sensitization process and chronification of migraine, in addition to the phenomena discussed in the previous sections. The SGCs constitute >90% of the total number of cells in the TG, and are linked to neurons via gap junctions (e.g. connexin) implicated in cell-cell signaling.226,229 Connexin, but also pannexin are involved in neuronal excitability and synaptic plasticity in the CNS forming gap junctions/hemichannels facilitating neuronal transmission.230,231 Experimental CSD provoked opening of pannexin1 channels, which was suggested to affect activation of the trigeminovascular system.
232
Ashina et al.
145
suggested that migraineurs suffer from abnormal neurogenic vascular control enduringly or permanently. Figure 5 gives an overview of possible pathophysiological effects, which occur due to migraine susceptible gene pools and external and internal headache triggers, all resulting in altered CBF.
Genetic headache predisposition, headache triggers and cerebral blood flow (CBF). Potential effects of and link between genetic headache susceptibility (input: green), extrinsic and intrinsic headache triggers (input: green) and CBF (output: red) through multiple pathophysiological steps (simplified flow diagram).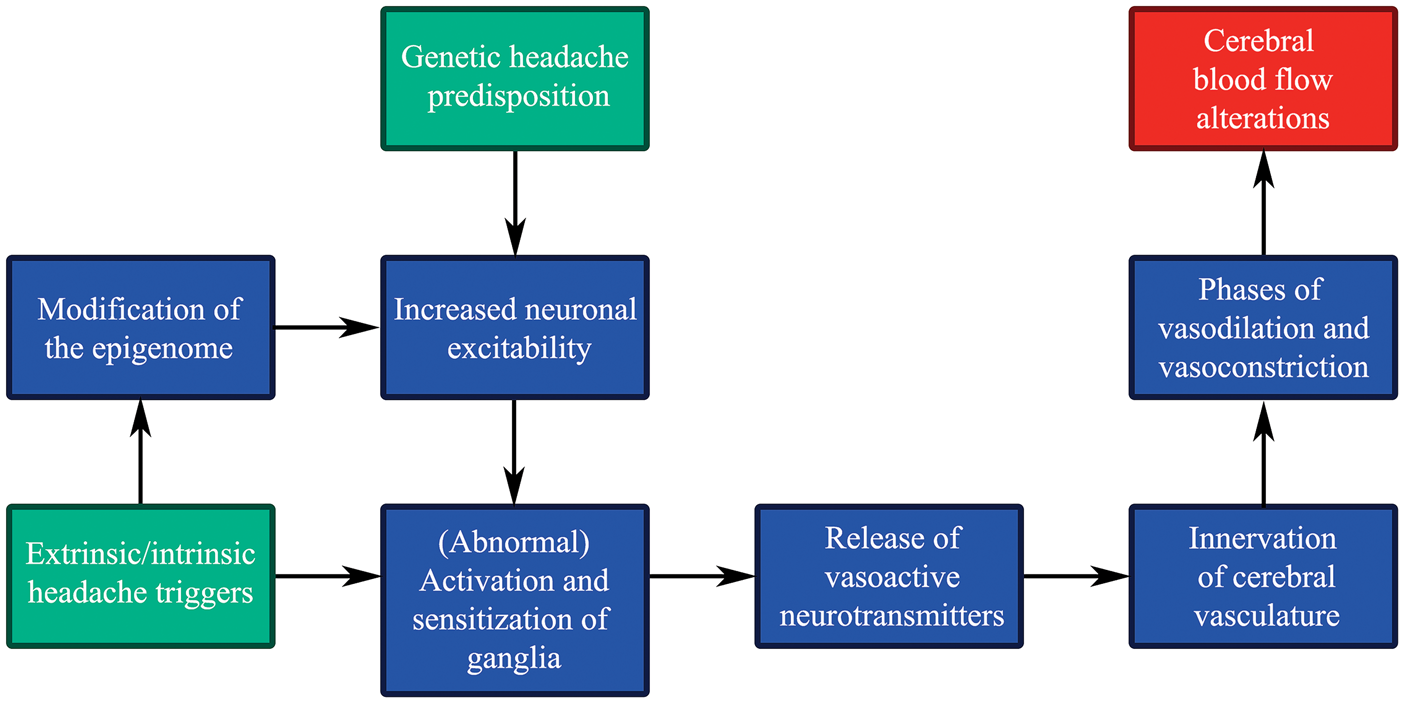
Primary headaches and sensitization
There are several primary headache disorders; migraine and trigeminal autonomic cephalgias (cluster headache, chronic paroxysmal hemicranias, inter alia) of which all are characterized by pain varying in duration, location and quality with accompanied autonomic and sensory neurological signs/symptoms depending on the diagnosis. 2 Cluster headache patients experience autonomic symptoms such as rhinorrhea, lacrimation and abnormal sweating (parasympathetic influence) and miosis and ptosis (sympathetic influence).2,233 Cranial sensory symptoms include experience of allodynia, and abnormalities in sensory perception of visual, olfactory and auditory stimuli. 234 This implies involvement of the parasympathetic, sympathetic and sensory nervous systems which form complex functional units implicated in processing of impulses. 235
During the headache attack phase, ganglia such as the TG and SPG, located outside the BBB, are activated and sensitized. The discovery of the sensitization process was pioneered by Burstein et al. 236 Basically, upon repeated activation/stimulation (heat, cold or pressure stimuli), the subsequent response to the same stimulus was enhanced, being referred to as sensitization. Sensitization has been associated with release of neurotransmitters, including CGRP, and could be a part of the mechanism behind allodynia. 236 Despite that the sensory trigeminal system contains several messenger molecules (e.g. CGRP, SP/NKA, PACAP and NO), only CGRP seems to be clearly associated with headache in migraine attacks.156,237,238 So far, release of CGRP/PACAP has only been positively linked to acute attacks of migraine, or in bouts of cluster headache.12,150,152,153,156,157 Moreover, patients with nasal congestion and rhinorrhea show release of VIP, 237 which is observed in chronic migraine as well. 143 Putatively, other molecules such as NO and cytokines are involved.149,239
Perivascular nerve terminals originating from sensory, parasympathetic and sympathetic ganglia, believed to be involved in primary headaches, have been observed on cerebral arteries and arterioles. 47 In addition to the involvement of multiple neurotransmitters in CBF regulation, they are part of: (i) the neurovascular unit in the brain parenchyma, and (ii) larger arteries belonging to the circle of Willis and pial cortex arterioles.
Chronification of pain in primary headaches
Migraine experienced at least 15 days per month for >3 months is considered chronic. Chronic cluster headache, on the other hand, should last for at least a year with or without remission periods (<1 month). 2 Chronic headache and headache frequency correlated significantly in adults 240 possibly due to the detrimental effects of pathophysiological changes (e.g. trigeminovascular activation and sensitization) arising during headache attacks. In coherence, chronic migraine seems to increase with age.241,242 Trigeminovascular activation has also been observed as a response to inflammation, which could be involved in headache progression. 243 At a cellular level, systemic inflammation of L4/5 DRG, involved in transmission of sensory signal, seems to modify the SGCs. Abnormal growth, formation of new gap junctions and increased ATP sensitivity of SGCs could potentially contribute to pain. 244 Structural and functional modification of SGCs might be connected to cellular stress and inflammation.190,243,245 Other findings suggest that SGC activation, as a response to inflammation, results in modulated excitability of neurons in TG and DRG.244,246 We hypothesize that neurogenic inflammation might have a reinforcing effect on neuronal hyperexcitability, a feed-forward loop, probably contributing to development of chronic headache.
The chronic pain state of the brain is believed to result in further structural, functional and chemical modifications. Chronic pain might arise due to impairment of pathways in the CNS, but also of the peripheral nervous system, which collectively manifest phenotypically as altered sensation, emotion and cognition or modulated pain response. 247 These changes are believed to be involved in disease progression from the episodic to chronic state. Therefore, it appears to be important for the future migraine management strategy to treat primary headaches early to avoid chronification bearing in mind that many primary headaches are considered complex polygenic disorders.
Episodic to chronic headache: Neurogenic inflammation and chronification
It is often argued, based on clinical observations, that a “migraine highway” is present in many patients. In the beginning, occasional attacks occur which, over the years, become frequent episodic migraine with up to 14 migraine headache days per month (according to ICHD-III beta 2 ). This may, in some cases, expand even more, sometimes associated with migraine medication overuse.248,249 The mechanisms involved are still not clear. Vasodilation, release of vasoactive peptides and plasma-protein extravasation constitute factors that are involved in development of inflammatory responses, also referred to as neurogenic inflammation,76,139,250 which can be triggered by electrical stimulation of ganglia. 76 Research shows that PACAP-38 is involved in central sensitization, 154 neuronal and satellite glial cyclic adenosine monophosphate (cAMP) production in TG, 251 and neurogenic inflammation. 252 CGRP and VIP are associated with cAMP accumulation occurring simultaneously with cerebral vasodilation. 112 Sarchielli et al. 10 showed that cAMP and cGMP release occurred after release of CGRP, NKA and nitrites in migraine attacks measured in internal jugular venous blood. In addition, both CGRP and cAMP induced NO and inducible NOS release from TG glial cells in culture. 226 However, single nucleotide polymorphisms in the neuronal NOS gene do not seem to increase the risk of migraine. 253 Moreover, in migraineurs, release of other inflammatory markers such as interleukin (IL) subtypes (e.g. IL-1β, IL-6, IL-10) and TNF have been observed,239,254 which seems to be most likely in frequent or chronic migraine. A tendency of higher lymphocyte number in the peripheral blood was observed in chronic migraine compared to episodic migraine suggested to be linked to the inflammatory state of the patients. 255 In the calvarial periosteum of chronic migraineurs, gene expression of proinflammatory markers was increased (e.g. IL-6, IL-1 receptor type 2 (IL1R2), C-C motif chemokine ligand 2 (CCL2), C-X3-C motif chemokine ligand 1 (CX3CL1)), whereas gene expression of markers involved in suppression of inflammation was decreased. 256 Some of these markers might affect nociception. In addition, results show that application of IL-1β to the meninges is involved in activation and sensitization of nociceptors, which was not observed to the same degree for IL-6. 257
Summary
Primary headaches are considered cerebral neurovascular disorders of which abnormal brain activity and CBF changes have been observed. CBF changes might occur as a response to pathophysiological changes such as neuronal hyperexcitability and autoregulatory impairment. GWAS studies, investigating the susceptibility to develop migraine, revealed suggestive candidate genes related to vascular diseases/mechanisms. One might postulate that some headache attacks are linked to underlying undiscovered vascular diseases. On the other hand, neuronal abnormalities have an effect on vascular mechanisms and function. Even though the original vascular theory is considered to be secondary in nature, these findings still emphasize the importance of the vasculature in primary headaches.
To summarize, this review gives rise to the following connections between primary headaches and CBF: (i) headache attacks start in the CNS and result in activated and sensitized ganglia and neurotransmitter release (e.g. CGRP, PACAP), (ii) the intracranial cerebral vasculature (large cerebral arteries and arterioles on the surface of the brain) is supplied with perivascular nerve fibers originating from the sensory, sympathetic and parasympathetic nervous systems, (iii) neurovascular activation putatively gives rise to vasoconstrictive/vasodilative phases and altered CBF and metabolism in the affected brain regions, (iv) neurotransmitter release seems to affect cAMP levels followed by inflammatory responses, but also plasma–protein extravasation in certain areas such as the dura mater (if SP is released), and (v) inflammation might reinforce neuronal hyperexcitability, pathological alterations and cellular dysfunctions through a feed-forward loop and thus contribute to chronification from the episodic state.
Finally, from a treatment perspective, it is important to detect and treat primary headache disorders in time in order to avoid chronification. Furthermore, new treatment strategies are required, which take into account that some individuals are genetically predisposed to develop primary headaches while others, primarily, might develop it as a response to environmental stimuli (e.g. stress).
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by grants from the Lundbeck foundation (Denmark), Swedish research council (grant no 5958) and Heart-Lung foundation.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.