
Abstract
Astrocytes express neurotransmitter receptors that serve as sensors of synaptic activity and initiate signals leading to activity-dependent local vasodilation and increases in blood flow. We previously showed that arteriolar vasodilation produced by activation of cortical astrocytes is dependent on endothelial nitric oxide synthase (eNOS) and endogenous agonists of N-methyl-D-aspartate (NMDA) receptors. Here, we tested the hypothesis that these effects are mediated by NMDA receptors expressed by brain endothelial cells. Primary endothelial cultures expressed NMDA receptor subunits and produced nitric oxide in response to co-agonists, glutamate and D-serine. In cerebral cortex in situ, immunoelectron microscopy revealed that endothelial cells express the GluN1 NMDA receptor subunit at basolateral membrane surfaces in an orientation suitable for receiving intercellular messengers from brain cells. In cortical slices, activation of astrocytes by two-photon flash photolysis of a caged Ca2+ compound or application of a metabotropic glutamate receptor agonist caused endothelial NO generation and local vasodilation. These effects were mitigated by NMDA receptor antagonists and conditional gene silencing of endothelial GluN1, indicating at least partial dependence on endothelial NMDA receptors. Our observations identify a novel astrocyte-endothelial vasodilatory signaling axis that could contribute to endothelium-dependent vasodilation in brain functional hyperemia.
Introduction
The brain is only 2% of body weight but receives 10% of cardiac output and consumes 20% of blood glucose and oxygen. 1 High energy demand and limited energy reserves create a critically important need for mechanisms that maintain organ perfusion and regulate regional blood supply to support dynamic fluctuations in metabolic demand. Autoregulation ensures that brain blood flow is maintained despite changes in blood pressure. Functional hyperemia directs regional brain blood flow to areas of greatest need. 2 Functional hyperemia is driven by neurovascular coupling, which is intercellular signaling among cells of the neurovascular unit in response to synaptic activity that results in local arteriole or capillary lumen diameter changes. The neurovascular unit is composed of cells at the brain–vascular interface, including neurons, astrocytes, endothelial cells, smooth muscle cells, and pericytes, as well as extracellular matrix and basement membrane. 3 Neurovascular coupling in the cerebral cortex is in part mediated by direct intrinsic innervation by interneurons. 4 In addition, astrocytes sense synaptic activity and respond by releasing factors that influence cerebrovascular lumen diameter. The excitatory neurotransmitter glutamate stimulates astrocyte-dependent and independent neurovascular coupling. By activating N-methyl-D-aspartate (NMDA) receptors expressed by interneurons, glutamate can enhance neuronal nitric oxide synthase (nNOS) activity and vasodilatory nitric oxide (NO) generation.4–6 Cortical glutamate can also activate astrocytic glutamate receptors, leading to enhanced intracellular Ca2+ levels and release of vasomotor effectors. 7 Prostaglandin E2 has been shown to cause vasodilation mediated by hyperemic astrocytic signaling.8–13 There is also evidence for other mediators, including K+, glutamate, D-serine, and carbon monoxide.11,14,15
Brain hyperemia consists of a coordinated series of vasomotor events, including dilation of local penetrating arterioles and capillaries, upstream pial vasodilation to increase zonal blood flow, and diversion of blood supply away from areas with ample supply of energy substrates or low activity. 16 Focal neurovascular coupling leads to distal effects by vasodilatory conduction, 17 which is a process dependent on an intact brain endothelial syncytium. 18 Despite evidence that the endothelium is an important contributor to brain hyperemia, the nature of how synaptic and/or astrocytic signals reach the endothelium in neurovascular coupling remains unknown. One possibility is that perivascular astrocytes release substances that activate receptors localized to proximal basolateral endothelial membranes, leading to endothelium-dependent vasodilation. However, direct astrocyte-endothelial vasodilatory signaling is yet to be identified.
NMDA receptors are heterotetrameric transmembrane cation channels that have been extensively characterized in neurons. Subunits are drawn from a pool of seven distinct gene products (GluN1, GluN2A-D, and GluN3A and B), 19 and resulting complexes are essential for several critical central nervous system functions, including functional hyperemia linked to nNOS activation.5,20 GluN1 is an obligatory NMDA receptor subunit essential for receptor assembly, trafficking of other subunits and functional activity. 21 Activation of most NMDA receptor channels requires binding of glutamate to GluN2 and co-agonist, glycine or D-serine, to GluN1. 19 But while there is clear dominant recognition of NMDA receptors as neuronal in nature, they have also been identified in a broad range of other cells and tissues,22,23 including endothelial cells.24–28 Endothelial NMDA receptors are likely important regulators of blood–brain barrier function,24,29,30 oxidative stress, mitochondrial dysfunction28,31 and NO generation. 25 They may also contribute to ensuring adequate energy substrate delivery from blood to brain cells by regulating glucose transport. 32 We 33 and others34,35 have demonstrated that NMDA receptor agonists dilate isolated arteries free of neural circuitry by activating eNOS. This raises the possibility that NMDA receptors expressed by the cerebrovascular endothelium could mediate vasodilation directly. The objective of the current study was to determine whether astrocytes communicate directly with the cerebrovascular endothelium to produce eNOS activity and cortical vasodilation dependent on endothelial NMDA receptors.
Materials and methods
Chemicals and animals
All chemicals were purchased from Sigma-Aldrich, unless otherwise noted. Procedures involving live animals were approved by the University of Manitoba Animal Care Committee in compliance with the policies of the Canadian Council on Animal Care. All experiments are reported in compliance with ARRIVE guidelines for how to report animal experiments. Conditional silencing of endothelial GluN1 was approached by breeding mice containing dual grin1 alleles flanked by loxP excision sites (grin1fl/fl; JAX 005246), with mice expressing Cre-recombinase driven by the endothelial promoter Tie-2 (Tek-cre; JAX 008863). Grin1fl/fl mice expressing Cre-recombinase (grin1fl/fl · Cre+/−) have reduced endothelial GluN1 expression, while littermate grin1fl/fl mice lacking Cre-recombinase (grin1fl/fl · Cre−/−) were used as controls. Animals were housed in pairs with a 12-h light/dark cycle and ad libitum access to water and standard chow.
Brain endothelial cell cultures
Brains from neonatal (14–21 days) male and female mice were digested with 0.05% collagenase, re-suspended in 17% dextran and centrifuged at 10,000 × g (30 min, 4℃). Isolated cells were collected on glass beads and cultured on Type I collagen-coated culture dishes in Dulbecco’s Modified Eagle Medium containing 10% fetal bovine serum, 2 mM L-glutamine, 1% penicillin-streptomycin, 40 µg/ml heparin and 150 µg/ml endothelial cell growth supplement. Cells were supplemented with 4 µg/ml puromycin for the first 2.5 days, and passaged twice before reaching confluence.
Western blot
Cells or tissues were homogenized in RIPA lysis buffer (50 mM Tris-base, 150 mM NaCl, 1% Nonidet P-40, 1 mM EDTA, 0.25% Na-deoxycholate with protease inhibitors; pH 7.4) and protein concentrations determined using a bicinchoninic acid assay (GE HealthCare). Proteins were separated using sodium dodecylsulfate (SDS)-polyacrylamide (7.5%) gel electrophoresis and transferred to polyvinylidene difluoride membranes. Membranes were exposed to a blocking solution of 5% non-fat powdered milk in TBS with 0.1% Tween-20. Primary antibodies were applied for 12 h (4℃) and included goat anti-GluN1 (1:300, Santa Cruz Biotechnologies), rabbit anti-GluN1 (1:300, Millipore) and goat anti-GluN2C (1:300; Santa Cruz Biotechnologies). Donkey anti-goat or anti-rabbit IgG secondary antibodies, conjugated to horseradish peroxidase (1:1000, Cell Signaling) were applied for 1 h (room temperature), and immunoreactive products visualized using the ECL Plus chemiluminescence kit (GE Life Sciences). Densitometry was performed using ImageJ software (NIH). Values were normalized to protein loading markers.
Immunoprecipitation
Cultured cells were washed with phosphate-buffered saline (PBS) and lysed in RIPA buffer at 4℃ (30 min). Lysates (0.5–1.0 mg) were incubated with 2 µg anti-GluN1 or anti-GluN2C antibodies (Santa Cruz Biotechnologies) at 4℃ (12 h). Protein A/G agarose beads (Thermo Scientific) were added and mixed gently at 4℃ (4 hours). Beads were then precipitated (2500 × g for 3 min) and washed with RIPA buffer. Complexes were then eluted with 50 µl RIPA buffer containing 2.5% SDS and boiled at 95℃ (30 min). Proteins were resolved using SDS-polyacrylamide gel electrophoresis and analyzed by Western blot.
Immunogold electron microscopy
Immunoelectron microscopy was performed as described previously. 36 Neonatal (14–21 day old) mice were anesthetized with ketamine (150 mg/kg) and xylazine (15 mg/kg), and perfused through the left ventricle with PBS, followed by 4% paraformaldehyde (PFA). Cortical volumes (1 mm3) were cryoprotected in sequentially higher glycerol concentrations (10–30% wt./vol), and plunge-frozen in liquid ethane (−180℃). Tissue was fixed en bloc with 1.5% uranyl acetate in methanol (−90℃, 30 h), and methanol progressively replaced by HM-20 embedding resin (Electron Microscopy Sciences) over 48 h (−45℃). Resin was then UV-polymerized for 48 h, and ultrathin sections were cut and collected on formvar-coated grids. Grids were etched with a solution of 0.1% sodium borohydride, followed by 50 mM glycine, washed in TBST (50 mM Tris-base, 150 mM NaCl, 0.1% Triton-X; pH 7.4), and blocked with 2% (wt./vol) bovine serum albumin in TBST. Grids were incubated overnight with primary antibodies (rabbit anti-GluN1, Cell Signaling; mouse anti-PSD-95, BD Biosciences), washed with TBST and incubated for 2 h with gold bead-conjugated secondary antibodies (anti-mouse, 15 nm; anti-rabbit 10 nm; Aurion). Grids were lightly counterstained with 2% (wt./vol) uranyl acetate and Reynold’s lead. GluN1-labeled gold particles within 30 nm of either cell membrane were considered to be associated with that structure.
Measurement of intracellular NO production
Endothelial cells were treated with glutamate and D-serine for 5 h, washed with modified Hanks-Buffered Saline Solution (HBSS; 140 mM NaCl, 3 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM Glucose, 20 mM HEPES) and loaded with the NO fluorescent indicator, 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM, 4 µM; Invitrogen) for 50 min (37℃). DAF signal was assessed using a Zeiss laser scanning microscope with 20× objective and an argon laser delivering an excitation wavelength of 495 nm. Emitted signal (515 nm) was captured and analyzed using ImageJ. NO production was expressed as relative fluorescence (F/F0) of DAF-FM, where F is fluorescence intensity of treatment groups and F0 is baseline fluorescence intensity of untreated cells.
Quantitative PCR
Total RNA was isolated using the TRIzol Reagent (ThermoFisher Scientific), and analyzed using the iScript One-Step RT-PCR Kit with SYBR Green dye (Bio-Rad) and an Applied Biosystems 7300 RT-PCR system. β-actin (internal control) and GluN1 were amplified using the following primers: β-actin forward, 5′-GGGCTATGCTCTCCCTCACG-3′; β-actin reverse, 5′-GTCACGCACGATTTCCCTCTC-3′; GluN1 forward, 5′-GTGCCAAACTTGCCATCTGC-3′; GluN1 reverse, 5′-CGGGGCCTAATGACACATCC-3′. PCR parameters were as follows: 50℃ for 10 min, 95℃ for 1 min, followed by 50 cycles at 95℃ for 15 s and 60℃ for 60 s. Relative amount of target gene mRNA was normalized to β-actin mRNA. Standard curves were generated and the relative amount of target gene mRNA was normalized to β-actin mRNA. Specificity was verified by melt curve analysis.
Brain slices and two-photon laser scanning microscopy
Brains from day 14–19 postnatal male and female mice were placed in ice-cold cutting buffer (2.5 mM KCl, 1.25 mM NaH2PO4, 10 mM MgSO4, 5 mM CaCl2, 26 mM NaHCO3, 10 mM glucose, 230 mM sucrose) with 95% O2 and 5% CO2. Slices (350 µm) were cut using a vibrating blade, and maintained in artificial cerebrospinal fluid (aCSF: 126 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 2 mM MgCl2, 2 mM CaCl2, 26 mM NaHCO3, 10 mM glucose) equilibrated with 95% O2 (35℃). Slices were treated with the Ca2+ indicator dye, rhodamine-2 AM (rhod-2, 10 µM; Invitrogen), with or without the caged Ca2+ compound, o-nitrophenyl-EGTA AM (NP-EGTA, 10 µM; Invitrogen) and/or Griffonia simplicifolia 1 Isolectin B4 tagged with Alexa Fluor 488 (5 µg/ml; Invitrogen) for 1 h before imaging (37℃). In some experiments, DAF-FM diacetate (25 µM, Invitrogen) was bath-applied in aCSF for 30 min prior to rhod-2 exposure in addition to the rhod-2 incubation period. For imaging, slices were superfused in open chambers with aCSF equilibrated with 20% O2, 5% CO2, and 75% N2. Slices were imaged using a two-photon laser scanning microscope (TPLSM) with excitation wavelength of 800 nm delivered by a Ti-sapphire laser (Coherent) through an Ultima multiphoton scan head with dual galvanometers for imaging and uncaging (Bruker). Emitted signal was collected by photomultiplier tubes at 488 nm (DAF-FM and isolectin) or 580 nm (rhod-2).
In selected experiments, glutamate and D-serine (1 mM each, 0.2 µl) were delivered by 40 ms picospritzer pressure ejection (10 kPa) from glass micropipettes to discrete areas of cortical penetrating arterioles. Micropipette tips with an internal tip diameter of 1–10 µm were positioned 10 µm from arteriolar walls. For astrocytic Ca2+ uncaging, functional mapping software (TriggerSync, Bruker) was used to guide a 700 nm/200 ms laser pulse through a voltage-controlled pockels cell (laser power 15–20 mW). Images were analyzed every 15 s using Prairie View software to determine relative changes in lumen diameter, marked by isolectin staining and DODT laser contrast images. All vasodilatory responses were assessed with no agent provided to enhance baseline smooth muscle tone. Responses were similar to those observed in slices pre-treated with 2 µM norepinephrine (Supplemental Material).
PGE2 and 20-HETE quantification
Acute brain slices were incubated in aCSF with 20% O2/5% CO2 and exposed to tACPD (100 µM, Tocris Bioscience) or glutamate with D-serine (10 µM each) for 5 min. 11 PGE2 release over 5 min was measured using a PGE2 enzyme immunoassay Kit (Cayman). Tissue 20-HETE content was quantified using a 20-HETE ELISA Kit (Detroit R&D).
Statistics
All data are presented as means ± standard error of the mean (SEM). Statistical analysis was performed throughout using GraphPad Prism version 5.0. Unpaired, two-tailed t-tests were used to compare two groups with a single variable. One-way ANOVA with Newman–Keuls post hoc test was used to compare multiple groups. Area under curve (AUC) was calculated from 5-min time course assessments for vessel diameter changes, rhod-2 signal and DAF-FM signal. AUCs were compared using t-tests for two groups. Projected sample sizes were calculated assuming a standard deviation of 15% and a minimum detectable mean difference of 30%. Acceptable alpha error was set at 0.05 and the acceptable beta error at 0.80. Data collectors were blinded to genotype and treatment groups in all immuno-EM experiments, where manual counting was required.
Results
Brain endothelial cells express NMDA receptors linked to eNOS activity
Immunoreactivity for the pan-NMDA receptor subunit, GluN1, was detected in primary brain microvascular endothelial cells. Anti-GluN1 (Santa Cruz) was used to immunoprecipitate (IP) total homogenates from endothelial cultures and cerebral cortex, and immunoblotted products were detected with a separate anti-GluN1 (EMD Millipore, Figure 1(a)). IP products were also immunoreactive for GluN2C (Figure 1(a)), indicating a physical linkage between GluN1 and GluN2C. These results were confirmed using reciprocal IP experiments in which GluN2C IP products were immunoreactive for GluN1. To determine whether expression of NMDA receptor subunits correlates with functional NMDA receptors, endothelial cultures were loaded with the NO-sensitive dye, DAF-FM, and exposed to acetylcholine or NMDA receptor co-agonists, glutamate and D-serine (Figure 1(b), green). Glutamate enhanced DAF-FM fluorescence in the presence of 10 µM D-serine but not alone (Figure 1(c)). Glutamate concentrations as low as 3 µM significantly elevated DAF-FM signal (143 ± 11% baseline), with a maximum effect of 264 ± 21% at 300 µM. The NOS inhibitor, L-NIO, virtually eliminated DAF-FM signal induced by glutamate (30 µM) and D-serine (10 µM), as well as by acetylcholine (Figure 1(d)). The glutamate/D-serine effect on NO generation was also significantly reduced by NMDA receptor glutamate site antagonist, D-2-amino-5-phosphonopentanoate (AP5), and glycine site antagonist, 5,7-dichlorokynurenic acid (DCKA, Figure 1(e)). In contrast, neither AP5 nor DCKA affected NO generation produced by acetylcholine. NO production in response to glutamate/D-serine and acetylcholine was also mitigated by limiting accumulation of intracellular Ca2+ (Figure 1(f)). Loading cultures with the Ca2+-chelator, BAPTA [1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis]-AM, reduced NO accumulation from 184 ± 10% to 131 ± 10% control, and in response to acetylcholine from 234 ± 14% to 107 ± 5% control. Similarly, agonist treatment in Ca2+-free media with 0.1 mM EGTA reduced the effect of glutamate/D-serine to 101 ± 5% control and the effect of acetylcholine to 93 ± 12% control. Together these data show that primary brain endothelial cultures express NMDA receptors capable of activating NOS in a Ca2+-dependent fashion.
NMDA receptors induce nitric oxide production in brain endothelial cultures. (a) Homogenates from mouse cortex (Cx) or primary mouse brain endothelial cultures (Endo) were incubated with bead-conjugated anti-GluN1 or anti-GluN2C, and immunoprecipitates analyzed by Western immunoblot (IB). Homogenates pulled down by anti-GluN1 from both cortex and endothelial cells were immunopositive for GluN1 and GluN2C (i). In agreement, reciprocal immunoprecipitation with anti-GluN2C yielded homogenates detected by Western blots using anti-GluN2C and anti-GluN1 (ii). Ctrl indicates omission of primary IP antibody. (b) Brain endothelial cultures were loaded with DAF-FM. Exposure to acetylcholine (Ach) or increasing concentrations of glutamate (Glu) with D-serine, caused enhancement of DAF fluorescent signal (5 h, pseudo-colored green). (c) Quantification of DAF fluorescence intensity revealed dose-dependent increases in NO production in response to Glu in the presence of D-serine but not alone. *p < 0.05, ***p < 0.001 compared to baseline controls, using two-way ANOVA with the Bonferroni multiple comparison test (n = 4–5). (d) The eNOS inhibitor, L-NIO, mitigated NO production (3 h) induced by Ach or combined Glu and D-serine (D-ser). (e) Competitive NMDA receptor antagonists, AP5 and DCKA, inhibited NO production (5 h) induced by Glu and D-serine but not Ach. (f) Chelation of intracellular Ca2+ using BAPTA-AM, and nominally Ca2+-free medium both impaired NO responses to Ach and combined Glu/D-serine. For (d–f) *p < 0.05, **p < 0.01, ***p < 0.001 compared to agonist only groups, using one-way ANOVA with Tukey’s multiple comparison test (n = 3–5).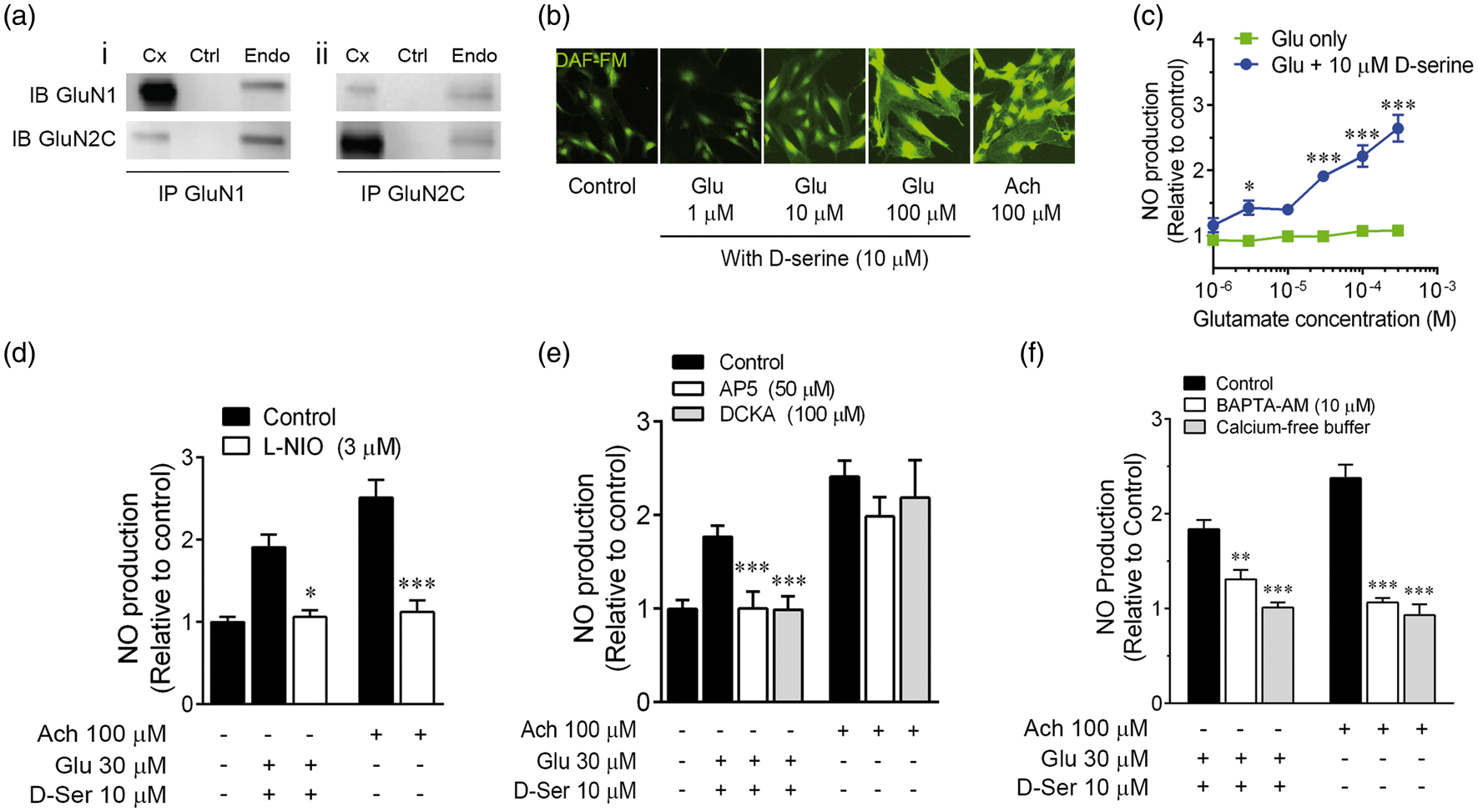
GluN1 is localized to basolateral brain endothelial membrane surfaces
We used immunoelectron microscopy to examine the subcellular endothelial GluN1 distribution in cortical slices. Anti-GluN1 was conjugated to gold beads with 10 nm diameter to create a detection system that indicates the presence of target epitope within a 30 nm radius (Figure 2(a)).
36
Cortical synapses were first examined to confirm GluN1 detection. Tissues were exposed to both anti-GluN1 (10 nm bead) and an antibody against PSD-95 (15 nm bead), which is a synaptic protein that associates with the GluN1 C-terminus.
37
GluN1-labeled cortical synapses were dominantly asymmetric (97.7%) and exhibited strong association with PSD-95 (Figure 2(b)). The vast majority of GluN1 was localized to within 30 nm of postsynaptic membranes (Figure 2(c)), verifying that anti-GluN1 faithfully labels NMDA receptors. We next quantified GluN1 beads within 30 nm of basolateral endothelial membranes. GluN1 was detected at basolateral and apical endothelial membranes (Figure 2(d) to (g)). For cortical arteries, arterioles and capillaries, apical GluN1 expression was significantly less, proportionally, relative to basolateral expression. Basolateral expression represented 73 ± 15%, 77 ± 4% and 71 ± 4% of GluN1 immunoreactivity in arteries, arterioles and capillaries, respectively (Figure 2(h)). These data demonstrate that NMDA receptor subunits are expressed with membrane polarity suitable for direct communication with the brain compartment.
GluN1 is localized to basolateral cortical endothelial membrane surfaces in situ. GluN1 was detected in fixed cortical slices by immunoelectron microscopy. (a) Representation of the approximate size scale of immunogold complexes relative to the GluN1 cognate target. (b) Representative electron micrograph of a cortical excitatory (asymmetric) synapse with double post-synaptic immunogold-labeling of GluN1 (10 nm beads, blue arrows) and PSD-95 (15 nm beads, green arrows). Scale bar is 100 nm. (c) Histogram reflecting distances of GluN1-immunogold beads from putative excitatory synaptic membranes. (d) Representative electron micrograph of penetrating cortical arteriole with excitatory glutamatergic synapse (blue frame) in the same field of view (scale bar = 500 nm). (e) Red frame is a high-magnification image (scale bar, 100 nm) of the segment of basolateral endothelial membrane (yellow line) in (d). GluN1 immunoreactivity was observed at basolateral endothelial membranes (blue arrows). (f) Representative electron micrograph of penetrating cortical arteriole with landmark endothelial tight junction (TJ) in the same field of view (scale bar = 500 nm). (g) Red frame is a high-magnification image (scale bar, 100 nm) of the segment of basolateral endothelial membrane (yellow line) in (f). (h) Manual counting indicated GluN1 immunoreactivity is preferentially distributed to basolateral endothelial membrane, relative to apical membranes for all vessel sizes. **p < 0.01 compared to basolateral groups for all vessel sizes using a two-way ANOVA with the Bonferroni multiple comparison test (n = 5 mice; two sections per mouse and ≥ 6 vessels per section).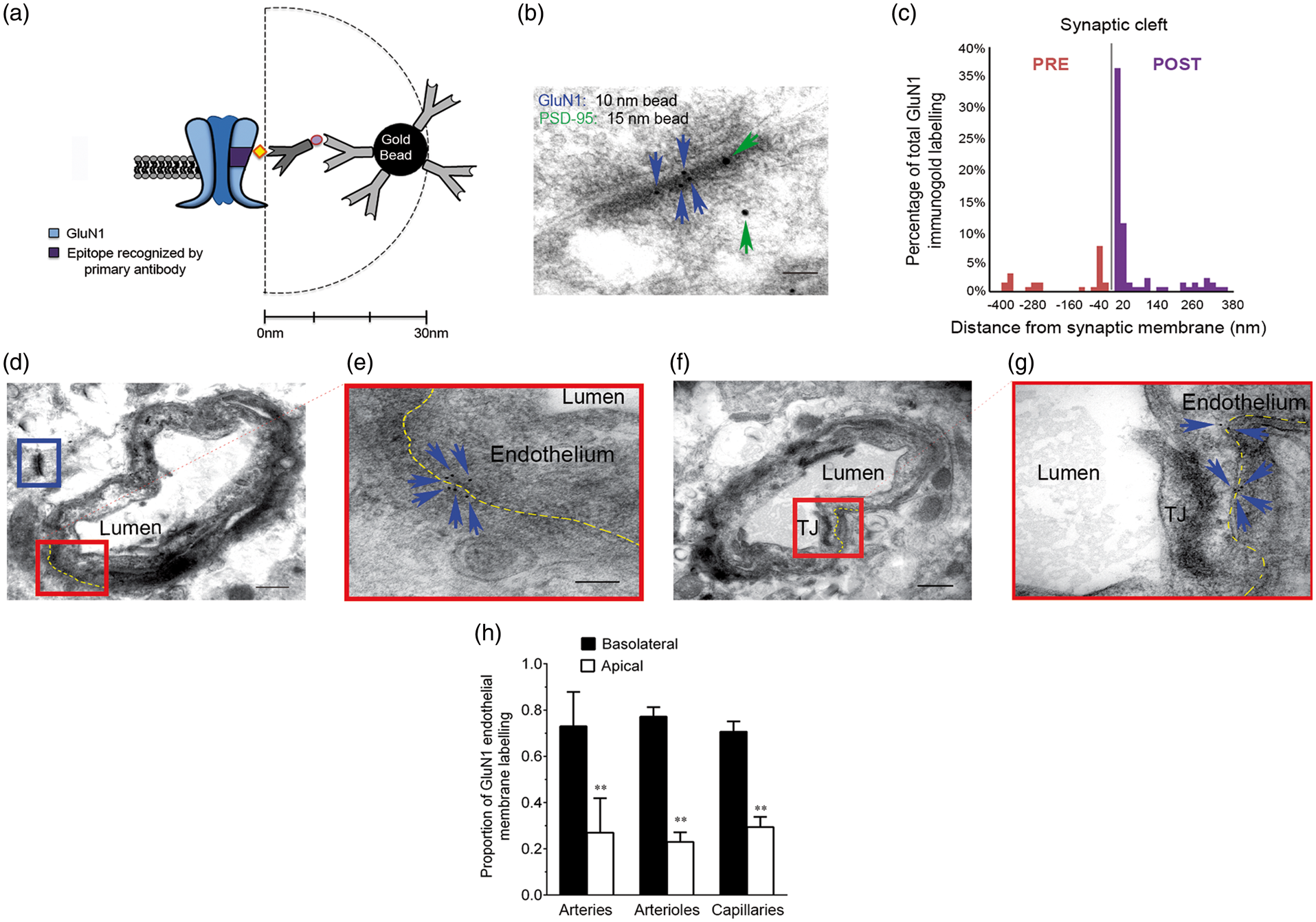
Astrocytic activation leads to cortical vasodilation mediated by endothelial NMDA receptors
In neonatal cortical slices, hyperemic signaling can be simulated by bath-applying trans-1-aminocyclo-pentane-1,3-dicarboxylic acid (tACPD), which activates astrocytic mGluR5 receptors and leads to astrocyte-mediated local vasodilation.8,11 Cortical layer II/III arteriolar segments with astrocytic endfeet were chosen for analysis by two-photon laser scanning microscopy (TPLSM) in acute cortical slices (Figure 3(a)). Astrocytic Ca2+ (Figure 3(b); rhodamine-2, green) and segmental arteriole lumen diameter were assessed every 15 s prior to and after tACPD application. Lumen diameter increased steadily to a peak of 5.2 ± 0.7% over baseline diameter at 105 s, followed by a return to baseline by 5 min (Figure 3(c)). In a cumulative analysis of this 5 min interval, assessed as area under the curve (AUC), the competitive glutamate site NMDA receptor antagonist, AP5 (50 µM), reduced the vasodilatory response by 74% (from 825 ± 135 to 211 ± 81, Figure 3(d)). Similarly, the co-agonist site NMDA receptor antagonist, DCKA (100 µM), attenuated tACPD-induced vasodilation by 67% (from 1063 ± 186 to 349 ± 121).
Metabotropic glutamate receptors cause NMDA receptor-dependent cortical vasodilation. Mouse cortical slices were isolated and layer II/III penetrating cortical arterioles with astrocytic process contacts (schematic representation in a) analyzed using two-photon microscopy. Slices were co-incubated with a Ca2+-sensitive dye (rhodamine-2, green, b) and the endothelial marker, isolectin B4 (red, b). Astrocyte Ca2+ levels (rhodamine-2 intensity) and local arteriole lumen diameter (marked by white box in b) were measured in response to bath-applied metabotropic glutamate receptor agonist, t-ACPD (100 µM). (c) Arteriolar lumen diameter increased in response to tACPD and this effect was mitigated in the presence of the NMDA receptor antagonist, D-(-)-2-Amino-5-phosphonopentanoic acid (AP5, 50 µM). (d) Quantification of cumulative responses over 5 min (area under curve, AUC) revealed a significant inhibition of vasodilatory response to tACPD by both AP5 and the glycine/D-serine NMDA receptor co-agonist antagonist, 5,7-dichlorokynurenic acid (DCKA, 100 µM). Data are all mean ± SEM. *p < 0.05 and **p < 0.01 compared to respective tACPD controls using one-way ANOVA with Tukey’s multiple comparison test. Scale bar in b is 20 µm.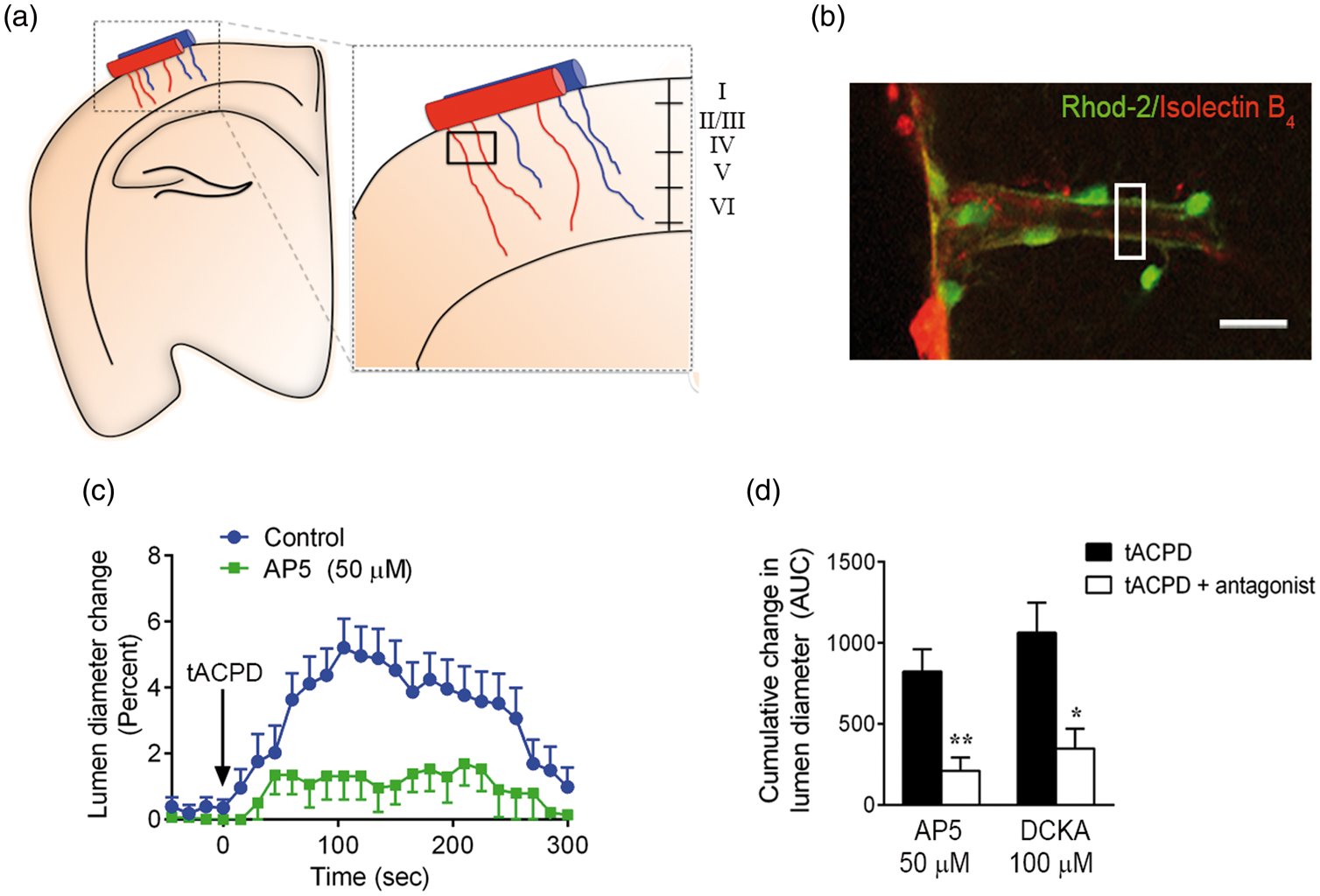
To more directly understand the role of endothelial NMDA receptors in neurovascular coupling, we developed a selective endothelial loss of function model by crossing floxed grin1 (gene for GluN1, grin1fl/fl) mice with mice expressing Cre-recombinase driven by the Tie-2 promoter (Tie-2-Cre+/−). Grin1fl/fl · Cre+/− offspring (Cre+) are animals with conditional GluN1 silencing, while littermate grin1fl/fl · Cre−/− offspring (Cre−) are controls. In Cre+ brain endothelial cultures, mRNA for GluN1 was reduced to 27 ± 10% control levels (Figure 4(a)), and GluN1 immunoreactivity was reduced to 49 ± 10% of control expression (Figure 4(b)). In agreement, NO generation in cultures treated with glutamate and D-serine was reduced from 209 ± 65% control in Cre−cultures, to 96 ± 6% control in Cre+ cultures (Figure 4(c)). By contrast, GluN1 loss of function did not affect NO generation induced by acetylcholine. Using quantitative PCR in cortical homogenates, we found no difference in GluN1 transcript levels between Cre+ and Cre− mice, likely reflecting a proportional dominance of neuronal GluN1 expression in brain tissue (Figure 4(d)). We also used mesenteric artery as a peripheral vascular tissue free of neuronal GluN1 and found that Tie-2-Cre expression reduced GluN1 transcript to 7.5 ± 0.5% of control Cre− tissues (Figure 4(d)). Synaptic and endothelial GluN1 immunoreactivities were examined using immunoelectron microscopy. GluN1-positive gold bead density in the basolateral endothelium declined from 1.24 ± 0.06 beads/µm of membrane surface area in control cortex, to 0.543 ± 0.001 in Cre+ cortex (Figure 4(e)), whereas synaptic GluN1 immunoreactivity did not change. These results indicate selective, partial loss (>50%, protein level) of GluN1 in endothelial cells.
GluN1 expression is selectively reduced in endothelial cells by crossing grin1fl/fl mice with a Tie-2-Cre recombinase driver line. (a) GluN1 mRNA levels were significantly reduced in brain endothelial cultures prepared from Cre-positive (Cre+) mice, relative to cultures prepared from Cre-negative (Cre−) littermate controls. p = 0.04 compared to Cre− control group using two-tailed t-test (n = 3–5) (b) GluN1 immunoreactivity was significantly reduced in brain endothelial cultures prepared from Cre-positive (Cre+) mice, relative to cultures prepared from Cre-negative (Cre−) littermate controls. p = 0.03 compared to Cre− control group using two-tailed t-test (n = 3–5) (c) In agreement, NO production in response to NMDA receptor agonists (glutamate and D-serine, 30 µM), but not acetylcholine (Ach, 100 µM), was significantly inhibited in Cre+, compared to Cre− controls. (d) Quantitative PCR indicated a dramatic loss of GluN1 mRNA in homogenates from Cre+ mesenteric artery homogenates, relative to Cre− arteries. Relative message levels were 30-fold higher in cortex, reflecting high neuronal expression. There was no silencing of overall cortical GluN1 in tissue from Cre+ mice. (e) GluN1 immunoelectron microscopy, followed by manual counting of immunogold beads, revealed significant silencing of GluN1 at basolateral endothelium but not synaptic membranes. For (c–e) **p < 0.01, ***p < 0.001 compared to respective Cre+ groups using two-way ANOVA with the Bonferroni multiple comparison test (n = 3–5).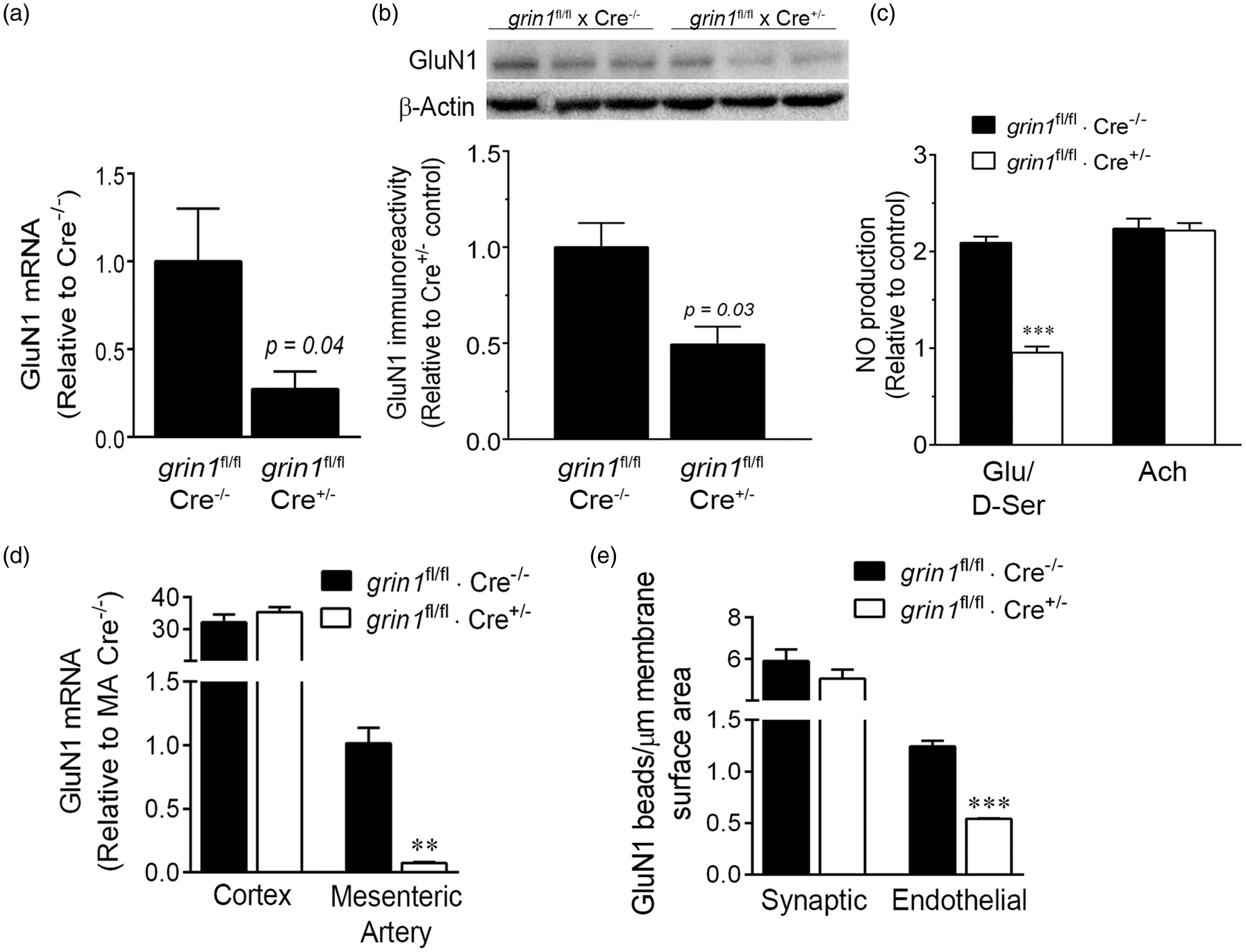
In Cre− control brain slices exposed to bath-applied tACPD, arteriolar lumen diameter increased to a peak of 5.0 ± 1.3% over baseline at 225 s, followed by a return to baseline by 5 min (Figure 5(a)). Cumulatively, vasodilatory responses (AUC) were reduced by 54 ± 9% in Cre+ slices with endothelial NMDA receptor loss of function (Figure 5(b)). By contrast, astrocytic Ca2+ levels increased rapidly in both Cre+ and Cre− slices in response to tACPD (Figure 5(c)), with no significant difference in cumulative responses between groups (Figure 5(d)). This indicates differences in vasodilatory effects result from events downstream of astrocytic activation. We also directly applied NMDA receptor co-agonists, glutamate and D-serine together, by pressure ejection 10 nm adjacent to cortical arterioles (1 mM each agonist). While this treatment initiated a peak vasodilatory response of 3.6 ± 0.8% of baseline in Cre+ slices, the peak effect was smaller and return to baseline more rapid than in Cre− wildtype controls (Figure 5(e)). This resulted in a significantly smaller cumulative vasodilatory effect in endothelial Cre+ slices (Figure 5(f)).
Selective endothelial GluN1 silencing mitigates cortical vasodilation. GluN1 expression was selectively reduced in endothelial cells by crossing grin1fl/fl mice with a Tie-2-Cre recombinase driver line. (a) Mouse cortical slices were isolated from mice Cre+ mice (grin1fl/fl × Cre+/−) or Cre− wildtype littermate controls (grin1fl/fl × Cre−/−), and loaded with rhodamine-2-AM and the endothelial marker, isolectin B4. Astrocyte Ca2+ levels (rhodamine-2 intensity) and local arteriole lumen diameter were measured in response to bath-applied metabotropic glutamate receptor agonist, t-ACPD (100 µM). (b) Quantification of cumulative responses over 5 min (area under curve, AUC) revealed significant inhibition of vasodilatory response in Cre+ slices; p = 0.006 compared to Cre−using a two-tailed t-test. (c) Rhodamine-2 intensity increased in perivascular astrocyte processes in response to tACPD exposure. (d) Endothelial GluN1 expression level did not affect the cumulative 5 min Ca2+ response in astrocytes (AUC); p = 0.58 compared to Cre− using two-tailed t-test. (e) Arteriolar lumen diameter increased in response to local pressure ejection or glutamate/D-serine (1 mM each, 10 µm adjacent to cortical arterioles). (f) Cumulative responses over 5 min (AUC) were significantly reduced in Cre+ slices; p = 0.04 compared to Cre− using two-tailed t-test. In (g) and (h), slices were loaded with the caged Ca2+ compound o-nitrophenyl-EGTA (NP-EGTA), rhodamine-2 (rhod-2), and the endothelial marker, isolectin B4. (g) Cortical arteriole lumen diameter was assessed in response to two-photon flash photolysis of NP-EGTA in perivascular astrocyte soma. (h) Astrocyte-induced vasodilatory responses were significantly inhibited in Cre+ slices. p = 0.04 compared to Cre− control group using two-tailed t-test (n = 20). Scale bars, 20 µM.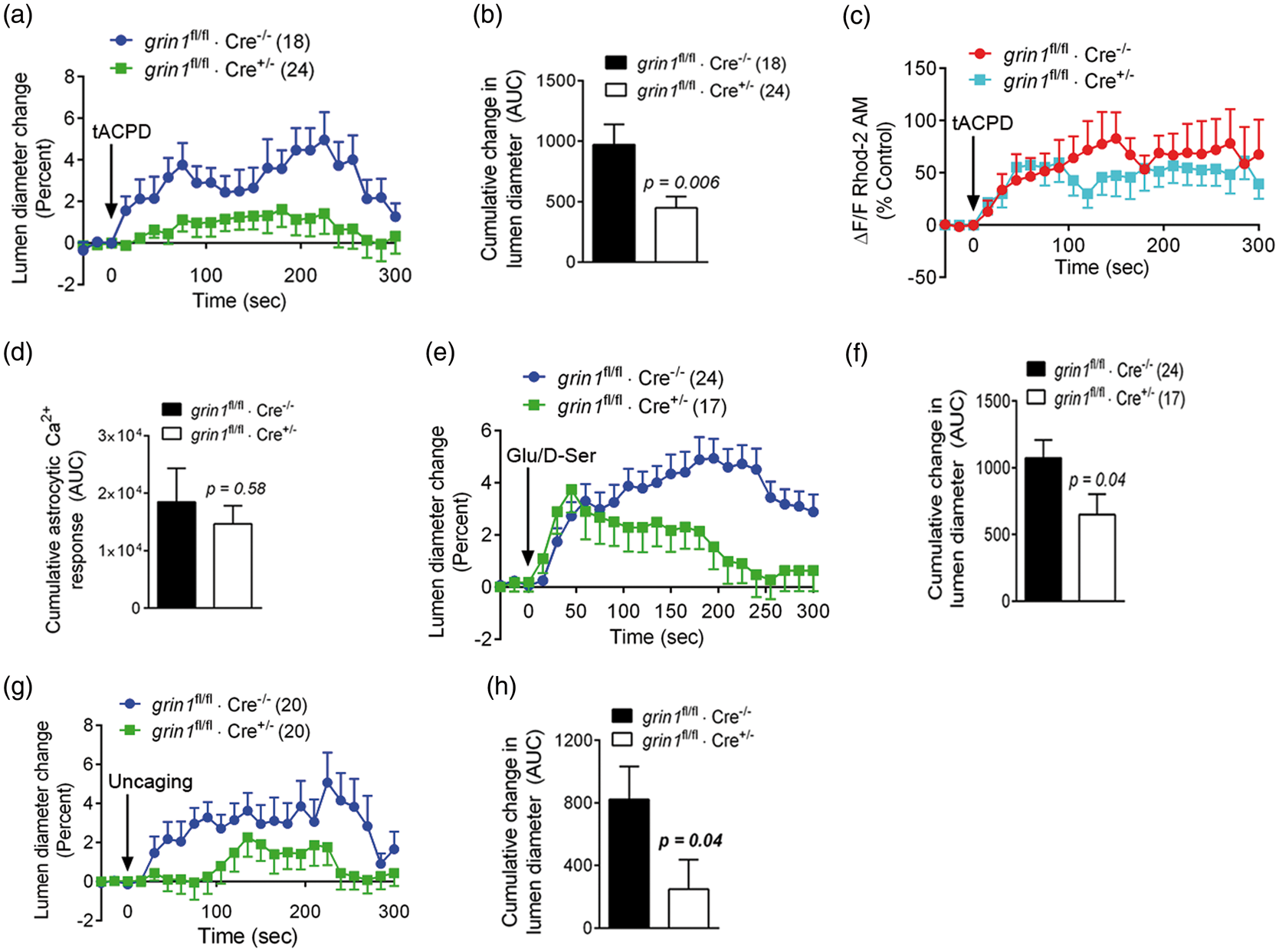
To more directly study linkage between astrocytic activity and endothelial NMDA receptors, acute cortical slices were loaded with the caged Ca2+ compound, NP-EGTA, and perivascular astrocytes targeted for two-photon flash photolysis (TPP). Similar to tACPD and NMDA receptor agonists, selective activation of single astrocytes increased lumen diameter in Cre− wildtype slices (Figure 5(g), Supplemental video 1). The cumulative vasodilatory response was reduced by 70 ± 18% with endothelial NMDA receptor silencing (Figure 5(h)). Overall these data demonstrate that mouse cortical slice arterioles with partial endothelial NMDA receptor silencing have diminished ability to dilate in response to multiple stimulation paradigms.
Direct activation of cortical astrocytes leads to endothelial NO generation dependent on endothelial NMDA receptors
To determine whether astrocytes communicate with the endothelium to produce NO, cortical slices were loaded with DAF-FM, NP-EGTA-AM and rhodamine-2-AM. Layer II/III cortical arterioles with adjacent perivascular astrocytes were then imaged by TPLSM. Single, rhodamine-2-labelled astrocytes were targeted for activation by TPP (Figure 6(a), white asterisks). TPP rapidly enhanced astrocytic rhodamine-2 fluorescence (Figure 6(a), green), reflecting increased intracellular Ca2+ levels. In control slices, TPP also enhanced DAF-FM signal intensity, indicating NO accumulation (Supplemental Video 2). This signal was observed in the endothelium, closer to the arteriolar lumen than the astrocytic rhod-2 signal. While indicative of NO accumulation, strong DAF-FM signals dampened vasodilatory efficacy and in some cases were associated with vasoconstriction, likely by inactivating NO (e.g. Supplemental Video 2). Arterioles were also identified that dilated in response to TPP but had weaker DAF-FM signals (e.g. Supplemental Video 3), suggesting vasodilatory activity was inversely related to DAF-FM loading in individual vessels. Astrocytic rhod-2 signal increased rapidly, rising to 161 ± 19% of unstimulated values within 3 s after TPP and to 196 ± 17% by 15 s (Figure 6(b)). Endothelial DAF-FM signal increased more slowly, reaching a maximum of 177 ± 33% of unstimulated control values by 225 s after TPP (Figure 6(a) and (b)). TPP was unable to generate an endothelial NO/DAF-FM signal in the presence of the NOS inhibitor, N5-(1-iminoethyl)-L-ornithine (L-NIO, 3 µM; Figure 6(c) and (d); also Supplemental video 3). These data confirm direct communication between astrocytes and endothelial cells, resulting in endothelial NO generation.
Direct astrocytic activation leads to endothelial NO production. (a) Mouse cortical slices were co-incubated with a Ca2+-sensitive dye (rhodamine-2, green), the NO-sensitive dye, 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM, red) and the caged Ca2+ compound, o-nitrophenyl-EGTA (NP-EGTA). Layer II/III penetrating cortical arterioles with astrocytic process contacts were imaged using two-photon microscopy. (b) Perivascular astrocytic soma (a, white asterisks) were targeted for two-photon flash photolysis of NP-EGTA, resulting in increased Ca2+-dependent rhodamine-2 fluorescence intensity in soma and processes. This was accompanied by enhanced DAF-FM fluorescence intensity in the arteriolar endothelium, reflecting endothelial NO accumulation. (c) 5-min time course of NO accumulation in the presence and absence of a NO synthase inhibitor (L-NIO, 3 µM). L-NIO inhibited NO production. (d) Cumulative 5 min NO responses (area under curve, AUC), showing that endothelial NO accumulation in response to astrocyte Ca2+ uncaging is eliminated by L-NIO. **p < 0.01 compared to control responses, using one-way ANOVA with Newman–Keuls multiple comparison test (n = 8–11). Scale bars = 20 µm.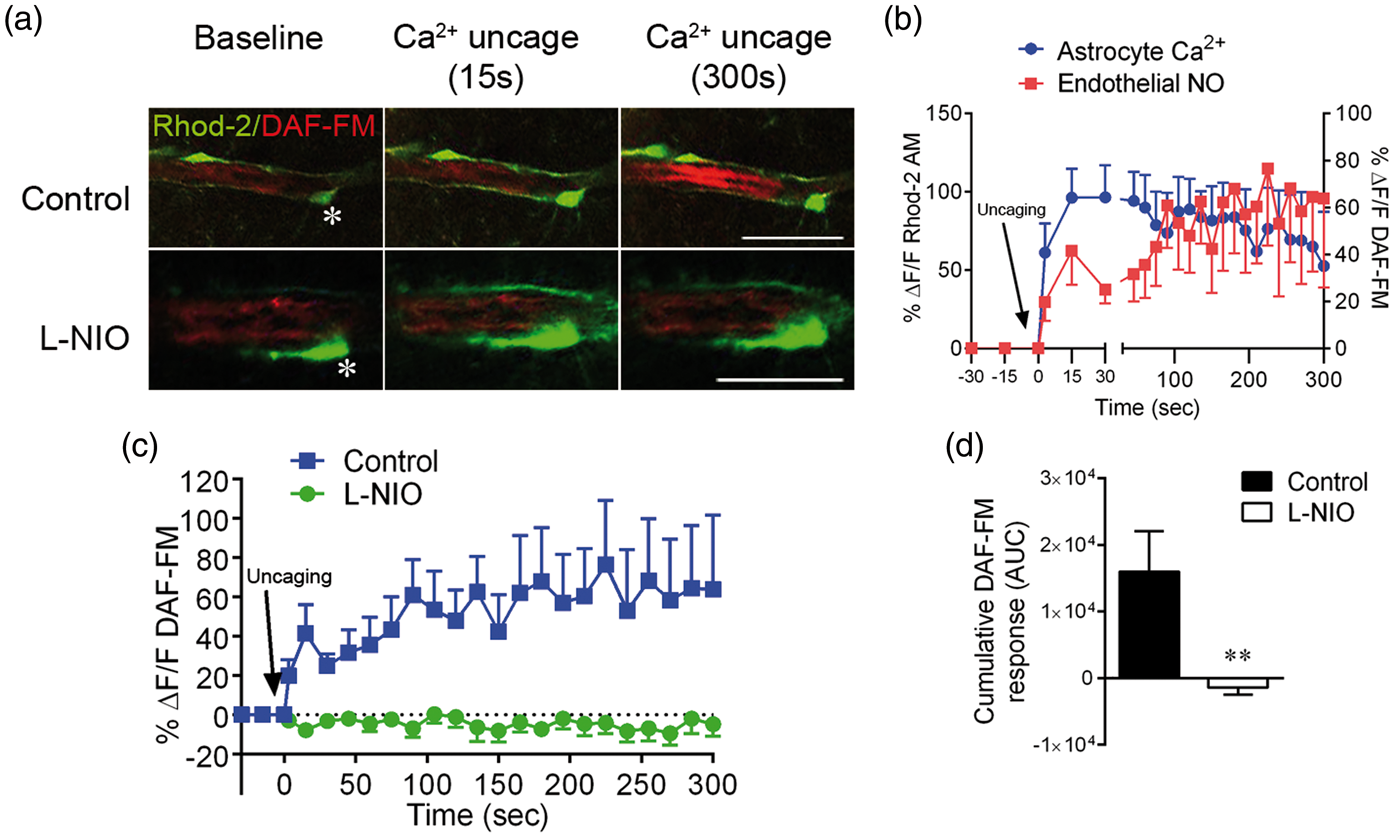
NO levels were assessed in response to astrocytic Ca2+ uncaging in slices from Cre− control and endothelial Cre+ mice. In Cre− slices loaded with DAF-FM, TPP induced a cumulative DAF-FM signal that was significantly inhibited by competitive NMDA receptor antagonists, AP5 and DCKA, and by Tie-2-Cre expression, but not by the AMPA receptor antagonist, CNQX (Figure 7(a)). Vasodilatory responses induced by TPP of NP-EGTA in astrocytes were significantly inhibited by the guanylyl cyclase inhibitor, 1 H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, 3 µM) in Cre− control cortical slices but not in Cre+ slices with endothelial NMDA receptor loss of function (Figure 7(b)). We also determined whether endothelial NMDA receptors affect the balance of arachidonic acid metabolites known to cause smooth muscle relaxation (PGE2) and constriction (20-HETE). Cortical slices were exposed to bath-applied tACPD (100 µM), and 20-HETE and PGE2 quantified by ELISA after 5 min. While 20-HETE levels were unaffected in Cre− controls, there was significant enhancement of 20-HETE in Cre+ slices with endothelial NMDA receptor silencing (Figure 7(c)). In contrast, PGE2 release was significantly elevated following tACPD treatment of Cre− wildtype slices, and this affect was not significant with more than 95% certainty in Cre+ slices (Figure 7(d)). There was also a significant decline in induced PGE2 levels when endothelial GluN1 was silenced, suggesting a source of PGE2 dependent on endothelial NMDA receptor activity. Vasodilatory responses to TPP of NP-EGTA in astrocytes were significantly reduced by the cyclooxygenase inhibitor, indomethacin (100 µM) in Cre− cortical slices, indicating that endothelial NMDA receptors affect arteriolar lumen diameter by a mechanism dependent on a vasodilatory arachidonic acid metabolite like PGE2 (Figure 7(e)). While the vasodilatory magnitude was dramatically reduced by endothelial GluN1 silencing, the remaining response was still significantly sensitive to inhibition by indomethacin.
Selective endothelial GluN1 silencing reduces endothelial NO production downstream vasodilatory signaling. Endothelial GluN1 expression was selectively reduced by crossing grin1fl/fl mice with a Tie-2-Cre recombinase driver line. Mouse cortical slices were isolated from mice Cre+ mice (grin1fl/fl × Cre+/−) or Cre− wildtype littermate controls (grin1fl/fl × Cre−/−). (a) Slices were loaded with the caged Ca2+ compound o-nitrophenyl-EGTA (NP-EGTA), rhodamine-2, the endothelial marker, isolectin B4, and the NO-sensitive dye, 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM). Endothelial NO accumulation (DAF-FM signal) was assessed in response to two-photon flash photolysis of NP-EGTA in perivascular astrocyte soma. Cumulative NO responses over 5 min (area under curve, AUC) were significantly reduced by AP5 (50 µM), DCKA (100 µM) and Cre-dependent endothelial GluN1 silencing, but not by the AMPA receptor antagonist, CNQX. *p < 0.05, **p < 0.01 compared to Cre− control using one-way ANOVA with Newman–Keuls multiple comparison test (n = 8–12). (b) Uncaging of astrocytic Ca2+ increases arteriolar diameter in a manner sensitive to guanylyl cyclase inhibitor, 1 H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, 3 µM), in Cre− but not Cre+ slices. *p < 0.05 compared to Cre− control and ††p < 0.01 as indicated using one-way ANOVA with Newman–Keuls multiple comparison test (n = 20–28). (c) tACPD or combined glutamate (glu) and D-serine (D-ser) were bath-applied to cortical slices and 20-HETE levels quantified. Both treatments significantly enhanced 20-HETE levels in Cre+ slices with GluN1 silencing but not littermate wildtype Cre− slices. *p < 0.05, **p < 0.01 compared to Cre+ control and †p < 0.05 as indicated using one-way ANOVA with Newman–Keuls multiple comparison test (n = 5–7). (d) tACPD or combined glu/D-ser (with 1 µM tetrodotoxin) were bath-applied to cortical slices and PGE2 levels quantified. Both treatments significantly enhanced PGE2 levels in Cre− wildtype control slices but not from littermate Cre+ slices with GluN1 silencing. *p < 0.05, **p < 0.01 compared to no-treatment baseline for each respective genotype and ††p < 0.01 as indicated, using one-way ANOVA with Newman–Keuls multiple comparison test (n = 5–8). (e) Uncaging of astrocytic Ca2+increases arteriolar diameter in a manner sensitive to cyclooxygenase inhibitor, indomethacin (100 µM), in Cre− and Cre+ slices. *p < 0.05 compared to respective Cre− or Cre+ controls lacking indomethacin; ††p < 0.01 as indicated using one-way ANOVA with Newman–Keuls multiple comparison test (n = 20–28).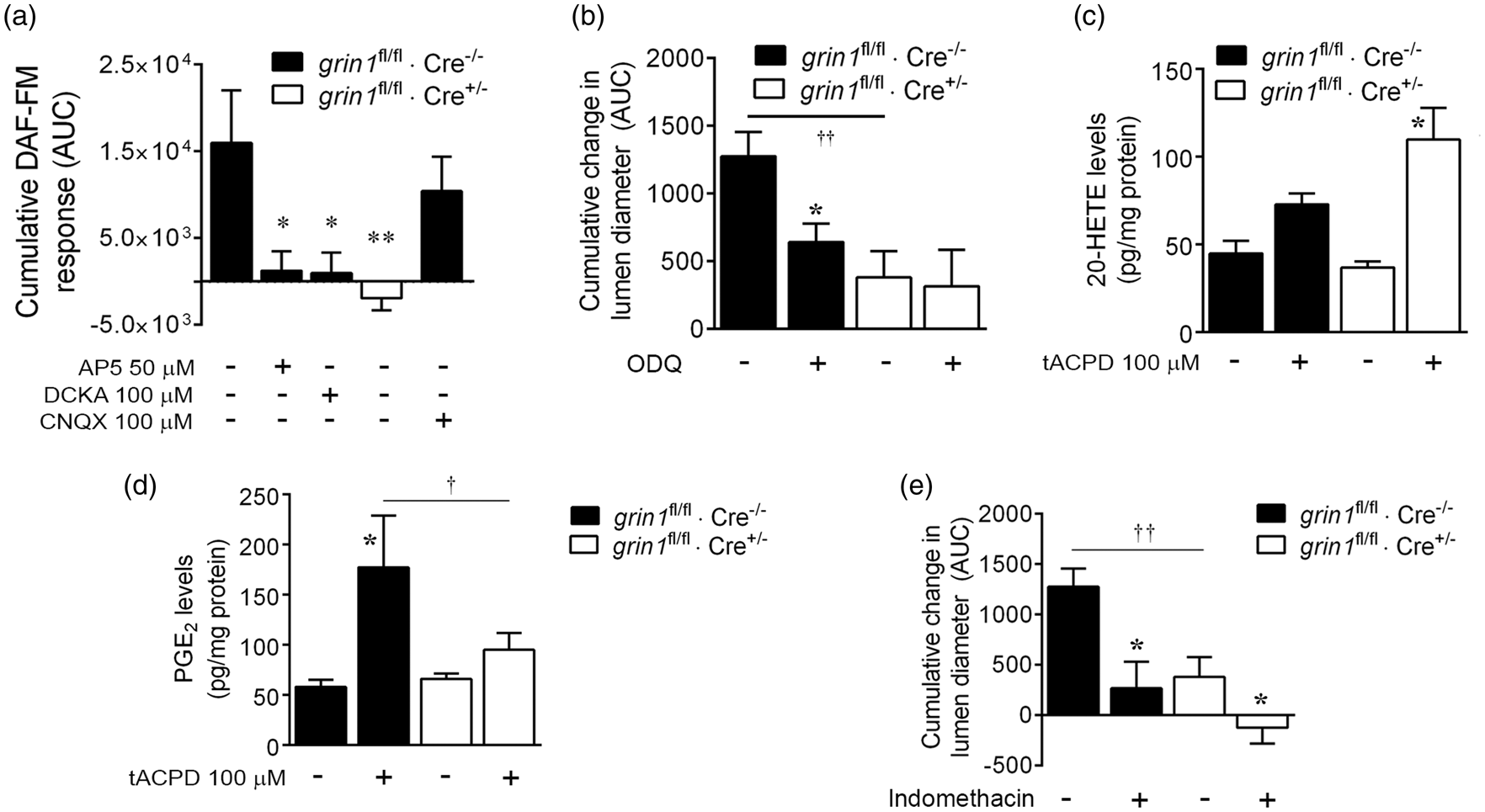
Discussion
Our results demonstrate an endogenous pathway that functionally connects astrocytic activity with vasodilatory signaling initiated by the cerebrovascular endothelium. The pan-NMDA receptor subunit, GluN1, is expressed at basolateral endothelial membranes in an orientation suited to receive chemical messengers from astrocytes. When cortical astrocytes were activated by agonist activity or direct TPP of caged Ca2+, enhanced endothelial NO, vasodilatory arachidonic acid metabolism and vasodilation were all observed in a manner sensitive to inhibition by conditional endothelial NMDA receptor loss of function. Together these results represent a previously unidentified mechanism of neurovascular coupling.
Several lines of evidence support brain endothelial expression of NMDA receptor subunits. In primary brain endothelial cultures, we detected immunoreactivity for the pan-NMDA receptor subunit, GluN1. This is consistent with a substantial body of evidence showing that endothelial cultures or isolated capillaries express either GluN1 message or immunoreactivity.24,25,27,32,35 We also detected immunoreactivity for the GluN2C subunit in input homogenates and in samples pulled down by anti-GluN1 immunoprecipitation. There are reports that mRNAs encoding NMDA receptor subunits other than GluN1 are found in endothelial cultures27,32,38, with some evidence for GluN2A or GluN2B immunoreactivity.24,30 However, our findings demonstrate a structural association between GluN1 and another subunit in endothelial cells. This is consistent with our previous finding that GluN2C is co-expressed with endothelial markers in cortical arterioles, 33 but verification of a functional requirement for GluN2C is yet to be made. Beyond culture systems, there is very little evidence of endothelial NMDA expression in situ. GluN1 immunoreactivity was found co-localized with endothelial markers in brain arterioles of mice exposed to a model of experimental autoimmune encephalomyelitis, 39 but it was not clear whether this could be demonstrated in healthy mice. GluN1 co-localization with an endothelial marker was also reported in healthy mouse brain, 40 but there was insufficient resolution to rule out expression by other neurovascular unit cells types. Our initial attempts to examine neurovascular GluN1 distribution by immunofluorescence were obscured by the dominance of neuronal GluN1 expression. We therefore used immunoelectron microscopy to achieve resolution that allows precise subcellular delineations. This approach confirmed that GluN1 expressed by the cortical endothelium in large arteries, arterioles, and capillaries. In addition, our hypothesis was that chemical messengers produced by brain cells can activate endothelial NMDA receptors. For this to be true, GluN1 should be expressed at basolateral endothelial membranes. While immunoelectron microscopy revealed GluN1 expression at apical endothelial membranes and tight junctions as well, expression at basolateral endothelial membranes was proportionally dominant, supporting an orientation suitable for receiving brain signals. It should be noted that despite this and other support, there are also reports that have not detected either expression of NMDA receptor subunits or responses to NMDA receptor agonists.6,41–43 One reason for discrepancies could be that responses to NMDA or glutamate alone may have failed in the absence of a co-agonist. In agreement, we found an effect of glutamate on lumen diameter of isolated cerebral arteries and NO generation by endothelial cultures when applied with the co-agonist, D-serine, but not alone. The reasons NMDA receptor subunits were not detected in some systems are not clear.
An important conclusion of our study is that there is direct vasodilatory signaling between astrocytes and the endothelium mediated by endothelial NMDA receptors. We previously showed that NMDA receptor agonists initiate vasodilatory responses in isolated arteries and cortical slices mediated by eNOS.11,33 These studies, however, were not sufficiently powered to determine whether endogenous, astrocyte-mediated neurovascular coupling increases arteriole lumen diameter by activating NMDA receptors expressed specifically by endothelial cells. To distinguish endothelial NMDA receptors from those expressed by neurons or astrocytes, we used a conditional silencing approach based on co-expression of GluN1-coding grin1 alleles, flanked by loxP excision sites, and Cre-recombinase driven by the promoter, Tie-2. Over 50% loss of endothelial GluN1 was achieved, resulting in significant mitigation of cortical vasodilation and endothelial NO generation induced by astrocytic mGluR activation or direct astrocytic activation by TPP. This corresponded with reduced NO generation with selective endothelial GluN1 loss of function. This result strongly indicates that endothelial NMDA receptors are capable of initiating vasodilatory signaling, but an obvious question is, what is the source of endogenous receptor agonist? Our work suggests both glutamate and D-serine are required for endothelial NMDA receptor activation.11,33 We have induced endothelial NMDA receptor-dependent vasodilation by direct astrocytic Ca2+ uncaging, which isolates the astrocyte-vessel circuitry segment, and a substantial literature indicates that glutamate and D-serine can be released from astrocytes in a Ca2+-dependent fashion. 44 Thus, the most likely scenario is that astrocytic Ca2+ triggers regulated endfoot agonist release and activation of basolateral endothelial NMDA receptors. There are other possibilities, however, and future experiments must determine the precise steps spanning from synaptic activity to endothelial NMDA receptor activity.
Endothelial NMDA receptors were functionally linked to NOS activity in brain endothelial cultures and slices. Exposing cultures to NMDA receptor co-agonists caused cumulative DAF-FM fluorescence, indicative of NO generation. NMDA receptor antagonists and Cre-dependent silencing of GluN1 blocked NO production in response to glutamate/D-serine but not acetylcholine, verifying a distinct mechanism for NMDA receptor agonists. These results are consistent with previous reports showing increased peroxynitrite in response to glutamate in immortalized brain endothelial cells, 25 and increased NO generation in portal vein by endothelial NMDA receptors. 45 NOS activation in our hands was inhibited by chelating intracellular Ca2+ or by removing extracellular Ca2+. One interpretation is that NMDA receptors gate channel entry of Ca2+, leading to calmodulin-dependent NOS activity, however, a direct demonstration of this will be required. It also remains possible that alternate, channel-independent NMDA receptor signaling pathways could be involved. 46 NO was also generated in endothelial cells in brain slices in a manner sensitive to endothelial NMDA receptor loss of function. The catalytic source of endothelial NO generation is most likely eNOS. Although L-NIO is unable to distinguish eNOS from iNOS and nNOS, 47 we have shown previously that genetic deletion of eNOS significantly inhibits astrocyte-induced arteriolar vasodilation. 11 Endothelial NO can also be produced by the nNOS isoform, 48 leaving the possibility that both isoforms contribute to vasodilation in our paradigm. Used at a concentration with selectivity for nNOS, 49 the NOS inhibitor, 1-(2-trifluoromethylphenyl) imidazole (TRIM), significantly reduced vasodilatory responses (Supplemental material). This suggests involvement of both eNOS and nNOS and highlights the need to investigate the NO-mediated vasodilatory signaling mechanisms downstream of endothelial NMDA receptor activation in further detail.
NO may lead to cGMP-dependent vasodilation in brain by activating guanylyl cyclase 50 ; NO may also influence brain arteriolar lumen diameter by inhibiting the cytochrome P450 system that produces the smooth muscle constrictor, 20-HETE, from arachidonic acid. 9 The guanylyl cyclase inhibitor, ODQ, significantly reduced vasodilation caused by astrocytic Ca+ uncaging. Interestingly, sensitivity to ODQ was eliminated after endothelial NMDA receptor loss of function. This suggests that at least part of the endothelial NMDA receptor-mediated vasodilatory effect is dependent on cGMP generation but that the endothelial NMDA receptor-independent component is not. Astrocytic activation by tACPD caused vasodilation in Cre− control slices without significant changes in 20-HETE levels, but this effect was inhibited in Cre+ slices in a manner accompanied by a significant increase in tissue 20-HETE levels. This shows a correlation between 20-HETE levels and reduced vasodilation when endothelial NMDA receptor expression is partially silenced and implicates an increased 20-HETE:PGE2 balance in vasodilation initiated by endothelial NMDA receptors. For this to be true, we should also observe that PGE2 is responsible for at least some of the vasodilation we do see in Cre+ slices. In support of this, indomethacin mitigated vasodilation initiated by astrocytic Ca2+ uncaging in Cre− slices, and inhibited the effect remaining after endothelial NMDA receptor loss of function. Interestingly, endothelial NMDA receptor loss of function significantly reduced PGE2 levels induced by tACPD. This may indicate that there is a direct signaling pathway linking these receptors with cyclooxygenase activity in endothelial cells, and potentially that vasodilatory PGE2 in neurovascular coupling could be sourced from the endothelium. It is important to add that there is at least one report that PGE2 may not be involved in vasodilation, or may actually cause vasoconstriction in pressurized brain supply vessels. 51 Divergent PGE2 effects are likely related in part to the developmental stage of models used. Dabertrand et al. 51 made the point that EP2 and EP4 prostaglandin receptors responsible for vasodilation are expressed dominantly in neonatal developmental stages. Since slices in the current study are prepared during the first two postnatal weeks, it could be assumed as the reason why PGE2 induces smooth muscle relaxation in our hands. Overall, our data show that endothelial NMDA receptors drive NO production in endothelial cells, and that subsequent vasodilation is dependent on both guanylyl cyclase and influence on the 20-HETE/PGE2 balance.
There are two methodological issues that should be discussed. First, we created an endothelial NMDA receptor loss of function based on observations that promoter and enhancer elements from the Tie-2 genomic locus drive transgene expression uniformly in the vascular endothelium. 52 Neither total cortical GluN1 message nor synaptic GluN1 immunoreactivity were influenced by Tie-2-driven Cre expression, indicating neuronal GluN1 expression was not affected. In contrast, GluN1 mRNA and protein levels were dramatically reduced in mesenteric artery segments free from neuronal contamination, brain endothelial cultures and brain microvascular endothelial membranes in situ. These data demonstrate significant loss of GluN1 expression in endothelial cells but not neurons. Tie-2 also drives embryonic expression in erythro-myeloid progenitors that give rise to microglia. 53 NMDA receptor subunit expression has been detected in nascent and activated microglial cells in culture and may be involved in pro-inflammatory cytokine generation. 54 Thus, we cannot completely exclude the possibility that functional phenotypes we observed in grin1fl/fl·Tie-2-Cre+/− mice are partially attributable to loss of microglial NMDA receptors. Second, a common limitation of studying neurovascular coupling in brain slices is removal of arteriolar flow contributions to baseline smooth muscle tone. Replacement of tone is often simulated by adding a smooth muscle constrictor but this is also not ideal as these agents may interfere with the mechanisms investigators want to study. Most of the current study was performed without such an agent so it should be asked if vasodilation was assessed from a zero tone state and whether that would be meaningful. It is unlikely that brain slice arterioles are completely without tone given that tone is regulated by non-flow factors, including astrocytes. 55 Moreover, supplemental experiments using norepinephrine to further increase baseline tone showed that vasodilatory results were similar for selected experiments, including the effect of Cre-dependent endothelial NMDA receptor silencing. We thus believe that our results are a useful representation of real vasodilatory responses in the slice model.
While there is a sizeable literature supporting a strong role for astrocytes in neurovascular coupling leading to functional hyperemia, all reports of astrocyte-induced vasodilation are restricted to local effects and appear to be endothelium-independent. Thus, the requirement of brain endothelium for full coordinated responses beyond the local site of neuronal activity 18 has left an unexplained conceptual gap. Our results provide a potential mechanism for connecting local vasodilatory responses with endothelium-dependent vasodilatory conduction. At the local level, we found that endothelial NMDA receptors mediated endothelial NO generation and arteriolar vasodilation. eNOS signal dissipates rapidly and is not likely responsible for conducted vasodilation. 56 This could explain why eNOS does not appear to mediate functional hyperemia evoked by whisker stimulation or hypercapnia.57,58 However, Ca2+ increases sufficient to activate eNOS are likely also associated with activation of KCa2.3 and KCa3.1, producing a hyperpolarization impulse capable of relaxing vascular smooth muscle and long-distance electrotonic endothelial conduction. 59 Further experiments directly assessing the role of endothelial NMDA receptors, Ca2+ dynamics and K+ channels on conducted vasodilation and coordinated hyperemia responses will be important to fully identify the critical steps between astrocytic activity and distal gating of blood flow to brain regions in need.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research was supported by an operating grant from the Canadian Institutes of Health Research (CMA; CIHR, MOP 102681). CMA is a Manitoba Research Chair in Neurodegeneration. ADH was supported by a student award from Research Manitoba. PL and LL were supported by postdoctoral fellowships from Research Manitoba.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
AK, PL, SB and JI made substantial contributions to experimental design and acquisition, analysis and interpretation of data. LL, AH and CA contributed to experimental design, data acquisition, analysis and interpretation, and writing the manuscript. All authors approved the final version of the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.