
Abstract
Metabolic regulation of cerebrovascular tone directs blood flow to areas of increased neuronal activity and during disease states partially compensates for insufficient perfusion by enhancing blood flow in collateral blood vessels. Acid–base disturbances frequently occur as result of enhanced metabolism or insufficient blood supply, but despite definitive evidence that acid–base disturbances alter arterial tone, effects of individual acid–base equivalents and the underlying signaling mechanisms are still being debated. H+ is an important intra- and extracellular messenger that modifies cerebrovascular tone. In addition, low extracellular [HCO3–] promotes cerebrovascular contraction through an endothelium-dependent mechanism. CO2 alters arterial tone development via changes in intra- and extracellular pH but it is still controversial whether CO2 also has direct vasomotor effects. Vasocontractile responses to low extracellular [HCO3–] and acute CO2-induced decreases in intracellular pH can counteract H+-mediated vasorelaxation during metabolic and respiratory acidosis, respectively, and may thereby reduce the risk of capillary damage and cerebral edema that could be consequences of unopposed vasodilation. In this review, the signaling mechanisms for acid–base equivalents in cerebral arteries and the mechanisms of intracellular pH control in the arterial wall are discussed in the context of metabolic regulation of cerebrovascular tone and local perfusion.
Neurovascular coupling matches local blood flow to the metabolic demand of the tissue and the requirement for waste product elimination. This regulation of cerebrovascular tone in response to changes in metabolic activity supports normal neuronal function and reduces the consequences of cerebrovascular pathologies. Responses to acid–base deviations are also important for physiological and pathophysiological adaptations during systemic disturbances, for instance, in patients with pulmonary, renal, or metabolic disease.
Outside the nervous system, crosstalk between perivascular tissue and arteries has been demonstrated for coronary,1,2 skeletal muscle, 3 subcutaneous, 4 and mesenteric5,6 arteries and probably is of general importance in the resistance vasculature—albeit with some variation in the magnitude of influence between different vascular beds. Signaling between perivascular tissue and arteries can be bi-directional involving paracrine factors released, among others, from neurons, glial cells, cardiomyocytes, skeletal muscle, and adipocytes in the perivascular tissue1,7–10 but also from endothelial cells in the vascular wall. 11 Moreover, metabolites in the interstitial space are potential bi-directional signals as their concentrations are sensitive to the local metabolic rate and also respond to changes in perfusion that alter the delivery of nutrients and lead to local washout or accumulation of waste products. Deregulated local communication has been implicated in neurological (e.g. epilepsy 12 and Alzheimer’s disease 13 ) and metabolic (e.g. diabetes 2 and obesity4,14) disorders. Although the cause-effect relationship is still largely unclear, the homeostatic imbalance resulting from disturbed signaling between perivascular tissue and arteries likely contributes to disease development. Unravelling disease-related alterations in metabolic blood flow regulation remains an important current challenge. 15
Mechanisms of cerebrovascular control include responses to acid–base disturbances, hypoxia, and extracellularly accumulated adenosine and K+, and relies in part on vasoactive neurotransmitters and local paracrine factors (e.g. gasotransmitters and prostanoids). 16 Cellular responses to acid–base disturbances have become increasingly understood through the identification of putative intra- and extracellular sensors of acid–base equivalents. The function of many ion channels17,18 and enzymes19–22 is modulated by local extracellular (o) and/or intracellular (i) pH. In addition, G-protein-coupled receptors sensitive to pHo 23 and membrane-bound or intracellular enzymes sensitive to [HCO3–]o or [HCO3–]i24–26 have been identified. Molecular interventions to knockout, downregulate or overexpress the novel acid–base sensors in cultured cells, isolated tissues, and in vivo have proven valuable in the initial mapping of downstream signaling consequences:24,27–29 by selectively eliminating effects of individual putative acid–base sensors, it is possible to dissect functional roles and involved signaling cascades despite parallel activation of interdigitating messenger systems.
The molecular physiology of the signaling pathways modified by acid–base equivalents has been intensively studied but difficult to establish conclusively because of the spontaneous chemical reactions: CO2 + H2O ⇄ H2CO3 ⇄ HCO3– + H+ that interlink the CO2/HCO3– buffer components. Under standard experimental conditions—where the CO2/HCO3– buffer is in equilibrium—it is not possible to vary one of the parameters [HCO3–], pCO2, or pH while keeping the other two constant. However, because of the relatively slow equilibration of the spontaneous reaction CO2 + H2O ⇄ H2CO3, the interdependency of [HCO3–], pCO2, and pH can be circumvented using out-of-equilibrium solutions that are based on continuous rapid mixing and delivery of solutions with different HCO3–/CO2/H+ composition. 30 The out-of-equilibrium technique is an essential tool for mechanistic studies of acid–base sensing as it allows investigators to subject cerebrovascular preparations to experimental solutions with virtually any desired combination of [HCO3–], pCO2, and pH.24,31
Although the highlighted technical advances and new molecular information have furthered our understanding of acid–base transport and sensing in the cerebrovascular wall, additional functional and molecular information is required in order to accelerate the rational development of therapeutics for diverse cerebrovascular disease manifestations such as stroke and neurodegenerative disorders. In particular, in vivo investigations are required to understand how intrinsic contractile responses of the vascular wall are influenced by nerve activity, circulating hormones, and paracrine signals from perivascular tissue and immune cells. Much of our current knowledge derives from in vitro studies of isolated arteries. These studies present a risk of surgical damage during blood vessel isolation and are too simplistic to take into account the numerous interactions occurring in the intact organism. Still, in vitro studies have a number of technical advantages: (a) common cardiovascular side-effects of anesthetics required under in vivo conditions 32 are avoided in studies of isolated tissues, (b) acid–base parameters are much more easily manipulated than under in vivo conditions where local perfusion and metabolism as well as systemic renal and respiratory functions need to be monitored and controlled, (c) conclusive evidence regarding molecular mechanisms can more easily be reached, for instance, based on out-of-equilibrium technology,24,31 and (d) cell type-specific recordings of intracellular ion dynamics (e.g. pHi and [Ca2+]i) and membrane potentials can be performed by advanced imaging techniques19,33 more easily than in vivo where transgenic models expressing genetically encoded fluorophores may be required to ensure proper restriction of dyes to the desired cell type. 34 With the current technical limitations, in vivo and in vitro studies must generally be combined to accomplish the dual goal of identifying molecular mechanisms of acid–base-mediated vasomotor control and extrapolating their importance to integrated physiological and pathophysiological conditions.
The current review highlights and discusses the mechanisms and importance of acid–base regulation and sensing in the cerebrovascular wall in the context of metabolic regulation of cerebrovascular tone and development of cardiovascular disease.
Acid–base regulation in the vascular wall
Due to the inside-negative membrane potential and net metabolic production of acid equivalents, cells—including those of the vascular wall—are usually prone to developing intracellular acidification. At a membrane potential of –50 mV and pHo 7.4 ([H+]≈40 nM), H+ is at equilibrium across the plasma membrane when pHi is around 6.6 ([H+]≈260 nM). The typical resting pHi in endothelial cells (ECs) and vascular smooth muscle cells (VSMCs), however, is around 7.1–7.3 ([H+]≈50–80 nM),19,35–38 and hence net acid extrusion is needed to maintain normal intracellular acid–base homeostasis.
When arteries are exposed to extracellular acid–base disturbances, the intracellular environment of the cells of the vascular wall is affected to varying degrees: typically, the change in VSMC pHi in rat and mouse resistance arteries is up to 70% of the change in pHo when exposed to respiratory acidosis caused by elevated pCO2 and around 50% when exposed to metabolic acidosis accompanied by reduced [HCO3–]o.24,39,40 The plasma membrane is relatively impermeable to passive movement of H+, yet extracellular acidification inhibits the major secondary active acid extruding transporters (see the section Net acid extrusion below). 40 As a consequence, pHi falls during extracellular acidosis until a new steady-state is reached where net acid extrusion—which increases as function of decreasing pHi—again matches the metabolic acid production, H+ leaks through the plasma membrane, and any net base extrusion. The pHi value corresponding to this new steady-state depends on the pHo- and pHi-sensitivities of the involved membrane acid–base transporters and metabolic pathways, which explains the variation between different cell types. VSMCs of mouse basilar arteries are better protected against intracellular acidification than intracellular alkalinization during equivalent changes in pHo. 24 The greater tendency for changes in pCO2 than [HCO3–]o to affect VSMC pHi 40 and the vasocontractile effect of lowering [HCO3–]o per se 24 may explain why hyper- and hypocapnia with parallel changes in [HCO3–]o in some—albeit not all—studies are found to change vasomotor tone beyond the expected based on the change in pHo. 41 Indeed, current evidence supports that vasomotor adaptations in response to extracellular acidification are partly explained by the associated decrease in pHi,24,42 which—as discussed later—can lead to vasoconstriction in the acute phase31,43 but attenuates the Ca2+-sensitivity of the VSMC contractile machinery and relaxes arteries at steady-state.19,35,38,44 The effects of pHi-changes on vasomotor function highlight the need for cells to regulate intracellular acid–base conditions.
Net acid extrusion
Membrane proteins—particularly Na+,HCO3–-cotransporters of the Slc4-family and Na+/H+-exchangers of the Slc9-family—contribute to net elimination of intracellular acid equivalents from cells. Net acid extrusion from VSMCs and ECs in resistance arteries takes place predominantly via NBCn1 (Slc4a7)-mediated Na+,HCO3–-cotransport, and NHE1 (Slc9a1)-mediated Na+/H+-exchange.19,33,35,38,44 These secondary active transporters use the electrochemical gradient for Na+ to export intracellular acid equivalents45–47 (Figure 1). Whereas NHE1 has high capacity and quantitatively dominates net acid extrusion from VSMCs at low pHi, NBCn1 predominates in the near-neutral pHi range and is particularly important for maintaining steady-state pHi.19,35,38 The strong increase of Na+/H+-exchange activity as function of decreasing pHi has been explained by an intracellular allosteric H+-modifier site on the Na+/H+-exchangers.48,49 In comparison, activation of NBCn1 with decreasing pHi is more linear.33,40,50 In the cerebrovascular circulation, the importance of NBCn1-mediated Na+,HCO3–-cotransport for net acid extrusion and steady-state pHi control has been demonstrated in middle cerebral arteries.35,44 The requirement for net acid extrusion from VSMCs increases during vasoconstriction because of the contraction-associated increase in intracellular acid loading.35,51,52 The source of the increased intracellular acid load during VSMC contractions is not fully understood, but metabolic acid production and H+ imported via the plasma membrane Ca2+-ATPase51,53 (PMCA, Figure 2) likely contribute. The increased need for net acid extrusion in VSMCs during contractions is met by augmented Na+,HCO3–-cotransport.52,54 The rise in VSMC [Ca2+]i stimulates NBCn1 through activation of the serine-threonine phosphatase calcineurin that interacts with the N-terminal cytosolic domain of NBCn1 via the PTVVIH motif of splice cassette II.54,55
Schematic showing the major pathways for net cellular acid and base extrusion in the vascular wall. Net acid extrusion takes place via NBCn1-mediated Na+,HCO3–-cotransport and NHE1-mediated Na+/H+-exchange. Net base extrusion takes place primarily via Cl–/HCO3–-exchange. Schematic showing known and putative sensors for H+ and HCO3– in the vascular wall, relationships to acid–base transporters, and relevant downstream signaling pathways for regulation of arterial tone. The Ca2+ and H+ buffers partially overlap,
138
which allows acute changes in pHi to change intracellular [Ca2+] and hence arterial tone. H+ has variable effects on different K+ conductances, with BK-channels inhibited and KATP-channels activated during intracellular acidification. Regulation of gap junction communication by pHi is complex as some connexins are inhibited by both intracellular acidification and alkalinization.59,139–142 BK: large-conductance Ca2+-activated K+-channel; CA: carbonic anhydrase; KATP: ATP-sensitive K+-channel; NOS: NO-synthase; PMCA: plasma membrane Ca2+-ATPase; VGCC: voltage-gated Ca2+-channels; Vm: membrane potential. The figure is adapted from Boedtkjer et al.
47
; ©The International Union of Physiological Sciences and The American Physiological Society.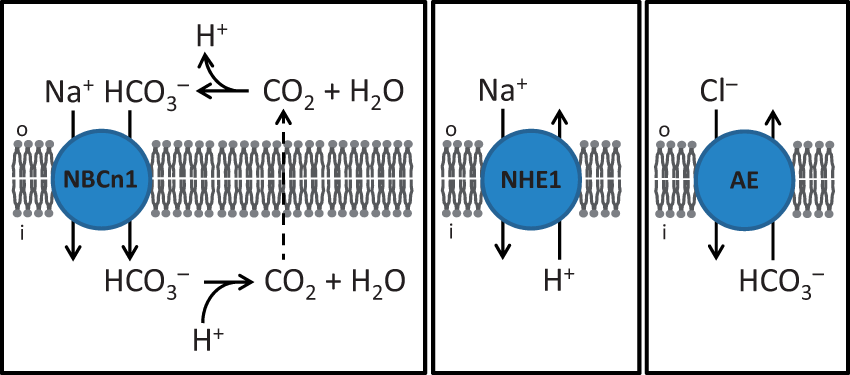
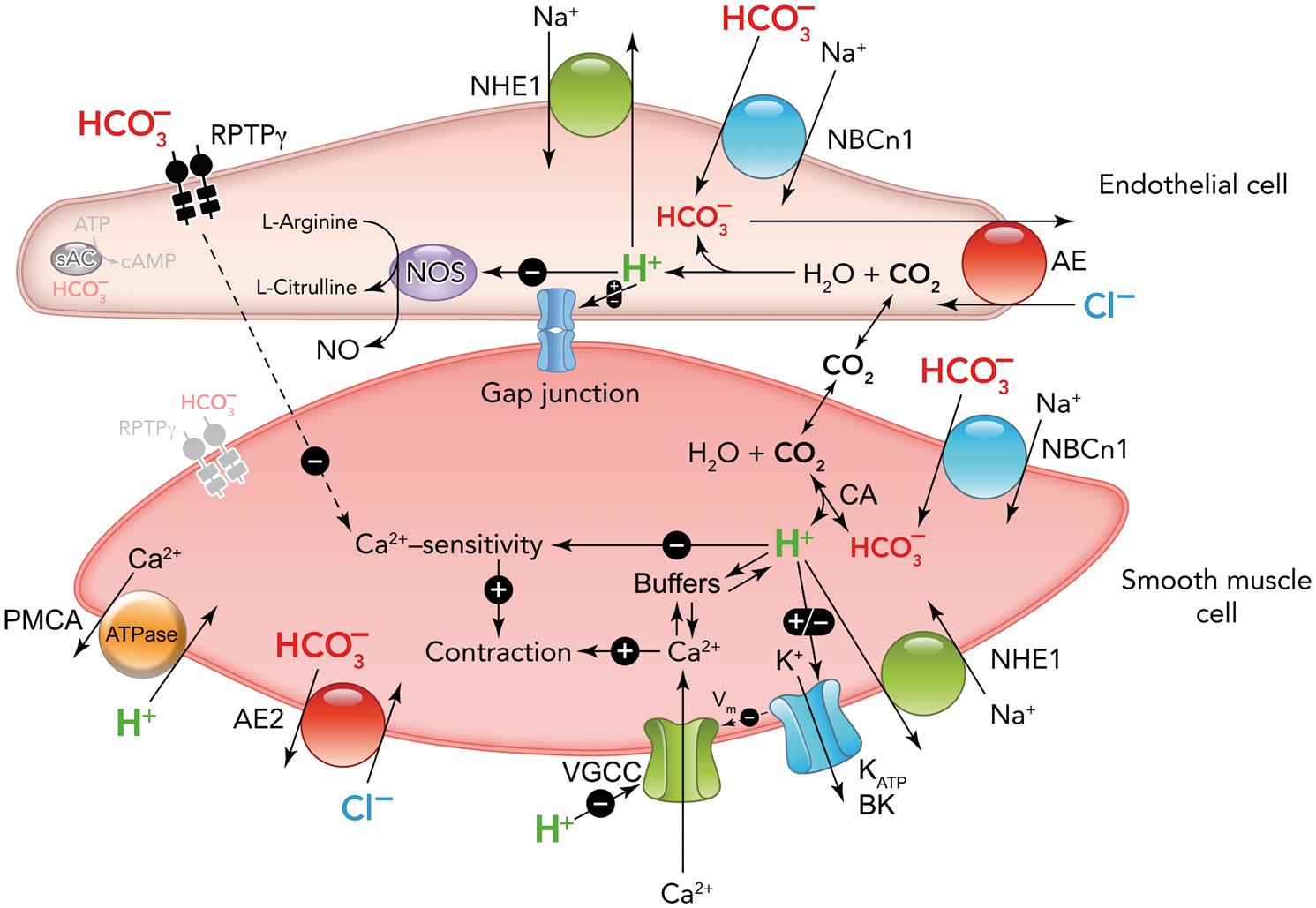
Cellular net acid extrusion is closely coupled to Na+-uptake, and it has been proposed that cellular Na+-overload with subsequent Ca2+-overload (e.g. via altered Na+/Ca2+-exchange activity) contributes to cell damage, for instance, during development of hypoxia-reoxygenation-induced endothelial dysfunction. 56 During ischemia, low pHo and increased levels of reactive oxygen species inhibit net acid extrusion activity,33,40 but the concomitantly low ATP levels reduce Na+/K+-ATPase activity and the ability to maintain normal cellular ion gradients. As a result, Na+ influx during ischemia likely exceeds the efflux capacity of the Na+/K+-ATPase. In accordance, inhibition of Na+/H+-exchange—in order to reduce the cellular Na+ load—has been found to minimize EC damage during hypoxia-reoxygenation.56,57 It is likely that reactive oxygen species 33 and low pHo 40 serve as endogenous signals that inhibit Na+-dependent net acid extrusion during ischemia. It is interesting that during extracellular acidosis, cells of the vascular wall appear to prefer control of [Na+]i over control of pHi, 40 which may be explained by the serious and irreversible damage (e.g. cell death) caused by cellular Na+- and Ca2+-overload compared to the largely reversible effects of low pHi even after long duration. 19
In addition to their contribution to global pHi regulation, it is increasingly clear—as reviewed elsewhere 58 —that acid–base transporters can establish pH micro-domains within and surrounding cells in areas with restricted diffusion. The transport mechanisms that control pH in local domains of VSMCs and ECs need further investigation but recent evidence supports that NBCn1—in a functional interaction with mobile buffer systems and carbonic anhydrases—can establish pHi gradients along the length of filopodia and thereby promote VSMC migration. 50
Net base extrusion
The mechanisms governing net elimination of intracellular base in the vascular wall are not fully understood. There is little doubt that the most important mechanism of cellular net base extrusion is Cl–/HCO3–-exchange59,60 (Figure 1) but the molecular characterization is still incomplete. Expression of AE2 (Slc4a2) and AE3 (Slc4a3) has been reported in arteries but further studies are needed to support their functional importance and the consequences of their dysfunction. 61 In addition to eliminating intracellular HCO3– during conditions of intracellular alkalinization, Cl–/HCO3–-exchange in VSMCs—and maybe ECs—is also important for maintaining [Cl–]i above electrochemical equilibrium 62 and thus for allowing Cl– channel opening to cause membrane depolarization. 47
Carbonic anhydrases
Carbonic anhydrases catalyze equilibration of the CO2/HCO3– buffer system via the reaction CO2 + OH– ⇄ HCO3–. Transcripts for carbonic anhydrase isoforms with demonstrated cytosolic (CAII, CAIII, and CAVII), mitochondrial (CAVβ), and extracellular (CAIV, CAIX, CAXII, and CAXIV) expression as well as CAVI typically secreted from cells are detectable in rat middle cerebral arteries. 31 Although their function in the vascular wall is still not comprehensively understood, carbonic anhydrases can minimize pH gradients originating from local production or transmembrane flux of acid or base50,63 and amplify pH transients in response to acute CO2/HCO3– disturbances. 31
By equilibrating the pH/CO2/HCO3– conditions, carbonic anhydrases may modify sensing of distinct buffer components and contribute to vascular tone regulation. Under in vivo conditions, carbonic anhydrase inhibitors affect systemic acid–base handling—decreasing urinary net acid excretion 64 and interfering with ventilatory control 65 —and the resulting acidosis dilates cerebral arteries. On the basis of its vasodilatory effect in vivo, the carbonic anhydrase inhibitor acetazolamide has been used for estimating the cerebral perfusion reserve in patients with cerebrovascular disease. 66 Direct cerebrovascular effects of carbonic anhydrase inhibitors are controversial.31,67–71 In rat middle cerebral arteries, carbonic anhydrase inhibition minimizes pHi transients and cerebrovascular tone development during fluctuations in the CO2/HCO3– buffer system but has no effect on pHi or artery tone at steady-state. 31 When applied at concentrations magnitudes above what is needed for carbonic anhydrase inhibition, acetazolamide and dorzolamide relax porcine retinal arteries through pH-independent mechanisms.71,72 In contrast to these findings, inhibition of carbonic anhydrase activity in guinea pig mesenteric arteries elevates pHi and decreases vascular tone also under steady-state conditions. 73 Thus, further evaluation is still required to definitively settle the cerebrovascular functions of carbonic anhydrases and the direct vasomotor effects of pharmacological inhibitors.
Carbonic anhydrase inhibition has relatively modest consequences for global pHi regulation in contractile, spindle-shaped VSMCs in the vascular wall. 31 In contrast, carbonic anhydrases play important roles for local pHi regulation in filopodia of migrating VSMCs. 50 As discussed in the section Acid–base equivalents as second messengers in vasomotor control and structural adaptations, current evidence supports that carbonic anhydrases play a key role by facilitating mobility of acid–base equivalents in the diffusion-restricted intracellular compartment of filopodia. 50
Sensing of acid–base disturbances
Protonation is an important reversible post-translational modification that affects the function and structure of most proteins. Changes in the concentration of acids and bases, for instance HCO3–, can also alter protein function independently of pH. 26 The proteins showing largest changes in activity in response to fluctuating acid–base conditions are likely candidates for acid–base sensors as they can translate even modest perturbations in for example pH or [HCO3–] into functionally relevant changes in cell and tissue function and initiate adaptive responses that reestablish homeostasis.
Acid–base disturbances adjust the function of VSMCs and ECs with consequences for vasoconstriction, vasorelaxation, and local tissue perfusion. The mechanisms by which acid–base disturbances affect cerebrovascular tone are still controversial, and the current evidence for cellular acid–base sensing and signaling mechanisms (Figure 2) will be discussed below. As discussed in the section Acid–base regulation in the vascular wall above, pHi fluctuations are typically of smaller magnitude than the underlying pHo variations during acid–base disturbances. However, the relative contribution of extracellular versus intracellular acid–base equivalents to sensing of metabolic disturbances and initiation of homeostatic responses is not yet completely clear.
Extracellular H+
The most obvious acid–base parameter to consider for regulation of vascular tone is extracellular H+, the concentration of which is increased during acidosis and decreased during alkalosis independently of the mechanistic cause. Direct vasorelaxant actions of H+—rather than secondary effects of CO2 and/or HCO3–—are supported by the observations that vasorelaxation in response to acidosis is evident also when cerebral arteries are investigated in CO2/HCO3–-free solutions 74 or under out-of-equilibrium conditions where pHo is modified while pCO2 and [HCO3–]o are kept constant at normal physiological levels (5% and 22 mM, respectively). 24 The predominant role of extracellular H+ is further supported by the finding that reduction of rat cerebral artery tension is generally more pronounced in response to extracellular than intracellular acidification. 39 Still, as discussed in more detail below (see the section Intracellular H+), several lines of evidence support that extracellular acidification can in part relax arteries via decreases in VSMC pHi.24,75–77
Inhibition of voltage-gated Ca2+ channels with consequently decreased Ca2+ uptake into VSMCs and attenuated vasoconstriction seems to be the main functional effect of a pHo decline (Figure 2). Evidence for pHo-mediated gating of L-type Ca2+-channels comes from patch clamp experiments on isolated myocytes;18,78 and the relevance for contraction of cerebral arteries is supported by reduced vasocontraction in response to depolarization with elevated extracellular [K+] at low pHo also when pCO2 and [HCO3–]o are kept constant under out-of-equilibrium conditions. 24 Inhibition of agonist-induced vasocontraction by low pHo is associated with the expected attenuation of the intracellular Ca2+ response. 24 Because L-type Ca2+-channels in many vascular preparations are absolutely necessary for sustained arterial contractions,79,80 it is meaningless to compare vasorelaxant responses to extracellular acidification under control conditions and after inhibition of L-type Ca2+-channels. As a consequence, it has been difficult to establish conclusive evidence for the role of Ca2+-channel inhibition by extracellular H+ during myogenic and agonist-induced contractions. Glutamate residues in the pore region of the L-type Ca2+ channels have been suggested to contribute to the H+-mediated inhibition based on mutagenesis studies in oocyte expression systems. 81 Future studies of H+-insensitive channel mutants in transgenic mouse models could provide more decisive evidence for the contribution of L-type Ca2+-channels to vasorelaxation induced by extracellular acidification.
Interestingly, H+ has also been suggested as a neurotransmitter contributing to neurovascular control: H+ is typically up-concentrated in synaptic vesicles and may therefore act as a co-transmitter released, for instance, from perivascular nerve endings. 82
Intracellular H+
Intracellular acidification in the vascular wall can occur by several mechanisms. Extracellular acid–base disturbances lead to parallel shifts in intracellular acid–base conditions, and intracellular acidification and alkalinization can therefore be consequences of systemic disorders (e.g. respiratory, metabolic, or renal disease) or local changes in perfusion (e.g. ischemia). Experimental or disease-related changes in the cellular acid extrusion machinery can also cause significant changes in pHi even under normal extracellular acid–base conditions: the consequences of intracellular acid–base disturbances caused by altered expression of acid–base transporters have been studied most intensively based on vascular preparations from transgenic mice,19,38 but there is also evidence that the pHi regulatory function can vary in human VSMCs and ECs based on polymorphism in SLC4A7 coding for NBCn183 and in animal models of hypertension.84–86
Intracellular acidification modifies artery tone via different pathways depending on the magnitude of the acidification and the rate of pHi-change. Abrupt VSMC acidification of large magnitude (e.g. 0.5 units within tens of seconds) consistently causes vasoconstriction of resistance arteries independent of whether it is induced by hypercapnia or by addition or washout of acid–base pairs with differing membrane permeabilities (e.g. NH4+/NH3 or CH3COOH/CH3COO–).36,43,87–89 The mechanism mediating the transient vasoconstriction caused by acute VSMC acidification is not yet completely clear. Most likely, contraction results from an increase in [Ca2+]i elicited by competition between H+ and Ca2+ for intracellular buffer binding (Figure 2).90,91 It is also possible that altered membrane excitability and transport of Ca2+ across cell or organelle membranes contribute to the vasocontractile response to acute VSMC acidification.17,43,92–96 Membrane depolarization during intracellular acidification may, for instance, be caused by inhibition of BK-channel activity (Figure 2). 17 Under physiological and most pathophysiological conditions, acidification most realistically develops over a time course of minutes to hours, and these slower intracellular decreases of VSMC pHi predominantly act by decreasing the Ca2+-sensitivity of the VSMC contractile machinery (Figure 2).19,24,35,38,44
Inhibition of net acid extrusion via NBCn1 (Figures 1 and 2) causes sustained intracellular acidification of ECs and VSMCs.19,35,44 In contrast, inhibition of NHE1 has sizable effects on steady-state pHi only when arteries are investigated in the absence of CO2/HCO3–. 38 When CO2/HCO3– is omitted from the experimental bath solutions, NBCn1 is inactive due to lack of substrate and the otherwise dominant effect of Na+,HCO3–-cotransport for setting steady-state pHi is eliminated. 19 Studying tissue from NHE1 knockout mice both in the presence and absence of CO2/HCO3– provides a valuable experimental tool for evaluating vasomotor effects of reversible intracellular acidification. 38 Acidification of ECs—by genetic disruption of NBCn1 in arteries investigated in the presence of CO2/HCO3– or NHE1 in arteries investigated in the absence of CO2/HCO3–—inhibits NO production (Figure 2).19,38,44 In congruence, cell-free assays show that the activity of the endothelial NO-synthase is pH-dependent with optimum around 7.5 and substantial inhibition when pH decreases below the physiological range. 20 NBCn1 knockout mice are mildly hypertensive at rest and display reduced blood pressure increases in response to NO-synthase inhibition, 19 which supports the conclusion that NO signaling is reduced in resistance arteries in vivo. Sustained acidification of VSMCs reduces the Ca2+-sensitivity of the contractile machinery in a rho-kinase-dependent manner;19,38,44 and in accordance, the blood pressure of NBCn1 knockout mice is less sensitive to in vivo infusion of angiotensin II compared to corresponding wild type mice. 19 Whereas the impact of NBCn1 in the vascular wall is well-established, more work is required to elucidate the role of NBCn1 in other blood pressure relevant organs such as the kidneys, nervous, and immune system. 97 In mouse middle cerebral arteries, knockout of NBCn1 decreases the NO-mediated inhibition and rho-kinase-mediated augmentation of myogenic tone development. 44 The attenuated vasomotor responses and reduced ability of cerebral arteries to respond to transmural pressure changes will likely lead to inappropriate fluctuations in cerebral perfusion and capillary pressure during variations in blood pressure. Lack of NBCn1 also inhibits the rhythmic, oscillatory contractile pattern, known as vasomotion, in middle cerebral arteries. 44 Although the physiological and/or pathophysiological significance of vasomotion is still unclear, reduced tissue oxygenation in individuals showing attenuated vasomotion has been proposed 98 and vasomotion may be impaired in Alzheimer’s disease. 99 Supporting the human cardiovascular relevance of Na+,HCO3–-cotransport and pHi in the vascular wall, single nucleotide polymorphisms in SLC4A7 (rs13082711 and rs820430) are associated with human blood pressure variation,100,101 increased NBCn1 expression,83,102 and elevated Na+,HCO3–-cotransport activity and steady-state pHi in VSMCs. 83
In addition to direct effects of extracellular H+ (see the section Extracellular H+ above), increasing evidence supports that extracellular acidification can relax arteries via secondary changes in pHi (Figure 2). The decrease in VSMC Ca2+-sensitivity demonstrated during sustained intracellular acidification under standard conditions of pHo 7.419,38,44 is also observed in mouse basilar arteries during extracellular acidification as evidenced by a greater reduction in artery tone than would be predicted based on the decrease in [Ca2+]i alone. 24 Furthermore, complex effects of pHi are reported for ryanodine receptor-mediated release of Ca2+ from intracellular stores: 103 in brain parenchymal arteries, extracellular acidosis—presumably via lowering of pHi—was found to modulate sarcoplasmic reticulum Ca2+-release from a pattern dominated by Ca2+ waves at normal pH to mostly Ca2+ sparks at low pH. Hence, low pH was suggested to cause vasorelaxation by indirectly activating large-conductance Ca2+-activated K+-channels (BKCa-channels) via enhanced ryanodine-receptor-mediated Ca2+ release. 75 This effect is in contrast to the direct inhibitory effect of low pHi on BK-channel activity observed in patch clamp experiments where [Ca2+]i is kept constant. 17 An additional proposed mechanism by which intracellular acidification—as primary disturbance or secondary to extracellular acidosis—can cause vasorelaxation is via activation of ATP-sensitive K+-channels (KATP channels)76,77 that contain an intracellular activator site for H+ binding (Figure 2). 104
The contrasting vasomotor responses elicited by acute and sustained intracellular acidification are consistent with a major effect of H+ in the acute phase releasing Ca2+ as it shifts the protonation of intracellular buffers:90,91 acute acidification has been found to cause a transient, membrane potential-independent increase in [Ca2+]i that leads to contraction, 92 but when the buffers have reached their new equilibrium, they provide a more or less normal buffering function as evidenced by unaffected Ca2+ responses during sustained acidification.19,38,44 The more subtle effects of H+ on the Ca2+-sensitivity of the VSMC contractile machinery inhibit contractions during sustained acidification19,38,44 but are most likely overwhelmed by the massive increase in intracellular [Ca2+] observed in the acute phase (Figure 2).
VSMC pHi in basilar arteries is more sensitive to intracellular alkalinization than acidification, as pHi changes are approximately 3-fold larger when the arteries are exposed to the same pHo disturbance in the alkaline compared to the acidic direction. 24 Not much is known about the sensitivity of EC pHi to changes in extracellular acid–base conditions. In ECs of mouse mesenteric arteries, intracellular alkalinization inhibits gap junctional communication between neighboring ECs and between ECs and VSMCs (Figure 2). 59 Because gap junctions in many vascular beds are important for conducted responses—typically, hyperpolarization traveling from capillaries or arterioles through cell-cell contacts to larger upstream feed arteries 105 —and endothelium-dependent hyperpolarizations,106–108 regulation of gap junctions by pHi has potential implications for small artery function and local control of tissue perfusion. Indeed, alkalinization of ECs in mouse mesenteric arteries inhibits endothelium-dependent vasorelaxation by decreasing endothelium-dependent hyperpolarizations of VSMCs. 59 Inhibition of current transfer between ECs and VSMCs at elevated pHi is confirmed by the finding that VSMC hyperpolarizations are absent, or very substantially reduced, despite normal EC hyperpolarizations. 59
Extracellular HCO3–
Sensing of acid–base parameters other than pH provides a mechanism for adjusting vascular resistance according to the cause of the acid–base disturbance. When [HCO3–] and/or pCO2 are sensed in addition to pH, metabolic and respiratory disturbances give rise to separate downstream signaling events and differentiated vascular responses may be initiated, for instance, in response to fully or partially compensated systemic acid–base disturbances compared to local acid–base disturbances developed due to mismatch between local blood flow and metabolism.
Vasocontractile responses to decreases in [HCO3–]o were recently demonstrated under out-of-equilibrium conditions, i.e. with pHo maintained at 7.4 and CO2 kept constant at 5%. 24 The regulation of vascular tone by extracellular HCO3– requires the single-pass transmembrane protein receptor protein tyrosine phosphatase (RPTP)γ, which is encoded by Ptprg and expressed in cerebral arteries. 24 Based on sequence similarity between the extracellular carbonic anhydrase-like domain of RPTPγ and the active site of the carbonic anhydrases, it is proposed that HCO3– binds to the extracellular aspect of RPTPγ and initiates cellular signaling responses via the intracellular phosphatase domain (Figure 2). RPTPγ is not expected to possess carbonic anhydrase activity as the carbonic anhydrase-like domain of RPTPγ lacks critical histidine residues required for catalytic activity. 109 Consistent with predominant Ptprg promoter activity in the vascular endothelium, the vasomotor effect of RPTPγ is endothelium-dependent. 24 The intra- and intercellular signaling pathways downstream of RPTPγ are still to be unraveled.
The vasocontractile response to decreases in [HCO3–]o counteracts the predominant vasorelaxation caused by the reduction in pHo during metabolic acidosis. 24 As discussed in more detail later (see the section Braking actions on vasorelaxation in response to extracellular acidosis), limiting vasorelaxation during acidosis could be important for tailoring the increase in capillary pressure, which if unopposed may lead to edema formation and capillary damage and thereby worsen the consequences of local inadequate perfusion.
Intracellular HCO3–
During acid–base disturbances, [HCO3–]i changes as function of pHi and pCO2. 24 Although experimental verification is needed, it is possible that changes in [HCO3–]i can modify vascular tone by altering the activity of the HCO3–-sensitive soluble adenylyl cyclase (sAC), which is expressed in vascular ECs (Figure 2). 110 It is not yet clear that HCO3–-stimulated cAMP production has functional effects in the vascular wall but cAMP has been shown to modify intercellular coupling 111 and cause vasorelaxation at least partly through enhanced endothelial NO production. 112
CO2
The potential direct signaling role of molecular CO2 for cerebrovascular relaxation during hypercapnic acidosis is still debated. 41 Whereas experimental increases in pCO2 under equilibrated conditions can be used to mimic respiratory acidosis, the net vasomotor response to hypercapnia is not evidence of a direct CO2-induced effect: under standard experimental conditions, primary changes in pCO2 inevitably lead to secondary changes in intra- and extracellular [H+] and/or [HCO3–]. Indeed, when pCO2 is raised selectively under out-of-equilibrium conditions—i.e. without concomitant changes in pHo or [HCO3–]o—no net vasomotor effect of hyper- or hypocapnia is observed at steady-state. 24 Yet, abrupt simultaneous increases of pCO2 and [HCO3–]o—with pHo maintained constant at 7.4—induce transient cerebrovascular contraction.31,43 This acute CO2/HCO3–-induced vasocontraction is attenuated by inhibition of intracellular carbonic anhydrases that—in the uninhibited state—increase the rate of CO2 hydration and thereby accelerate and amplify the imposed pHi decrease. 31 These findings support that vasocontraction induced by acute hypercapnia most likely results from CO2-induced intracellular acidification.
Taken together, the current evidence supports that the vasomotor effects of CO2 act via decreases in pHo and pHi. The apparent absence of a direct CO2-induced vasomotor effect is not contradictory to the well-established finding that respiratory acidosis causes vasorelaxation 41 or that hyperventilation-induced respiratory alkalosis via cerebrovascular constriction can reduce capillary pressure and filtration to acutely alleviate increased intracranial pressure. 113 Instead, changes in pH—perhaps with smaller contributions from concomitant changes in [HCO3–]—are the dominant signals leading to altered vascular tone during changes in pCO2. Because intracellular and extracellular acidification can have opposing effects on VSMC contractions (see the sections Extracellular H+ and Intracellular H+), the net contractile response to primary changes in pCO2 depends on the rate and magnitude of the secondary pHi and pHo fluctuations. Although CO2 has no net vasomotor effect at steady-state—i.e. when the transient pHi decrease has waned and the acid–base reactions have reached their new equilibria—we cannot fully exclude that CO2 modifies several signaling pathways: CO2 may for instance act both as a molecule per se and through pHi and pHo, and these individual effects could be opposing, resulting in no significant net effect.
Ion conductances sensitive to acid–base disturbances
A number of ion conductances are sensitive to local acid–base disturbances. As highlighted above (see the section Extracellular H+), pH-mediated inhibition of voltage-gated Ca2+-channels has been demonstrated in patch-clamp experiments and is considered a prominent underlying mechanism for vasorelaxation in response to extracellular acidosis.
K+-channels sensitive to pH (e.g. KATP-channels, BKCa-channels, and acid-sensing ion channels) have also been implicated in the local regulation of cerebrovascular tone.75,114–116 In particular, KATP-channels are suggested to play major roles for metabolic regulation of blood flow: activation of KATP-channels in presence of a low [ATP]/[ADP] ratio and in response to hypercapnic acidosis supports that insufficient nutrient levels can influence the VSMC membrane potential (Figure 2).117,118 The possibility that the VSMC membrane potential is affected by several acid–base parameters—e.g. extracellular and intracellular H+ as well as CO2/HCO3– buffer components—is supported by the observation that hypercapnic acidosis hyperpolarizes, whereas normocapnic acidosis depolarizes rat cerebral arteries. 42
Even though patch clamp experiments have identified multiple pHi-sensitive ion channels, our findings from isolated arteries support that sustained intracellular acid–base disturbances affect vascular tone mostly via changes in VSMC Ca2+-sensitivity independently of membrane excitability.19,38,44
Braking actions on vasorelaxation in response to extracellular acidosis
The vasorelaxant response to extracellular acidification increases cerebral perfusion, for instance, in the ischemic penumbra and thus improves the immediate oxygen delivery. However, the drop in pre-capillary vascular resistance also tends to increase capillary pressures and filtration, which may have detrimental consequences by worsening the edema typical for ischemic tissue. Increasing the interstitial pressures within the non-distensible skull can also limit cerebral perfusion. Cerebrovascular tone should therefore be very accurately controlled during ischemia in order to optimize local perfusion without causing tissue injury due to elevated intracranial pressure.
As described in the sections Extracellular H+ and Intracellular H+, vasorelaxation in response to extracellular acidification depends on reductions in pHo and sustained decreases in VSMC pHi. More recently, it has been demonstrated that changes in local concentrations of acid–base buffers, principally [HCO3–]o, modify the vasomotor response to pH disturbances. As discussed in the sections Extracellular HCO3– and CO2, recent studies show that acute CO2-dependent decreases in pHi 31 and reductions in [HCO3–]o 24 elicit vasocontraction; and it is predicted that these contractions counteract the vasorelaxant response to decreases in pHo during respiratory and metabolic acidosis, respectively. Effects of disturbed extracellular acid–base conditions on EC pHi have not yet been settled. However, inhibition of NO synthesis by low EC pHi19,20,38,44,119 represents a likely additional braking mechanism on vasorelaxation in response to extracellular acidosis.
The anti-relaxant influences of lowered [HCO3–]o, reduced EC pHi, and acutely decreased VSMC pHi may seem counterintuitive at first because they oppose the predominant vasorelaxant response observed during extracellular acidification. Parallel activation of opposing signaling pathways, however, is in line with the observation that many vasocontractile agonists (e.g. endothelin-1 and insulin) activate both vasocontractile and vasorelaxant signaling, which can be unmasked if individual signaling pathways are inhibited.120,121 Simultaneous activation of counteracting responses by the same agonist is typically explained by stimulation of multiple receptor subtypes and/or receptors in both ECs and VSMCs. Parallel activation of vasocontractile and vasorelaxant signaling pathways may prime the system to provide a more dynamic and accurate mode of vasomotor control than can be achieved with a simpler single-effector pathway.
Despite growing evidence that changes in pCO2 and [HCO3–]o limit the vasorelaxant response during acidosis either directly (e.g. HCO3– acting via RPTPγ) 24 or indirectly (e.g. CO2 acting via pHi), 31 more experimental work is needed to clarify the physiological and pathophysiological implications. Particularly, the in vivo consequences of RPTPγ knockout for vascular responses and cerebral perfusion during systemic (i.e. metabolic or respiratory) and local (e.g. ischemia-associated) acid–base disturbances should be defined in order to verify the concept that simultaneous activation of multiple cellular signaling pathways by acid–base equivalents optimizes local perfusion while limiting the risk of cerebral edema.
Acid–base equivalents as second messengers in vasomotor control and structural adaptations
Because pH-changes modify the activity of functional proteins, including enzymes and ion channels important for cellular signaling events, H+ has been proposed as a second messenger: for instance, bradykinin causes sustained intracellular alkalinization of cultured ECs, and this increase in pHi may serve as a persisting signal for activation of the intrinsically pH-sensitive endothelial NO-synthase when the transient intracellular Ca2+-response has waned. 20 Moreover, EC pHi-changes in response to shear stress122–124 may modify the activity of the endothelial NO-synthase and contribute to flow-mediated responses in artery tone. Rather than simply contributing to homeostasis, it is possible that local transport and sensing of acid–base equivalents can initiate dynamic tissue responses. Because the dynamic pHi effects of agonists and shear-stress have primarily been suggested based on ECs in culture, additional work is required to determine if they exist in arteries and are of consequence for control of vascular tone development.
Considering the widespread effects of pH on multiple proteins and the possibility of generating pHi and pHo gradients within diffusion-restricted compartments, it is probable that H+ serves a second messenger role by spatially coordinating the activity of enzymes and other functional and structural proteins. This may be important, for instance, during cell migration where longitudinal pH gradients are observed along the axis of migration. NBCn1 is responsible for establishing pHi gradients in migrating VSMCs and the migratory rate is substantially reduced when the pHi gradients are experimentally diminished. 50 NHE1 does not generate similar spatial pHi patterns under physiologically buffered conditions, but in the absence of CO2/HCO3–, pHi gradients re-appear in filopodia of VSMCs from NBCn1 knockout mice suggesting that NHE1 can generate spatial gradients for H+ when buffer capacity and buffer mobility are low. 50 NHE1 has also been suggested to facilitate cell migration and proliferation by serving as a scaffold for protein interactions and as an element in signaling cascades. 58 Consistent with a role for NHE1 in VSMC migration and arterial remodeling, mesenteric arteries from NHE1 knockout mice are thin-walled, 38 NHE1 knockout mice are protected against hypoxia-induced pulmonary hypertension and pulmonary artery remodeling, 125 and NHE1 knockdown has been found to inhibit proliferation and migration of isolated VSMCs. 126 So far, however, it remains unknown whether NBCn1 and/or NHE1 contribute to the plasticity of the cerebral vasculature, including generation of collateral blood vessels during hypoxia and in response to ischemic insults.
Other potential signals for metabolic blood flow regulation
In addition to changes in the concentrations of H+, CO2, and HCO3–, other signals of insufficient local perfusion include decreases in pO2 and increases in the concentrations of K+, lactate, and adenosine. 16 [K+]o has received particular interest because even moderate increases in the local concentration (e.g. to 10–15 mM, within the range observed in brain interstitial fluid during ischemia) 127 lead to vasorelaxation. 128 Confined increases in [K+]o can hyperpolarize VSMCs locally 129 or act on capillary ECs that initiate retrograde conducted responses into the resistance arteries; 130 in both cases, inward-rectifier K+-channels are believed to act as the molecular sensors that are activated in response to the local rise in [K+]o.
Lactate is generated in response to increased metabolism particularly under anaerobic conditions. The relevance of lactate as a signaling molecule in the vascular wall remains elusive but elevated concentrations of lactate have been found to reduce the responsiveness of VSMCs to vasoconstrictors. 131 Adenosine is released from tissue during metabolic activity and can cause hyperpolarization and relaxation of VSMCs locally and signal to upstream feed arteries via conducted responses in the VSMC layer.106,132,133 Thus, although vasomotor effects of lactate and adenosine need further investigation in the cerebrovascular wall, these markers of local increased, and possibly unmet, metabolic demand could contribute to metabolic regulation of cerebral blood flow.
As described above and illustrated in Figure 2, local signaling in the arterial wall involves a number of acid–base equivalents as well as other local waste products and signaling metabolites. The highlighted findings mostly relate to in vitro studies of artery function. Because the experimental conditions can be less precisely controlled under in vivo conditions, deciphering cellular signaling mechanisms in living animals is extremely challenging. It should be noted, however, that acid–base equivalents may also act on cells in the perivascular tissue and regulate vasomotor tone, for instance, via NO synthesized in perivascular nerves. 134 The relative contribution of the highlighted signals of insufficient local perfusion and other potential signaling molecules has not yet been established and more work—particularly integrated studies of cerebrovascular preparations from animals with targeted disruption of known and putative acid–base transporters and sensors (Figure 2)—is needed to clarify their physiological and pathophysiological importance.
Conclusions and perspectives
Responses to acid–base disturbances are complex with multiple pathways activated or inhibited by H+ and HCO3– acting inside or on the outside of VSMCs and ECs. The signaling pathways modulated by H+ and HCO3– act in parallel and have potential to fine-tune vascular tone and adapt blood flow in response to physiological and pathophysiological changes in the local metabolic demand. Recent evidence suggests that decreases in [HCO3–]o and acute reductions in VSMC pHi—possibly together with intracellular acidification of ECs—counteract the vasorelaxation induced by low pHo. In this manner, sensing of the CO2/HCO3– buffer composition can initiate downstream responses that protect against cerebral edema, which could otherwise be the consequence of unopposed vasodilation.
Understanding the mechanisms of acid–base-induced vasomotor control opens attractive prospects for new pharmacological approaches that can increase blood flow specifically in metabolically compromised tissue and reduce the consequences of ischemic cerebrovascular disease. Although suitable pharmacological tools against acid–base transporters and sensors are not yet available,45,135,136 it is probable that spatial specificity to the ischemic region can be achieved by use of acid-activated compounds or by targeting cellular signaling pathways with increased activity under the disturbed acid–base conditions. Drugs that specifically dilate arteries supplying ischemic brain tissue and the surrounding penumbra will likely be much more effective than agents inducing generalized systemic vasodilation: universal vasodilation can decrease the cerebral perfusion pressure and lead to vascular intracerebral steal phenomena 137 where blood is re-routed from ischemic tissue and brain regions with borderline perfusion to heathy parts of the circulation with larger blood flow reserve. Pharmacological treatment modalities that improve blood flow particularly in the ischemic penumbra may minimize the infarct size and buy the clinician enough time to implement relevant revascularization procedures.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Related work in the author’s laboratory was supported financially by the Danish Council for Independent Research (grants no. 10-094816 and 4183-00258B), the Lundbeck Foundation (grant no. R93-2011-8859), the Danish Heart Foundation (grants no. 08-10-R68-A2179-B719-22494 and 14-R97-A5321-22809), and the Novo Nordisk Foundation (grants no. 2131 and 7393).
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Ebbe Boedtkjer is inventor on a patent application describing tools for manipulating acid–base transport.