
Abstract
Aneurysmal subarachnoid hemorrhage (SAH) is a devastating cerebral event that kills or debilitates the majority of those afflicted. The blood that spills into the subarachnoid space stimulates profound cerebral artery vasoconstriction and consequently, cerebral ischemia. Thus, once the initial bleeding in SAH is appropriately managed, the clinical focus shifts to maintaining/improving cerebral perfusion. However, current therapeutic interventions largely fail to improve clinical outcome, because they do not effectively restore normal cerebral artery function. This review discusses emerging evidence that perturbed cerebrovascular “myogenic reactivity,” a crucial microvascular process that potently dictates cerebral perfusion, is the critical element underlying cerebral ischemia in SAH. In fact, the myogenic mechanism could be the reason why many therapeutic interventions, including “Triple H” therapy, fail to deliver benefit to patients. Understanding the molecular basis for myogenic reactivity changes in SAH holds the key to develop more effective therapeutic interventions; indeed, promising recent advancements fuel optimism that vascular dysfunction in SAH can be corrected to improve outcome.
Keywords
For neurologists tasked with saving patients suffering from subarachnoid hemorrhage (SAH), this review provides a mechanistic overview of the next frontier of therapeutic interventions. The insight provided calls attention to emerging evidence that perturbed cerebrovascular “myogenic reactivity” is a critical element underlying “delayed cerebral ischemia” (DCI) in SAH. Myogenic reactivity is a crucial microvascular process that potently dictates cerebral perfusion: indeed, this mechanism can explain why many current therapeutic interventions fail to improve outcome (e.g., triple H therapy) and is key to developing more targeted and successful interventions. As a caveat, this review's focus on myogenic mechanisms does not diminish the relevance of other pathological processes (e.g., blood barrier disruption, microthrombosis and cortical spreading) in the SAH injury process. 1 In fact, we speculate that the ischemia caused by perturbed myogenic reactivity either causes or exacerbates these complications.
A brief clinical overview of subarachnoid hemorrhage
SAH is a devastating cerebral event that kills or debilitates the majority of those affected.2,3 Although SAH accounts for only 5% of all strokes (∼10 in 100,000 persons per year), the young age of incidence and poor prognosis instills a substantial societal burden: SAH accounts for 27% of all stroke-related years lost before the age of 65 (a measure of premature mortality) and therefore, closely rivals the most frequent stroke subtype, ischemic stroke, in terms of productive life years lost (38% for ischemic stroke).4,5 SAH carries an extremely high fatality rate (about 50%) and approximately 30–50% of the patients who survive are seriously debilitated.2,3 As these grim statistics suggest, current medical interventions display limited efficacy, and thus, SAH remains broadly regarded as a “large unmet medical need”.
Almost 85% of spontaneous SAH cases (i.e. cases not due to trauma) are caused by a ruptured cerebral aneurysm. 5 In its initial phase, SAH is a medical emergency: bleeding into the subarachnoid space sharply elevates intracranial pressure, which ultimately causes cerebro-circulatory arrest if it is not swiftly relieved. Consequently, almost half of SAH patients die before reaching hospital. 5 However, much of the treatable mortality and morbidity in SAH is caused by a pronounced secondary ischemic event, aptly termed “delayed cerebral ischemia or DCI”, that emerges 4-12 days after the initial hemorrhage.6,7 Thus, once the initial bleeding in SAH is appropriately managed, the clinical focus shifts to maintaining cerebral perfusion.
The therapeutic failure of targeting angiographic vasospasm
Delayed cerebral ischemia in SAH has been traditionally attributed to “angiographic vasospasm,” a radiographically identifiable vasoconstriction of large/proximal cerebral arteries: vasospasm is a frequent complication in SAH, it clearly associates with poor clinical outcome, and it is an intuitive cause of deficient cerebral perfusion.2,8 Clinical studies implicated endothelin 1 (ET-1), a potent endothelial-derived vasoconstrictor,9,10 as a causative factor by demonstrating a strong correlation between plasma/cerebrospinal fluid ET-1 levels and angiographic vasospasm severity in SAH patients.11–13 Experimental studies that targeting ETA, the “vasoconstrictive” ET-1 receptor subtype, demonstrated a clear benefit in terms of mitigating angiographic vasospasm:
14
: these results fueled numerous clinical trials, most notably CONSCIOUS.15–17 Unfortunately, while these clinical trials (that to date, include over 2600 SAH patients) demonstrate that ETA antagonism (clazosentan) successfully curtails angiographic vasospasm in SAH, the intervention
In retrospect, the decision to initiate large randomized trials for ET-1 receptor antagonists was not supported by robust experimental data on clinically relevant outcomes. 14 Although angiographic vasospasm is a well-established predictor of cerebral ischemia/infarction in SAH,19–21 its sensitivity and specificity (both approximately 70%) are far from optimal. 19 A meta-analysis of 55 experimental studies determined that the majority failed to assess whether ET-1 receptor antagonism changed cerebral blood flow (CBF) in SAH (only 3 of the 27 studies measured CBF); none of the studies investigated neurofunctional outcomes. 14
Unfortunately, clinical trials are frequently initiated on limited, incomplete, and weak preclinical data. This longstanding issue has been recurrently addressed by the publication of scientific guidelines, with notable contributions from the Stroke Therapy Academic Industry Roundtable (STAIR)22,23 and Sena et al. 24 In general, ideal interventions should (i) demonstrate efficacy in at least two species and in at least two laboratories that use different models; (ii) involve adequate physiological monitoring, including CBF measurements; and (iii) assess multiple functional and outcome measures, including brain injury volume, and neurological function. Individually, studies should adhere to good scientific practices by utilizing proper sample sizes, randomization, allocation concealment, blinding and case fatality reporting.22–24 In this light, the CONSCIOUS trial failure would almost certainly have been averted had the available data been scrutinized against these guidelines: critical measures were clearly missing and the studies were generally weak according to STAIR criteria. 14 It is worrisome that scientific quality remains an underappreciated issue among those who conduct preclinical research,14,25 since there is no doubt that weak studies frequently overestimate drug efficacy. 25 Thus, as the SAH field presses forward in its search for new therapeutic interventions, a concerted effort to assess all relevant clinical endpoints (e.g. CBF, neuronal injury, etc.) with rigorous scientific methodology is imperative.
Although ET-1 antagonists failed in clinical trials, two crucial conclusions can be formulated: first and foremost, while angiographic vasospasm is an informative parameter that correlates with disease severity and predicts clinical outcome, it is itself not the central determinant of ischemic injury.14,26–28 Indeed, there is ample evidence that SAH reduces CBF in the absence of angiographic vasospasm,29–34 indicative of a significant dissociation between these two endpoints. Second, distinct mechanisms drive constriction in the proximal and distal regions of the cerebrovascular tree; consequently, angiographic vasospasm is an insufficient endpoint for assessing new therapeutic interventions. Ultimately, the failure of the CONSCIOUS clinical trials compels a shift in attention from large cerebral arteries (i.e. angiographic vasospasm) to the cerebral microcirculation. In fact, augmenting small resistance artery vasoconstriction 35 would compromise one of the most significant determinants of cerebral perfusion: CBF autoregulation.
Cerebral blood flow autoregulation
Without question, minimum perfusion levels must be maintained in the brain, because neurons have high metabolic demand and minimal energy reserves. Large perfusion reductions rapidly compromise neuronal function and lead to infarction; however, even in the absence of “symptomatic hypoperfusion” (i.e. reductions in blood flow that cause dizziness or other overt symptoms), inadequate cerebral perfusion slowly compromises neuronal function and drives cognitive decline. 36 While hypoperfusion has obvious negative consequences, the brain is also detrimentally impacted by hyperperfusion (appropriately termed “cerebral hyperperfusion syndrome”). 37 Specifically, insufficient vascular resistance (i) promotes hydrostatic edema formation, due to increased Starling forces 37 and (ii) can cause pressure-induced damage of the fragile capillary networks.38,39 Cerebral perfusion, therefore, must be tightly regulated with an exquisite level of precision.
Cerebral blood flow autoregulation refers to the intrinsic ability of the distal cerebral resistance arteries to match their flow resistance to the prevalent perfusion pressure, thereby maintaining constant perfusion over a wide range of systemic pressures. 40 Although the precise pressure range governed by autoregulation is subject to some dispute, 41 the widely accepted autoregulatory range in humans is 60-150 mmHg. Technically, the term autoregulation refers only to the maintenance of constant flow in response to changes in pressure; this process is achieved by a mechanism localized to resistance arteries (named “the myogenic response”). When neuronal metabolism necessitates an increase in perfusion, “metabolic” and “neurovascular” coupling mechanisms superimpose on autoregulation, to locally match perfusion to neuronal energy demand. As examples of the latter mechanisms, carbon dioxide tension, adenosine (a metabolic product), nitric oxide and sympathetic activity all locally influence vascular tone and hence, regional perfusion within the brain.42,43 Separating local influences from autoregulation in vivo is challenging: consequently, our understanding of how disease processes alter autoregulation is rather rudimentary. Non-invasive imaging studies clearly demonstrate that SAH compromises autoregulation,44–46 but the mechanistic basis requires an experimental system that characterizes a resistance artery's myogenic response (i.e. its pressure/diameter relationship) in vitro to eliminate the confounding influences of local metabolic and neurovascular mechanisms.
The myogenic response
Over a century ago, Sir William Bayliss made the seminal observation “when now the pressure was raised inside the artery it was seen at first to swell, but immediately, and while the mercury was still kept at its height, a powerful contraction took place.” 47 This observation, named the myogenic response,48–56 describes the dynamic adjustment of blood vessel diameter to changes in perfusion pressure that forms the basis of local blood flow autoregulation. Specifically, the myogenic response is vasoconstriction in response to an increase in transmural pressure (i.e. the pressure across the vessel wall) or vasodilation in response to a decrease in transmural pressure.
As the etymology of its name implies (myo = muscle, genic = generated), the myogenic response originates from the vascular wall smooth muscle layer.57–59 Vascular smooth muscle cells are continuously exposed to changes in mechanical load (i.e. circumferential stretch), due to changes in blood pressure: the smooth muscle cells respond to this stimulus by actively adjusting their contractile force and ultimately, the level of active constriction exhibited by a artery. Notably, a vessel will constrict to a diameter smaller than its initial diameter in response to an increase in transmural pressure,60,61 indicating that the controlled variable in myogenic signaling is not vessel diameter per se. Instead, in accordance with the law of Laplace (wall tension = transmural pressure gradient × vessel radius), myogenic vasoconstriction endeavors to normalize pressure-induced increases in vessel wall tension, a concept supported by a direct correlation between changes in vessel wall tension and intracellular myogenic vasoconstriction signaling events.60,62,63
Not all arteries within the vascular tree display myogenic reactivity: large conduit arteries, which are tasked with rapidly conducting large volumes of blood, do not display pressure-stimulated vasoconstriction. 64 Instead, it is the small, muscular resistance arteries (30–250 µm in diameter), located just proximal to the capillary beds, that actively control blood flow in response to pressure and integrate the modulatory influences of tissue-derived signaling molecules.65,66 Even here, resistance arteries display a gradient of myogenic reactivity that is inversely related to luminal diameter. For example, in the hamster cheek pouch, third-order resistance arteries (28 µm) exhibit ∼23% greater myogenic index (i.e., the slope of the pressure-diameter relationship) than the first-order arteries (81 µm). 67 This indicates that the smaller resistance arteries are of particular functional importance for blood pressure regulation,68,69 the protection of downstream capillary integrity38,39 and minimization of edema formation. 70
The molecular basis for the myogenic response has been extensively reviewed;52,53,55,56 since this is a substantial topic in its own right, this review will provide a simplified and abbreviated overview. The molecular signaling pathways that control the myogenic response involve multiple signaling streams, broadly separated into: (i) mechanosensor components that register the pressure/stretch stimulus and transduce physical forces into intracellular signals, (ii) the activation of calcium release and calcium sensitization mechanisms, and (iii) the convergence of these mechanisms to processes that phosphorylate the myosin light chain and initiate cross-bridge cycling.
No single mechanosensor has been established as the master regulator of myogenic reactivity: instead, there appears to be a diverse array of putative mechanosensors, including stretch-activated ion channels (e.g. transient receptor potential nonselective cation channels71–73), G-protein coupled receptors (e.g. angiotensin type I receptor, 74 integrins (e.g. α5β1 and αvβ3 heterodimers75,76) and most recently, membrane-bound tumor necrosis factor. 77 These mechanosensitive elements are redundantly organized, which likely preserves a fully functional myogenic mechanism in several settings.
Mechanosensor activation increases intracellular calcium levels and stimulates signaling processes that amplify responsiveness to calcium, termed calcium sensitization.52,78 Initially, vascular smooth muscle cells depolarize in response to increased wall tension,79–81 which stimulates calcium influx through the dihydropyridine-sensitive L-type voltage gated calcium channels; 81 calcium release from the sarcoplasmic stores also occurs. 82 Cytosolic calcium binds to calmodulin, leading to myosin light chain kinase (MLCK) activation and subsequent myosin light chain 20 (MLC20) phosphorylation. 55 MLC20 phosphorylation is the critical signaling event in pressure- and agonist-induced vasoconstriction62,83,84 that activates actin-myosin filament interaction and gliding. 55 Concurrent with these calcium-dependent mechanisms, calcium sensitization processes increase the level of MLC20 phosphorylation for a given concentration of cytosolic calcium: 52 this is ultimately achieved by inhibiting the activity of myosin light chain phosphatase (MLCP). 55 There are many signaling mechanisms meditating calcium sensitization: notable examples include Rho kinase and protein kinase C signaling.55,78,83,85–87
Subarachnoid hemorrhage augments myogenic reactivity
Surprisingly, despite its fundamental role in CBF autoregulation,40,88 only a handful of investigations have assessed myogenic reactivity in SAH. Although few, these studies clearly demonstrate that SAH augments cerebral artery myogenic tone in mouse, 35 rat,89,90 rabbit91,92 and canine93,94 models. Intuitively, augmented myogenic reactivity must reduce CBF; however, the mechanistic basis for these augmented responses and its relationship to critical in vivo parameters (e.g. CBF and neuronal injury) have not been extensively explored.
In mouse olfactory cerebral resistance arteries, SAH augments myogenic tone in a time-dependent manner: the enhancement is maximal at two days post-SAH induction; it then declines (but does not fully resolve) by five days post-SAH induction. 35 Remarkably, this time course is consistent with vasospasm severity reported by Lin and coworkers 95 in a similar mouse model. Myogenic tone does not increase when saline is injected into the intracranial space, confirming that the augmented myogenic tone is specifically caused by the presence of blood in the intracranial space. The preservation of a normal phenylephrine dose–response relationship indicates that the augmentation is specific to myogenic mechanisms, rather than an increase in general contractility. 35
Assuming that the olfactory artery reasonably replicates effects within the distal microcirculation, we can predict the effect on CBF by using Poiseuille's law. To review Poiseuille's law: F = π ΔP r4 / 8 η L, where F is the flow, ΔP is the pressure differential, r is the vessel radius, η is the fluid viscosity, and L is the length. Using the myogenic tone measures collected by Yagi and coworkers (the calculations here use the myogenic tone measured at 60 mmHg in Figure 1A from Yagi and coworkers),
35
we calculate the flow effect as follows, defining the maximum possible flow as “1” (i.e., the flow at maximum diameter of “1”):
The myogenic mechanism underlies the failure of current therapeutic interventions. Shown are schematic tracings of myogenic responses and cerebral autoregulation, as would be observed in the cerebral microcirculation prior to and following subarachnoid hemorrhage (SAH). The myogenic response adjusts vascular resistance proportionally to pressure, resulting in constant flow over a wide range of perfusion pressures. In SAH, myogenic reactivity is augmented: autoregulation persists, but perfusion is reduced. The reduced perfusion in SAH results in secondary ischemic injury. Therapeutic intervention with vasodilators overrides the myogenic response, which compromises autoregulation. Perfusion increases, but pressure is transmitted to the delicate microcirculation, resulting in microvascular injury and edema formation. Therapeutic intervention with “Triple H Therapy” (hypertension, hypervolemia and hemodilution) confers minimal benefit since: (i) within the autoregulatory range, the myogenic response counteracts the pressure increase, resulting in no flow change; or (ii) beyond the autoregulatory range (“breakthrough” pressures), pressure is transmitted to the delicate microcirculation, resulting in microvascular injury and edema formation. Restoring normal myogenic reactivity is the most effective means to improve perfusion without secondary injury.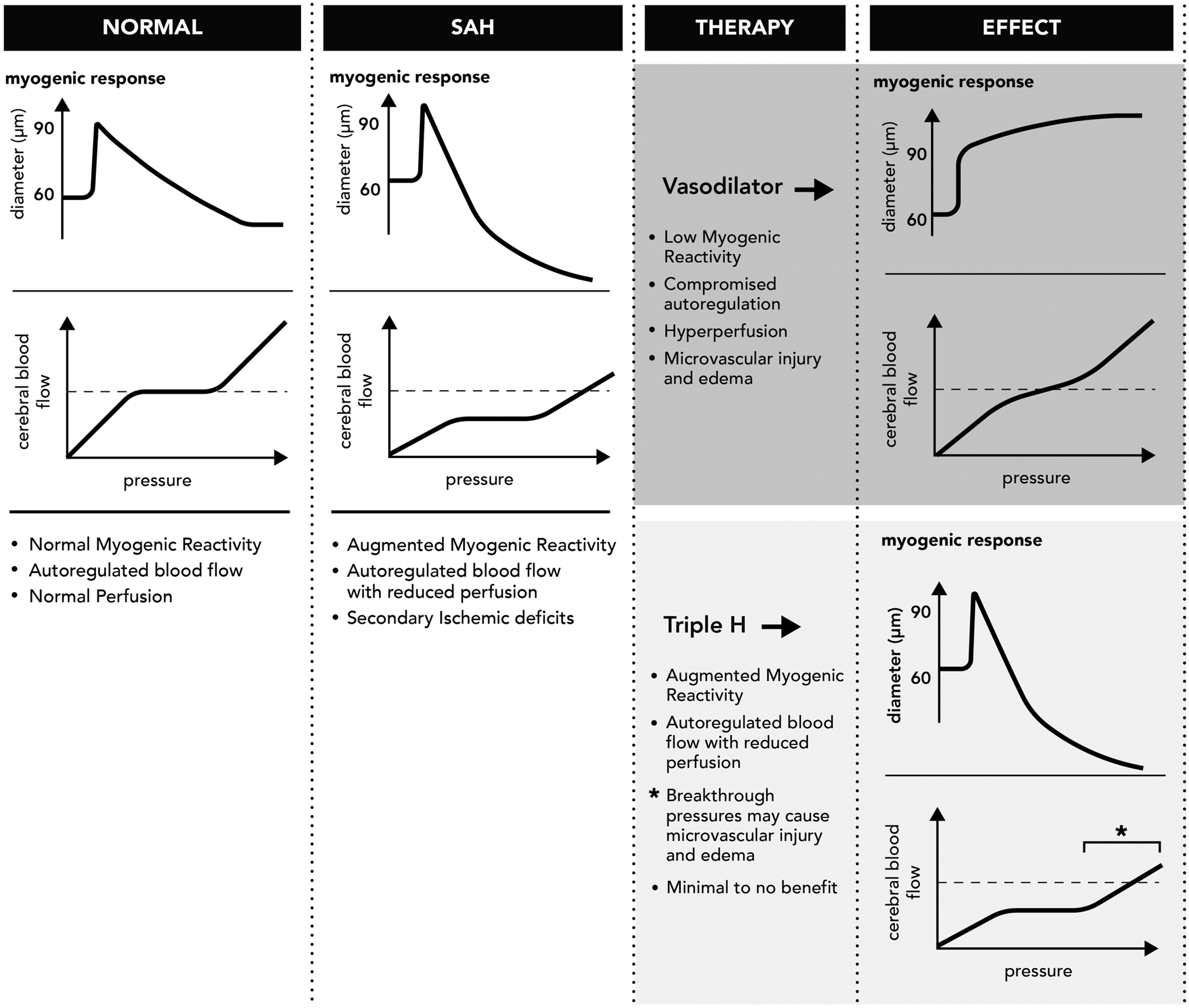
As predicted by the myogenic data, Yagi and coworkers observe that forebrain perfusion is, in fact, reduced by 56% in SAH 35. In addition to entirely explaining the CBF reduction, this calculation illustrates that relatively small changes in myogenic tone (i.e. from 13% to 29%) can cause serious perfusion impairment.
Setting perfusion thresholds that faithfully predict neuronal injury is extremely challenging. Ischemia is not defined by the single parameter of blood flow: rather, it represents the balance between blood supply and metabolic rate. Setting a singular threshold, therefore, ignores regional variation in metabolic need; by association, estimating a threshold from a single region does the same. Adding to this complexity, ischemic susceptibility varies across different species and even within different mouse strains.96,97 Defining a perfusion threshold for injury, therefore, is at best, an estimate.
Nevertheless, several experimental and clinical investigations have attempted to deduce a perfusion threshold, by assessing CBF in ischemic tissue (i.e. most often, the penumbra region of a focal infarct) and then discriminating the tissue that progresses to infarct (including the ischemic core) from the tissue that recovers. As a reasonable estimate, reducing CBF by >40% significantly risks permanent neuronal injury, with the severity proportional to both the degree and duration of hypoperfusion.98–101 Thus, the 56% CBF reduction measured by Yagi and coworkers is very likely to cause the neuronal apoptosis/degeneration and reduced neurofunctional scores observed at two days post-SAH induction. 35
In order to therapeutically correct exaggerated myogenic vasoconstriction in SAH, a significant investigative investment is required in order to understand the molecular basis for altered responses. However, several factors discourage this investment: the molecular mechanisms that control myogenic signaling are extraordinarily complex and display significant heterogeneity across different species and vascular beds; 56 the pressure myography assessment method is slow and labor intensive; and a high level of training is required to develop the necessary microsurgical skills. In the absence of the key mechanistic insight into how myogenic responsiveness changes in SAH, the current therapies utilized are generally ineffective and, in certain instances, are counterproductive.
Current therapeutic interventions in SAH
Impaired cerebral autoregulation is recognized as a strong independent predictor of both delayed cerebral ischemia 44 and negative outcome45,46 in SAH. This is not surprising, given that blood flow autoregulation is required to maintain the structural and functional integrity of the brain. However, correcting autoregulatory dysfunction necessitates the specific correction of perturbed myogenic mechanisms (Figure 1). In this regard, current therapeutic interventions are non-selective and attempt to improve cerebral perfusion by “forcibly increasing flow” (e.g. “Triple H” and hypertensive therapy), physically imposing dilation (e.g. angioplasty) or indiscriminately interfering with vasoconstrictor mechanisms (e.g. the calcium channel antagonist nimodipine) 102.
Triple-H Therapy, classically defined as hypertension, hypervolemia, and hemodilution, is intended to improve cerebral hemodynamic flow by increasing perfusion pressure, expanding intravascular volume, and decreasing blood viscosity.103,104 The rationale behind Triple H and hypertensive therapies is an assumption that cerebral vasospasm eliminates cerebral blood flow autoregulation.103,104 In this scenario, if the vasospasm cannot be therapeutically targeted, the only variables in Poiseuille's law that can be manipulated to improve perfusion are pressure (ΔP) and viscosity (η). However, the clear flaw in this rationale is that autoregulation, in fact, persists in SAH (i.e. increasing pressure stimulates vasoconstriction), with a shifted autoregulatory window that actively maintains CBF at an insufficient level.46,105,106 Thus, Triple-H therapy can be expected to increase CBF only at pressures higher than the autoregulatory window, a maneuver that risks microvascular damage to all microcirculatory beds exposed to systemic pressures.38,39 This presumably explains why Triple H therapy frequently fails to improve CBF or outcome in SAH (Figure 1).107–109 Despite the clear lack of objective evidence from controlled clinical trials that Triple-H therapy or any its separate components increase CBF, 109 current clinical guidelines 110 recommend treating delayed ischemia in SAH with hypertension, based primarily on anecdotal observations 110 and individual case reports.111,112 The methods and manner of administrating Triple-H therapy are highly variable 113 and the therapy can cause serious medical complications, including cardiopulmonary failure, renal dysfunction, exacerbation of cerebral edema, and the potential rupture of unsecured intracranial aneurysms.27,102,104 There is no consensus on how to administer the therapy, nor is there agreement on clinical endpoints that should guide the therapy. 113
Endovascular therapy (e.g. transluminal balloon angioplasty) may be employed prophylactically or as a “rescue procedure”, if severe angiographic vasospasm is quickly detected. 114 The therapeutic window is narrow 115 and the prospects of successful treatment are likely limited to the most severe cases, where proximal artery vasoconstriction reaches near-occlusive levels. For obvious reasons, endovascular therapy will not alleviate ischemia caused by distal cerebrovascular constriction: this may explain why prophylactic angioplasty reduces vasospasm, but generally fails to improve clinical outcome. 116 The evidence supporting angioplasty is sparse and no large, random controlled trials have been conducted.
Several Calcium Channel Blockers have been investigated as a therapeutic intervention in SAH, with nimodipine and nicardipine receiving the majority of attention.27,102 Nimodipine, a dihydropyridine that blocks L-type calcium channels, has purported selectivity for cerebral vascular smooth muscle and is the only medication approved by the FDA for treating “symptomatic vasospasm” (i.e. the symptoms of delayed cerebral ischemia in SAH). Orally administered nimodipine clearly improves clinical outcome;27,102,117–119 however, experimental 120 and clinical117,118,121,122 evidence indicate that the neurological improvement is not associated with a reversal of angiographic vasospasm or the normalization of CBF: the improved outcomes, therefore, are attributed to direct neuroprotection.123–126 Indeed, neurons express higher levels of L-type Ca2+ channels than vascular cells 127 and topical, intra-arterial nimodipine application at high doses appears to be the only means of reliably inducing vasodilation.128–130 Interestingly, antagonizing voltage-dependent calcium channels with magnesium fails to improve clinical outcome131,132 and clinical outcome results for nicardipine, an alternative dihydropyridine that blocks L-type calcium channels, have been mixed.133,134
The search for the mechanism responsible for augmented myogenic reactivity in subarachnoid hemorrhage
There is general agreement that the breakdown of blood in the subarachnoid space causes the delayed cerebral artery vasoconstriction and ischemia. 135 Several classic in vitro studies involving oxyhemoglobin and erythrocytes136–138 strongly support this premise, as does clinical evidence that delayed cerebral ischemia and vasospasm correlate with the volume of blood in the subarachnoid space.139,140 However, there are many ways that subarachnoid blood can elicit cerebral artery vasoconstriction: the most notable hypotheses involve nitric oxide scavenging,141,142 oxidant stress/free radical generation,141,143 the induction of inflammatory responses,141,144 and the stimulation of vasoconstrictor release (e.g. endothelin-1). 145 These proposed mechanisms are not mutually exclusive and they themselves elicit a myriad of pleiotropic effects. Thus, the task of selecting a single therapeutic target that will ameliorate ischemic injury in SAH is difficult, as exemplified by the dismal success rate of most new interventions in clinical trials. 146
As stated earlier, the most significant factor hindering the development of more precise therapeutic strategies to correct/prevent microvascular constriction is a distinct lack of mechanistic knowledge of how cerebral resistance artery reactivity changes in SAH. In fact, even within the limited number of reports utilizing isolated artery models, many of these investigations do not explore myogenic mechanisms per se (i.e. they do not directly address how SAH augments myogenic tone 91 or they study biomechanical/histological changes93,94). Nevertheless, several propositions, including endothelial dysfunction, reduced potassium channel activity, and increased 20-HETE signaling, are sound hypotheses: these mechanisms clearly modulate or drive myogenic reactivity and they appear to be profoundly perturbed in the pathological setting. Yet, several significant issues have hindered their development into therapeutic interventions: (i) although there are clear indications that SAH perturbs microvascular function,147,148 mechanisms promoting angiographic vasospasm (i.e. large artery vasoconstriction) have received an unbalanced share of clinical attention, as classic investigations149,150 concluded that vasospasm was the primary culprit responsible for DCI; (ii) there appears to be limited availability of clinically viable therapeutic interventions that modulate these pathways; and (iii) tested interventions have yet to be comprehensively studied with CBF and neuronal injury endpoints. To elaborate further, the next three subsections will briefly review the status of each of these proposed mechanisms.
Endothelial dysfunction in subarachnoid hemorrhage
SAH is known to profoundly alter endothelial function in cerebral arteries. 1 Since the endothelium constitutively releases vasodilating factors (e.g. endothelial derived relaxing and hyperpolarizing factors; EDRFs and EDHFs), it is reasonable to speculate that endothelial dysfunction augments myogenic reactivity. In this regard, there is ample evidence that SAH impairs endothelial-dependent vasodilator responses in both experimental151–154 and clinical155–157 settings. Whether these impaired responses are truly endothelial in origin, however, is controversial. While certain studies link attenuated endothelial responses to impaired vasodilator release,158,159 numerous others show that vasodilator deficits are, in fact, an inability of smooth muscle cells to transduce the endothelial signals.151–155These latter studies prompted two small clinical trials testing sildenafil, a cyclic guanosine monophosphate (cGMP)-specific phosphodiesterase type 5 (PDE5) inhibitor that prolongs endothelial-derived signals transduced by smooth muscle cells: their results suggest that sildenafil may alleviate angiographic vasospasm, 160 but does not improve brain perfusion. 161
With respect to myogenic reactivity, demonstrating that SAH alters vasodilator responsiveness is not, in and of itself, sufficient to implicate endothelial dysfunction as a causal mechanism augmenting myogenic vasoconstriction. To our knowledge, only one isolated resistance artery investigation has directly assessed the endothelial contribution to augmented myogenic reactivity in SAH. 89 In this study, SAH induced a myogenic tone augmentation that persisted following endothelial denudation, clearly demonstrating the existence of a smooth muscle cell mechanism. Since the degree of augmentation (i.e. the absolute tone difference between control and SAH) did not markedly diminish with denudation, this smooth muscle cell mechanism appears to be the predominant mechanism enhancing myogenic tone. 89 A second isolated resistance artery investigation has since provided indirect corroborating evidence, based on the preservation of phenylephrine dose-response sensitivity in SAH. 35 Although no direct assessments of endothelial contributions were made, the authors precluded an endothelial mechanism on the basis that compromised endothelial vasodilatory influences should similarly affect both myogenic reactivity and vasoconstrictor responses. 35 In fact, the preservation of “general contractility” observed in both studies (to potassium and phenylephrine, respectively)35,89 points to a specific effect on the myogenic mechanism. Consistent with this conclusion, “unpressurized” (5-10 mmHg) cerebral arteries from sham and SAH have similar membrane potentials and intracellular calcium levels, 89 while pressure-stimulated depolarization and calcium elevations are markedly enhanced in arterioles from SAH mice, relative to sham controls.89,92
Membrane potential and potassium channels in subarachnoid hemorrhage
Depolarization is a critical initiator of the myogenic response that activates voltage-sensitive calcium channels and leads to extracellular calcium influx. In this regard, potassium channels are prominent regulators of vascular tone, as only a handful of channels need to open in order to achieve meaningful changes in membrane potential.162,163 Opening potassium channels increases potassium conductance, causing potassium efflux and hyperpolarization (until the −85 mV equilibrium potential for potassium is reached). In cerebral arteries, voltage gated potassium channels (K v ) and large conductance calcium-activated potassium channels (BK) dominate potassium conductance in smooth muscle cells and play a key role in limiting depolarization. 163
Perturbed membrane potential regulation was one of the earliest hypotheses advanced to explain underlying basis of vascular constriction in SAH. Indeed, enhanced smooth muscle membrane potential depolarization clearly contributes to the augmented arteriolar tone observed in SAH.89,90,92,164,165 Several studies demonstrate that SAH reduces potassium currents in cerebral artery smooth muscle cells that, as expected, correlate with depolarization and enhanced constriction.90,164,165 Experiments using oxyhemoglobin to mimic the effect of the subarachnoid blood provide solid corroborating evidence.166–168 The precise channels affected, however, are less clear: Kv1.5, Kv2.1 and Kv2.2 transcripts and protein levels are reduced in SAH, 169 although functional measures suggest that Kv2 and Kv7 family members are the pertinent functional targets.164,170 The involvement of BK channels is contradictory: Koide and co-workers showed that SAH causes a distinct reduction in calcium spark-induced transient BK currents, 92 while Jahromi and co-workers found that virtually all BK channel characteristics are unaffected in SAH. 171
To our knowledge, Kv7 channels are the only K v channels that have been therapeutically targeted: Retigabine or Celecoxib both attenuate angiographic vasospasm in experimental SAH. 170 Neurological outcomes were not assessed in this study 170 ; however, both therapeutic entities are utilized clinically and Kv7 channels are known to regulate cerebral artery myogenic responses. 172 Thus, it will be intriguing to follow further developments in this field. One caveat to consider, however, is that potassium channel modulating therapeutics are unlikely to specifically target the pathological mechanism augmenting myogenic tone: these therapeutics will possess broad vasodilatory properties, and therefore likely suffer similar pitfalls as other general vasodilators. This may explain why calcitonin-gene-related peptide, a putative ATP-dependent potassium channel activator, 173 shows experimental promise, 174 but has yet to demonstrate a clear clinical benefit in small preliminary trials.175,176
20-HETE in subarachnoid hemorrhage
Arachidonic acid metabolites are well-established modulators of vascular tone. 177 The cytochrome P450 metabolite 20-hydroxyeicosatetraenoic acid (20-HETE) rose to prominence as a vasoconstrictor following the revelation that it plays a key role in hypertension. 178 Within the vascular wall, 20-HETE is synthesized predominantly by smooth muscle cells. 178 Many vascular beds, including the cerebral microcirculation, 179 utilize 20-HETE as a signaling molecule in smooth muscle contractility, proliferation, angiogenesis and inflammation.178,180 In cerebral arteries, 20-HETE potently stimulates vasoconstriction by promoting depolarization, calcium influx and the activation of calcium sensitization mechanisms, the latter of which notably involve protein kinase C (PKC) and Rho kinase activation.178,181,182 These vasoconstrictor mechanisms clearly overlap with known myogenic mechanisms, and thus, it is not surprising that 20-HETE is a prominent regulator of cerebral artery myogenic responsiveness.181,183,184
Observations that SAH stimulates fatty acid release185–187 fuelled speculation that 20-HETE could be a relevant metabolite that drives vasospasm and ischemic injury. In accordance with this hypothesis, cerebrospinal 20-HETE levels are elevated in SAH patients.188–190 However, its predictive value for putative vascular endpoints is rather poor. Specifically, while cerebrospinal 20-HETE clearly associates with neurological deterioration, its relationship to angiographic vasospasm, delayed cerebral ischemia, and infarction is weak.189,190 Siler and co-workers observed that elevated cerebrospinal 20-HETE levels measured within the first 96 h of admission predicts delayed cerebral ischemia; however, 20-HETE levels fall and are no longer predictive at the time when the ischemia actually occurs. 191 While cerebrospinal 20-HETE levels may be an appropriate biomarker to identify patients at high risk for unfavorable neurological outcomes, it remains unclear whether the 20-HETE profiles for cerebrospinal fluid and the vascular wall align. This could explain the discrepancy between these clinical results that, at best, weakly correlate 20-HETE to DCI and the strong experimental evidence demonstrating that 20-HETE synthesis blockade improves vasospasm and CBF reductions in SAH.184,192–194 Currently, there is paucity in experimental studies examining 20-HETE as a therapeutic target in SAH: directly assessing whether 20-HETE augments myogenic reactivity in SAH, therefore, represents a key priority.
Tumor necrosis factor augments myogenic reactivity in subarachnoid hemorrhage
Recently, Yagi and co-workers have made significant advancements in our molecular understanding for how myogenic reactivity changes in SAH.
35
Their study built on mechanistic insight gained from an etiologically distinct model of heart failure, where tumor necrosis factor (TNF) augments cerebral artery myogenic tone and disrupts CBF autoregulation.195,196 Briefly, previous work by this group demonstrates that myocardial infarction induces pathological vascular smooth muscle cell TNF expression,
196
which down-regulates the cystic fibrosis transmembrane conductance regulator (CFTR) by an autocrine/paracrine mechanism.
195
As a critical transporter involved in smooth muscle cell sphingosine-1-phosphate (S1P) degradation,
195
CFTR down-regulation enhances S1P signaling and pro-constrictive effects in the context of myogenic vasoconstriction.195,196 A composite of this signaling mechanism, which has been constructed over decades,35,39,52,56,85,195–200 is presented in Figure 2.
Current and proposed molecular mechanisms augmenting myogenic reactivity in subarachnoid hemorrhage. A mechanosensitive complex localized to the plasma membrane initiates the myogenic response, resulting in membrane potential depolarization, calcium entry and extracellular signal-regulated kinase 1 and 2 (ERK1/2) activation. ERK1/2 phosphorylates sphingosine kinase 1 (Sphk1), eliciting its translocation to the plasma membrane and synthesis of sphingosine-1-phosphate (S1P) from sphingosine. Intracellular S1P modulates pressure-stimulated calcium elevation, which activates calmodulin (CAM), myosin light chain kinase (MLCK) and the contraction apparatus. Extracellular S1P activates the S1P2 receptor subtype (S1P2R) to activate the RhoA/Rho kinase (ROCK) signaling pathway, which inhibits myosin light chain phosphatase (MLCP). Inhibiting MLCP enhances MLCK's activation of the contractile apparatus. Extracellular S1P is sequestered from S1P2R by the cystic fibrosis transmembrane conductance regulator (CFTR), which transports S1P across the plasma membrane for dephosphorylation by S1P phosphohydrolase 1 (SPP1). In subarachnoid hemorrhage (SAH), the induction of inflammatory tumor necrosis factor (TNF) signaling curtails CFTR gene expression (not shown) and accelerates CFTR degradation mechanisms: this results in higher S1P bioavailability for S1P2R signaling and increases activation of the contractile apparatus. In experimental settings, preventing TNF signaling (etanercept; ETN) or antagonizing S1P2R signaling (JTE013) successfully blocks this pathological process. Therapeutics that increases CFTR expression (Lumacaftor) or activity (Ivacaftor) would be expected to have similar efficacy in terms of blocking the pathological enhancement of S1P/S1P2R signaling: their value as a therapeutic intervention awaits experimental validation.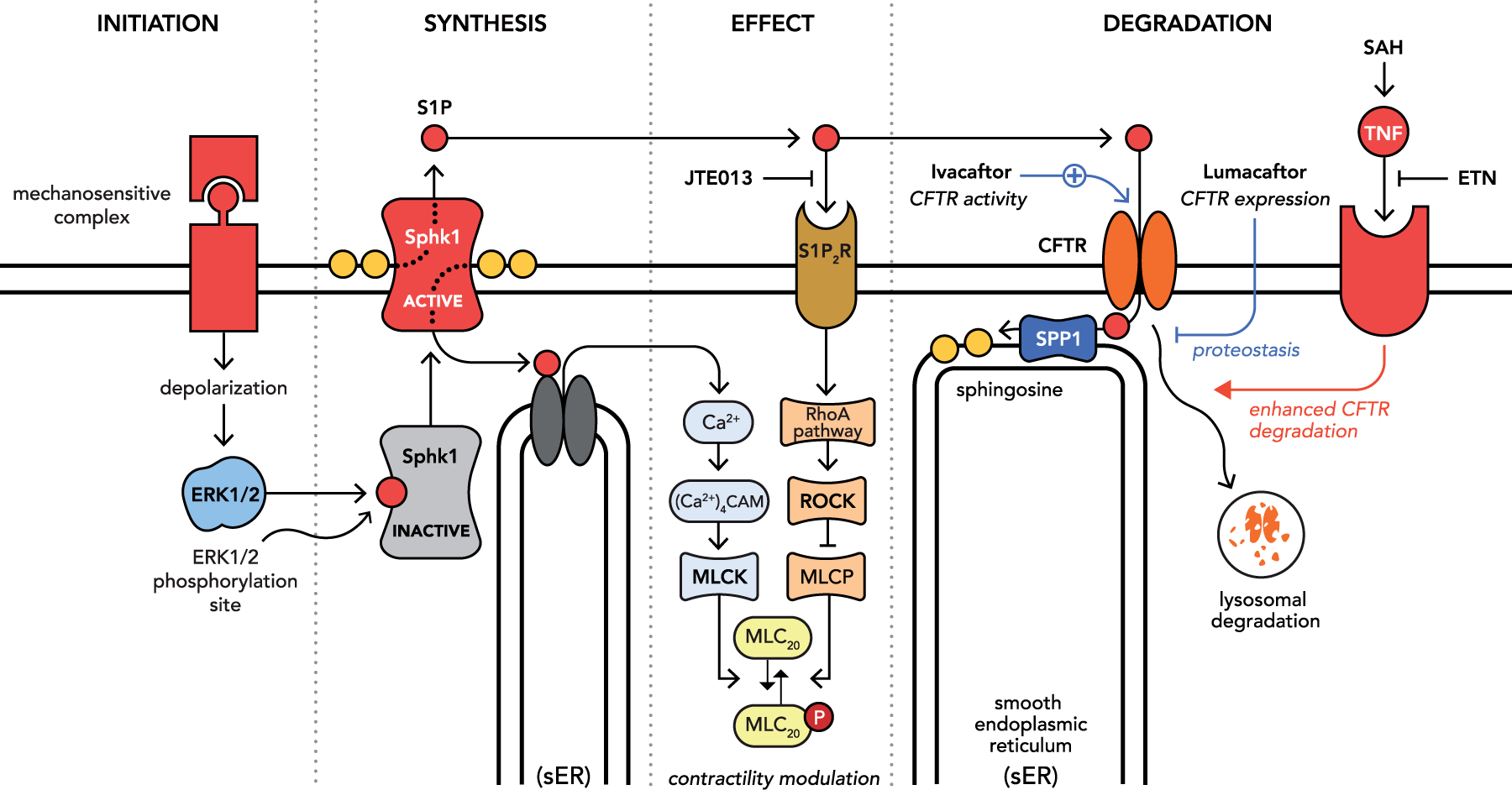
As a framework for how cerebrovascular reactivity changes in SAH, this mechanism is attractive for two key reasons: first, inflammation undoubtedly occurs in SAH 144 and thus it is conceivable that heart failure and SAH elicit the same functional effect on cerebral artery myogenic tone. Indeed, TNF levels peak in a timeframe consistent with delayed cerebral ischemia in SAH201,202 and associate with angiographic vasospasm, abnormal cerebral flow velocities, and poor clinical outcome.203–206 Second, S1P is a potent regulator of cerebral artery tone35,195,196,207–211 and prominently regulates and coordinates pressure-dependent calcium responses,56,85,197 putatively providing the upstream link to the pressure-stimulated depolarization and calcium elevations mentioned above.89,90,92,212
Consistent with the heart failure model, Yagi and co-workers observed that SAH induces robust vascular TNF expression in olfactory cerebral arteries, predominantly localized to the smooth muscle cells within the microvascular wall. 35 Using distinct inhibitory strategies, including smooth muscle cell-targeted TNF gene deletion, Yagi and co-workers conclusively demonstrated that smooth muscle cell TNF expression drives the SAH-mediated enhancement of myogenic vasoconstriction. 35 Therapeutically sequestering TNF (etanercept) eliminates the SAH-mediated myogenic tone augmentation, normalizes cerebral perfusion, reduces neuronal injury and prevents neurofunctional deficits. 35 These latter observations have since been independently corroborated: etanercept reduces apoptosis and improves neurological scores in a rat model of SAH. 213 Also consistent with the heart failure mechanism,195,196 antagonizing S1P receptors (JTE013) has the same therapeutic benefits as sequestering TNF: normalization of myogenic tone, attenuated neuronal injury, and the prevention of neurofunctional deficits. 35 Again, recent reports independently corroborate these therapeutic observations: fingolimod, a functional S1P receptor antagonist, 214 (i) preserves vascular reactivity and improves neurofunctional outcome in a rat SAH model 215 and (ii) improves clinical outcome in a similar type of hemorrhagic stroke (supratentorial intracerebral hemorrhage). 216
Two significant features of this work should not be overlooked. First, both therapeutic interventions (etanercept and JTE013) restricted their effect to the SAH-mediated enhancement myogenic tone: 35 neither intervention reduced myogenic tone below the sham level; they had no effect in sham mice; nor did they affect phenylephrine responses, indicating that general contractility was not compromised. This specificity confers clear advantages over conventional therapies that non-specifically blunt vasoconstriction and/or disrupt autoregulation. Second, both interventions affected all endpoints similarly, 35 strengthening the intuitive concept that myogenic tone, CBF, and neuronal injury are causally linked, rather than simple correlations.
The next frontier – CFTR as a vascular regulator
Although Yagi and co-workers conclude that anti-TNF therapy can be used to improve neurological outcome in SAH, 35 the immune system suppression associated with the intervention may further threaten these critically ill patients by increasing their risk of infection. 217 Similarly, S1P signaling is central to many cellular processes (e.g. endothelial barrier function218,219): S1P receptor antagonism, 220 therefore, may carry a significant risk of secondary effects. Of the therapeutic targets their proposed mechanistic concept identifies (Figure 2),35,195 the cystic fibrosis transmembrane conductance regulator (CFTR) emerges as an attractive and logical choice for correcting myogenic reactivity within the cerebral microcirculation for three key reasons: (i) it is the proposed mechanistic basis for TNF's tone-enhancing effect, 195 (ii) FDA-approved therapeutics that augment CFTR activity are available221,222 and (iii) CFTR therapeutics are not known to compromise immune function and likely have fewer secondary effects than S1P receptor antagonism.
CFTR is classically known for its critical role in lung epithelial chloride secretion; 223 however, as an ATP-binding cassette (ABC) transporter that functions as an ion channel, it possesses the undeniably unique feature that it consumes energy mediating the passive diffusion of chloride ions. 224 In many cells, the equilibrium potential for chloride is similar to the membrane potential: activating or inhibiting CFTR activity, therefore, does not induce significant chloride flux or change membrane potential. However, as reviewed by Bulley and Jaggar, smooth muscle cells possess high intracellular chloride levels, and the equilibrium potential for chloride is estimated to fall between -30 and -20 mV. 225 Here, opening CFTR channels will result in chloride efflux, except when the smooth muscle cells are already highly depolarized. Although chloride efflux should have a depolarizing effect, changing membrane potential will influence other channels involved in membrane potential maintenance (e.g. inward rectifier and voltage dependent potassium channels). Thus, it is not straightforward to predict how changes in CFTR activity impact membrane potential. Since CFTR's unitary conductance is relatively low (∼10 pS), 226 other ion channels (both cation and anion) likely dominate the control of membrane potential. This presumably permits CFTR's chloride independent functions (e.g. transporter activities) to have a more pronounced role in modulating vascular reactivity. In this regard, CFTR regulates the transport of several charged molecules,227–229 including S1P.195,230 Specifically, CFTR constitutively transports extracellular S1P across the plasma membrane and into the intracellular compartment. 195 This process diverts extracellular S1P from interacting with cell surface receptors, forming a rapid and effective basis to regulate S1P receptor activity.195,230 Increasing CFTR-mediated S1P transport sequesters S1P from its pro-constrictive S1P2 receptors, 195 which attenuates vasoconstriction
In both olfactory 35 and posterior 195 cerebral arteries, inhibiting CFTR activity (e.g. via gene deletion or inhibitory compounds) augments myogenic tone; this augmentation can be eliminated by S1P receptor antagonism (JTE013). These effects are consistent with CFTR's role in transporting/sequestering S1P, 195 as opposed to changes in chloride efflux. The loss of CFTR function does not alter phenylephrine responses,35,195 indicating restricted effects on myogenic mechanisms, rather than effects on general contractility. Protein expression analyses confirm that SAH down-regulates CFTR protein expression by a TNF-dependent mechanism. 35 The strong correlation between CFTR expression and cerebral artery myogenic tone35,195 drives the proposition that CFTR therapeutics may confer benefits in SAH treatment.
As a caveat, we must note that CFTR is also expressed in endothelial231,232 and neuronal233,234 cells, with functions relevant to glycocalyx swelling (endothelial), 231 vesicular transport (neuronal) 235 and neuronal excitability. 236 The presence of CFTR in these cell types raises the prospect that CFTR therapeutics could confer vascular reactivity and/or neuroprotective benefits in addition to the smooth muscle cell localized effect on S1P signaling. Thus, should CFTR therapeutics prove beneficial in SAH, follow-up studies utilizing cell-specific gene deletion models will be required to mechanistically localize the site of action. An obvious initial choice would be a smooth muscle cell-specific gene deletion model, which successfully characterized smooth muscle cell TNF as the relevant TNF pool augmenting myogenic reactivity in SAH. 35
Therapeutic strategies to restore deficient CFTR function
The incidence of cystic fibrosis in Western society has fuelled substantial CFTR research and therapeutic agent development. In general, mutations to CFTR alter cell surface expression, channel gating properties, or both. At present, there is insufficient information to rationally design chemical therapeutics: therefore, high throughput screening, a reliable, sensitive and cost-effective assay to screen libraries of drug-like compounds, is the strategy of choice to identify potential CFTR therapeutics. 237 High throughput screens have identified several compounds that reliably enhance CFTR function, although in many cases, the precise mechanisms of action have yet to be elucidated. 237
CFTR therapeutics is broadly segregated into two distinct classes: “CFTR potentiators” and “CFTR correctors”. Potentiators (e.g. Ivacaftor/Kalydeco®) enhance channel gating, 238 and thus only increase CFTR activity when the channels are present at cell membrane. Consequently, CFTR potentiators display limited efficacy when CFTR abundance is very low. 239 In contrast to potentiators, correctors interact with CFTR motifs and/or CFTR interacting proteins that orchestrate and control the processing of CFTR, its delivery to and expression at the apical membrane. 240 This therapeutic strategy is termed “proteostasis,” 241 because the aim is to treat cystic fibrosis by manipulating the stability and location of CFTR. Lumacaftor (VX-809) 222 is the best-characterized corrector compound to date: the FDA recently approved Lumacaftor/Ivacaftor combination therapy (Orkambi™) for treating cystic fibrosis stemming from the ΔF508 mutation (which profoundly disrupts CFTR trafficking and cell surface expression). 242 Combining potentiators and correctors (i.e. Orkambi) is an obvious pharmacological strategy that would maximally boost CFTR activity; however, to be viable as an SAH treatment, the respective entities would have to exert effects on non-mutant CFTR, since the wild-type form will be present in the vast majority of patients. Fortunately, both ivacaftor 226 and lumacaftor 243 exert their respective activity and expression effects on wild-type CFTR.
The availability of CFTR therapeutics that increase wild-type CFTR activity/expression presents an opportunity to repurpose the FDA-approved pharmaceutical Orkambi for SAH therapy. Without doubt, it would be premature to initiate clinical trials prior to undertaking the experimental animal studies. However, in anticipation that CFTR therapeutics will work experimentally, as the strong correlation between CFTR expression and vascular reactivity implies, 35 it is tempting to briefly comment on the limited clinical data available with respect to the safety of CFTR therapeutics in patients who do not have cystic fibrosis. At present, clinical trial data for lumacaftor, ivacaftor, and lumacaftor/ivacaftor in healthy subjects are proprietary and therefore, not publicly available. NIH clinical trial records indicate that initial safety studies in healthy subjects were completed in 2010 (NCT00966602) and 2012 (NCT01216046) and included the analyses of laboratory values (clinical chemistry, hematology, coagulation, and urinalysis), electrocardiograms, and vital signs. The fact that four clinical trials involving healthy subjects were subsequently completed (NCT01910415, NCT01768663, NCT01888393 NCT01899105) and a fifth is now underway (NCT02310789) is a strong indication that no serious adverse events have been encountered. It is therefore reasonable to assume that CFTR therapeutics do not pose an overt safety risk to patients with normal CFTR.
Summary
Summary of proposed mechanisms augmenting microvascular constriction in subarachnoid hemorrhage.
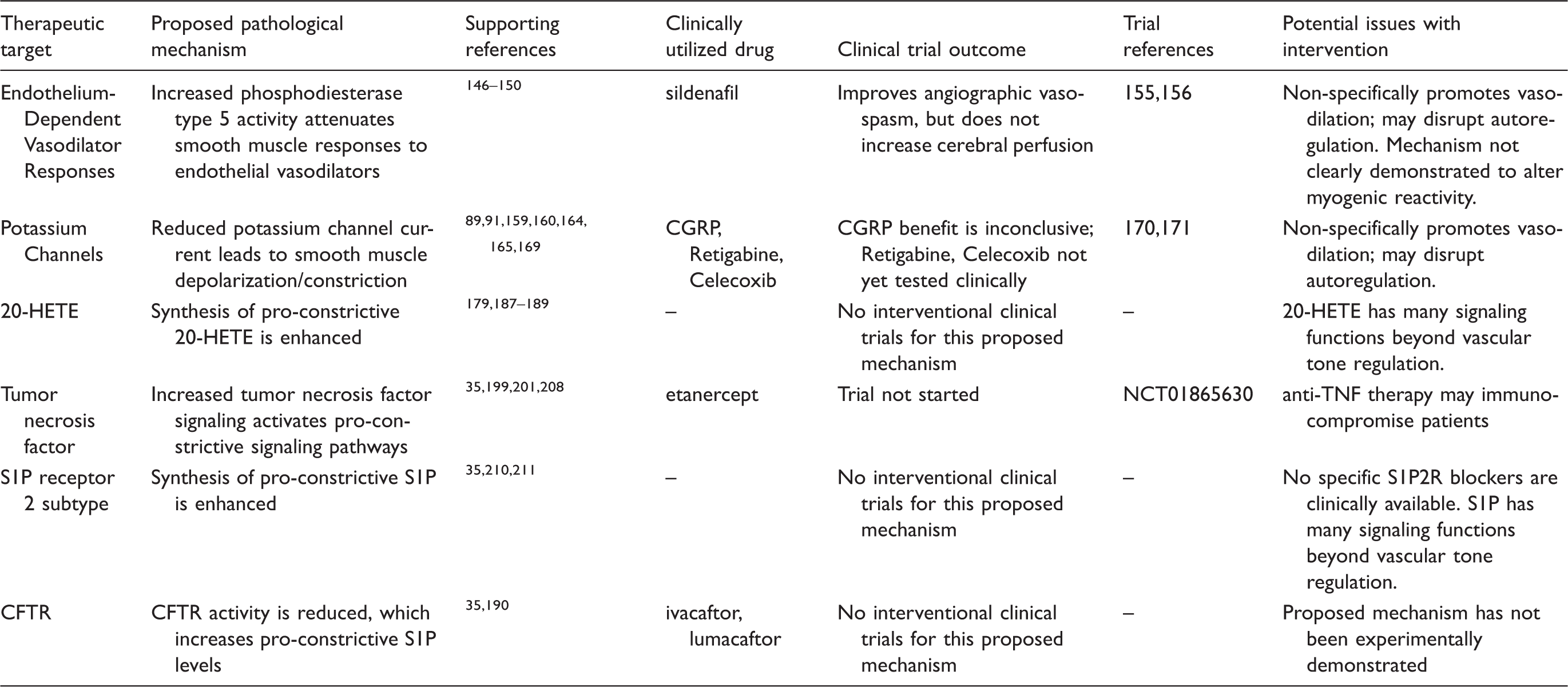
20-HETE: 20 hydroxyeicosatetraenoic acid; CGRP: calcitonin-gene-related peptide; S1P: sphingosine-1-phosphate; S1P2R: S1P receptor 2 subtype; CFTR: cystic fibrosis transmembrane conductance regulator.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We gratefully acknowledge funding support from a Heart and Stroke Foundation of Canada Grant-in-Aid (StSB; G16-00014175), a Heart and Stroke Foundation of Canada Career Investigator Award (StSB CI-7432) and the Brain Aneurysm Foundation Northwell Health – North Shore University Hospital Brain Aneurysm Centre Chair of Research Award (StSB).
Acknowledgement
The authors thank Alexandra Erin Papaelias for graphic design.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
All three authors (DL, JTK and StSB) significantly contributed to the conceptualization of this review and the writing/revision process.