
Abstract
Aging leads to a gradual decline in the fidelity of cerebral blood flow (CBF) responses to neuronal activation, resulting in an increased risk for stroke and dementia. However, it is currently unknown when age-related cerebrovascular dysfunction starts or which vascular components and functions are first affected. The aim of this study was to examine the function of microcirculation throughout aging in mice. Microcirculation was challenged by inhalation of 5% and 10% CO2 or by forepaw stimulation in 6-week, 8-month, and 12-month-old FVB/N mice. The resulting dilation of pial vessels and increase in CBF was measured by intravital fluorescence microscopy and laser Doppler fluxmetry, respectively. Neurovascular coupling and astrocytic endfoot Ca2+ were measured in acute brain slices from 18-month-old mice. We did not reveal any changes in CBF after CO2 reactivity up to an age of 12 months. However, direct visualization of pial vessels by in vivo microscopy showed a significant, age-dependent loss of CO2 reactivity starting at 8 months of age. At the same age neurovascular coupling was also significantly affected. These results suggest that aging does not affect cerebral vessel function simultaneously, but starts in pial microvessels months before global changes in CBF are detectable.
INTRODUCTION
Owing to the high energy demands of membrane potential repolarization, neuronal activation needs to be tightly matched to cerebral blood flow (CBF). Therefore, neurons, glia, and vascular cells of the neurovascular unit (NVU) interact to increase CBF in response to neuronal activation through a mechanism known as neurovascular coupling (NVC). 1 The resultant functional hyperemia ensures that neuronal energy demands are satisfied by the timely delivery of oxygen and glucose.
Aging is known to have profound effects on NVC and CBF, thereby contributing to an increased risk of stroke and possibly dementia.2,3 Resting CBF and neuronal activity-mediated increases in CBF have been reported to decrease with age,4,5 and decreases in resting CBF and cerebrovascular reactivity to neuronal activation are associated with an elevated risk of cerebrovascular disease. 6 Despite the importance of NVC for proper function of the brain and the profound effects of aging on CBF regulation, significant gaps remain in our knowledge of age-related cerebrovascular dysfunction (ACD). For instance, it is unknown if all cerebral vessels are affected by ACD simultaneously or if ACD starts in pial vessels and proceeds down to parenchymal vessels at a later stage as observed in animal models of small-vessel disease. 7 Therefore, the aim of the current study was to investigate the effect of aging on CBF regulation and reactivity of the cerebral microcirculation.
MATERIALS AND METHODS
SUBJECTS
Animal breeding, housing, and all experimental procedures were conducted according to institutional guidelines of the University of Munich and were approved by the Ethical Review Board of the Government of Upper Bavaria and the Institutional Animal Care and Use Committee of the University of Vermont. In vivo experiments were conducted on 6-week, 8-month, and 12-month-old male and female FVB/N mice bred at the Center for Neuropathology, University of Munich (Munich, Germany) and are reported according to the ARRIVE criteria.
Two- to three-month-old male C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, MA, USA) and 18-month-old male C57BL/6 mice were obtained from the National Institutes of Aging (USA). All animal cohorts were group housed and kept on a 12-hour light:dark cycle with ad libitum access to food and water.
Anesthesia and Physiologic Monitoring
For in vivo experiments on CO2 reactivity, anesthesia was induced by intraperitoneal injection of midazolam (5 mg/kg; Braun, Melsungen, Germany), fentanyl (0.05 mg/kg; Janssen-Cilag, Neuss, Germany), and medetomidine (0.5 mg/kg; Pfizer, Karlsruhe, Germany) and was maintained for up to 4 hours by hourly injections of one-quarter of the initial dose, as previously described.8–11
For in vivo experiments on NVC mice were initially anesthetized with 2% isoflurane in 70% N2O and 30% O2. Later on isoflurane was gradually reduced over the course of 10 minutes to a range of 0.5% to 0.9% in 70% room air and 30% O2, and at the same time, a continuous intraarterial infusion of ketamine (30 mg/kg/h, Inresa, Freiburg, Germany) was administrated.
Mice were orotracheally intubated and mechanically ventilated (Minivent, Hugo Sachs, Hugstetten, Germany). End-tidal pCO2 was measured continuously with a microcapnometer (Capnograph, Hugo Sachs, Hugstetten, Germany) and kept constant between 20 and 30 mm Hg by respective adjustments to the ventilation frequency to obtain arterial blood gas values physiologic for FVB/N mice, which are ~10 mm Hg lower than in C57BL/6 mice.8,12 A thermostatically regulated, feedback-controlled heating pad (FHC, Bowdoin, ME, USA) was used to maintain body temperature at 37°C. The left femoral artery was cannulated for continuous blood pressure monitoring and for infusion of 240 μL/h physiologic saline solution to prevent dehydration of the mice. Regional CBF (rCBF) was measured with a laser Doppler probe placed over the right somatosensory cortex (NVC) or over the territory of the left middle cerebral artery (CO2 reactivity).
Imaging of Cerebral Microvessels In Vivo
A cranial window (4 × 4 mm) was drilled under constant cooling above the right parietal cortex leaving the dura mater intact as previously described.11,13–16 Animals were placed under an epifluorescence microscope (Axio Scope Vario, Zeiss, Oberkochen, Germany) and the exposed dura mater was kept wet at all times with isotonic saline. The plasma was stained by an intraarterial injection of fluorescein isothiocyanate (FITC–dextran, molecular weight 150 kDa; 0.05 ml of a 0.5% solution; Sigma-Aldrich, Deisenhofen, Germany) and pial microvessels (diameter 20 to 40 μm) were visualized by epifluorescence imaging (excitation 470 nm; emission 527 nm) using a × 10 W N-Achroplan objective (Zeiss; numerical aperture (NA): 0.3). Four regions of interest containing arterioles, venules, and capillaries were investigated (Supplementary Figure S1A). Vessel diameters were quantified using a calibrated image analysis software (Zen, Zeiss) and expressed in percentage of baseline.
Neurovascular Reactivity to CO2
Cerebral blood flow and diameter of pial vessels were examined for at least 10 minutes under physiologic, stable conditions, and the mean values obtained were taken as a baseline. Thereafter, CBF and vessel diameter were observed during inhalation of 5% CO2 for 15 minutes as previously described.17,18 After a break of 15 minutes the procedure was repeated with 10% CO2 (Supplementary Figure S1B). The amount of inhaled CO2 was measured by microcapnometry (Supplementary Figure S1C).
Forepaw-Evoked Neurovascular Coupling
The left forepaw was stimulated with two subdermally inserted needle electrodes with a diameter of 0.2 mm (Hwato, Suzhou, China) at an intensity of 2 mA for 0.3 ms (Digitimer, Hertfordshire, England). One stimulation cycle contained 96 stimulations and lasted for 16 seconds (6 Hz). The interval between two stimulation cycles was 40 seconds (Figure 1A). To identify the exact location of the somatosensory cortex representing the left forepaw, a train of 10 stimuli was applied and the rCBF response (Figure 1D) was assessed at five different locations within the somatosensory cortex. The region with the strongest synchronous response to stimulation was used for analysis and further for assessment of the microvascular response by in vivo microscopy.
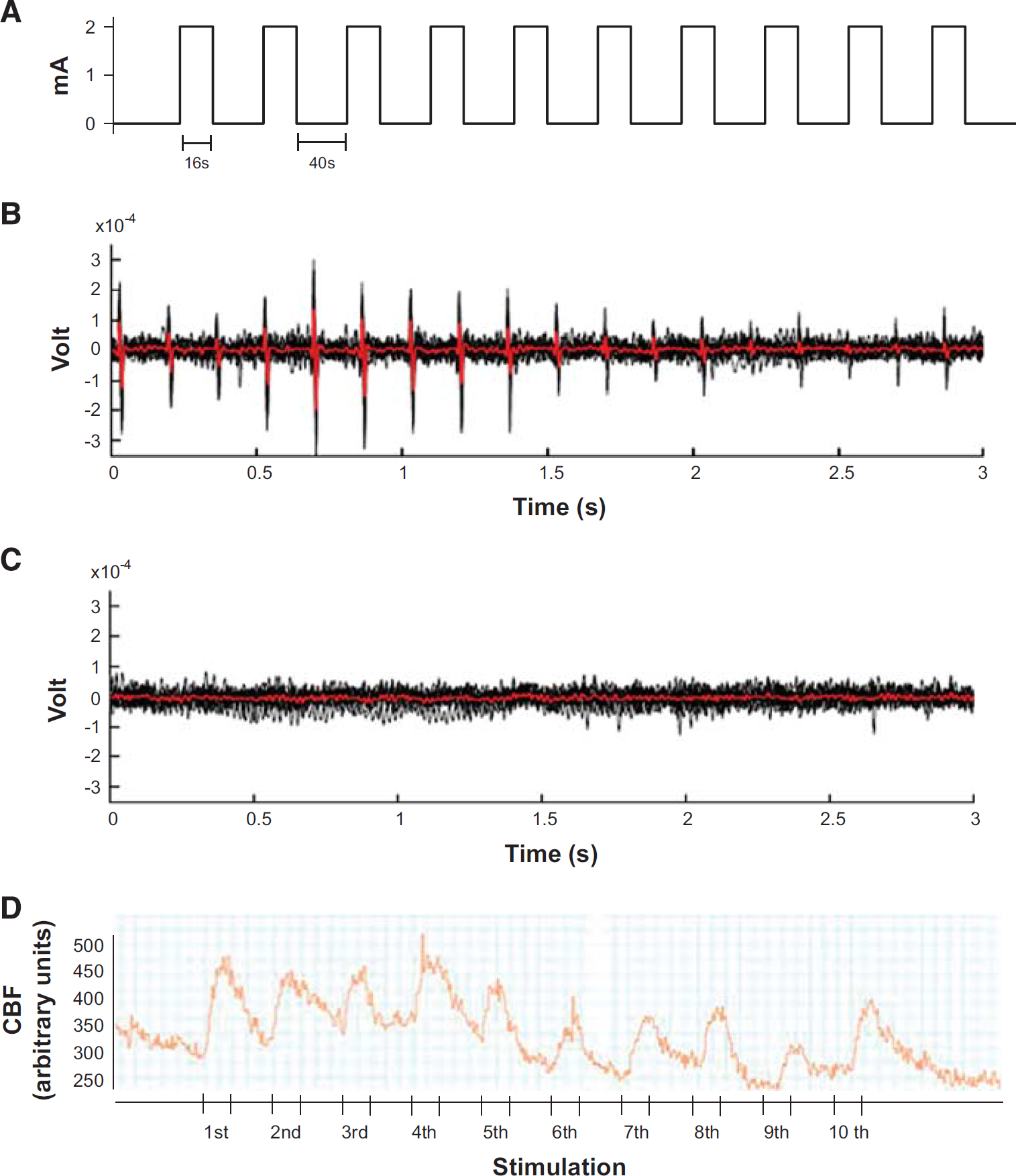
Experimental set up for the investigation of neurovascular coupling. (
To prove that our stimulation protocol indeed evoked neuronal activity, neuronal activation was assessed 3 seconds after the onset (Figure 1B) and 3 seconds after the end of each pulse train (Figure 1C) using custom-built cortical surface electrodes connected to an FE136 animal bioamplifier (ADInstruments, Oxford, UK). Only the recordings corresponding to the stimulus pulse trains showed the typical neuronal response to the single stimulus pulses with intervals of 167 ms.
Brain Slice Preparation and Imaging
Brain slices were prepared essentially as previously described. 19 Briefly, mice were killed by pentobarbital overdose and decapitated. The brain was rapidly removed into ice-cold artificial cerebrospinal fluid (aCSF) containing 124 mmol/L NaCl, 3 mmol/L KCl, 2 mmol/L CaCl2, 2 mmol/L MgCl2, 1.25 mmol/L NaH2PO4, 26 mmol/L NaHCO3, and 4 mmol/L glucose; the aCSF used during slice preparation also contained 0.4 mmol/L ascorbic acid. Slices (160 μm) were prepared using a Leica VT1000 S vibratome (Leica, Buffalo Grove, IL, USA) and stored in oxygenated aCSF. Slices were then loaded with 10 μmol/L Fluo-4 AM (Invitrogen, Waltham, MA, USA) in aCSF containing 2.5 μg/mL pluronic acid for 1.5 hours at 32°C, and arteriolar diameter was measured and Ca2+ imaging was performed as previously described. Experimental solutions—gassed with 20% O2, 5% CO2, and balance N2—contained 125 nm of the thromboxane analog U46619 to preconstrict arterioles (by 47% ± 4%, n = 9) and mimic physiologic tone. To enable offline quantification of endfoot [Ca2+]i, we treated a subgroup of slices with 10μmol/L ionomycin and 20 mmol/L [Ca2+]o to saturate loaded Fluo-4 AM (Invitrogen) and obtain a maximal fluorescence measurement. All other experiments were concluded by obtaining the passive vessel diameter by perfusing the slice with aCSF containing 50 μmol/L diltiazem, 200 μmol/L papaverine, 0 CaCl2 and 5 mmol/L EGTA (in the absence of U46619) to allow assessment of arteriolar tone. The intensity and pulse pattern of electrical field stimulation remained constant throughout all experiments (a 3-second train delivering a 20 V, 50 Hz alternating square pulse of 0.3 ms duration).
Immunofluorescence Staining
Anesthetized mice were injected intraarterially with 30 μL of FITC-labeled tomato lectin, which stains vessels by direct binding to endothelial glycoproteins. Five minutes thereafter mice were transcardially perfused with sodium chloride and 4% paraformaldehyde. Brains were removed and fixed overnight in 4% paraformaldehyde. Coronal sections (50 μm thick) were made using a vibratome (Leica VS1200, Leica, Nussloch, Germany) and collected in phosphate-buffered saline. Free-floating sections from the rostral, medial, and dorsal brain were used for immunostaining. Sections were blocked with 3% bovine serum albumin for 60 minutes and incubated overnight at 4°C with the primary antibody in blocking solution. The following primary antibodies were used: for smooth muscle cells, Cy3-conjugated antialpha smooth muscle actin (1:100; Sigma-Aldrich, Taufkirchen, Germany, C6198); for pericytes, goat anti-platelet-derived growth factor receptor β (PDGFRβ) (1:100, R&D Systems, Wiesbaden, Germany, AF1042); for astrocytes, mouse anti-glial fibrillary acidic protein (GFAP) (1:100; Sigma-Aldrich, G3893); and for plasma protein (albumin), mouse antialbumin (1:100; Sigma-Aldrich, A6684). Sections were then washed three times in phosphate-buffered saline, blocked with 3% bovine serum albumin for 30 minutes, and incubated with fluorophore-conjugated secondary antibodies for 2 hours at room temperature. To visualize PGDFRβ-positive pericytes, sections were incubated in secondary Cy3-conjugated donkey antigoat antibody (1:100, Jackson ImmunoResearch, Suffolk, UK, 705165147); to visualize GFAP-positive astrocytes, sections were incubated in secondary Cy3-conjugated donkey antimouse antibody (1:100, Jackson ImmunoResearch, 715165150); and to visualize albumin, sections were incubated in secondary Cy3-conjugated donkey antimouse antibody (1:100, Jackson ImmunoResearch, 715165150). Sections were subsequently washed and mounted on slides using Fluoromount mounting medium (Sigma-Aldrich).
Imaging Acquisition and Analysis
All mounted sections were examined under a fluorescent (Axiovert 200M, Zeiss) or a confocal microscope (Leica TCS SP5 II, Wetzlar, Germany). Quantitative image analysis was performed by a blinded investigator using the software ImageJ (National Institute of Health, Bethesda, MD, USA). In each mouse, four regions of interests from the cortex were analyzed. This analysis was performed in three nonadjacent sections 100 μm apart from six animals per group. Capillaries were identified by positive FITC-lectin labeling and capillary density was quantified by subtracting the background and counting all FITC-positive pixels on maximum intensity projection confocal images using ImageJ. Smooth muscle cell coverage was determined by calculating the number of alpha-smooth muscle actin (α-sma)-positive pixels as a percentage of FITC-lectin-positive pixels per field (410×410 μm). Pericyte coverage was evaluated in the same way by the area occupancy of PDGFβR and lectin signals, respectively. Pericyte number was obtained by counting the number of PDGFβR-positive pericytes per mm2 of selected field area. Astrocyte endfoot coverage was determined as a percentage of GFAP-positive astrocyte surface area covering lectin-positive capillary surface area per field. Extravascular albumin deposition was quantified on maximum intensity projection confocal images using ImageJ.
Statistical Analysis
Depending on whether data were normally distributed or not results are presented as mean±s.e.m. or as median ± 75/25 percentile, respectively, and respective statistical tests were used to test for differences between groups/time points.
For the in vivo study, statistical analysis was performed with a standard statistical software package (SigmaPlot 12.5; Systat Software, Erkrath, Germany). Differences across groups were evaluated using the Mann–Whitney rank sum test with the Bonferroni correction.
For the in situ study and immunohistologic experiments, statistical analyses were performed using Prism 5 (GraphPad Software, La Jolla, CA, USA). Time courses were analyzed using two-way repeated measures analysis of variance. Calcium concentration data were analyzed using one-way analysis of variance with Sidak's multiple-comparison test. Mean diameter data were analyzed using Student's unpaired t-test. Multiple group comparisons were analyzed using a one-way analysis of variance test followed by a Tukey's post hoc analysis. Data reported are mean±s.e. m. A value of P < 0.05 was considered to be statistically significant.
Randomization and Blinding
All animals were randomly assigned to the procedures; the surgical preparation and data analysis were performed by a researcher masked toward the treatment of the animals.
RESULTS
In Vivo Physiologic Parameters
Body temperature, systemic blood pressure, and blood gases—factors shown to have strong effects on CBF 20 —were carefully monitored in all investigated mice. Parameters did not differ between groups and were within the physiologic range for FVB/N mice (Supplementary Table 1). These mice have a significantly lower pCO2 than C57BL/6 mice anesthetized under the same conditions (Supplementary Figure S3) and a trend toward a lower arterial pH. 12 In keeping with this, we maintained endtidal pCO2 at values ~20 to 30 mm Hg, which correspond to an arterial pH of ~30 to 40 mm Hg, i.e., the physiologic value for FVB/N mice.
Effect of Age on Neurovascular Response to CO2 In Vivo
Inhalation of 5% or 10% CO2 elicited the expected rCBF response in young (6 weeks old) FVB/N mice (Figures 2A and 2D, white symbols). In these animals, rCBF increased maximally by 20% and by 70% during 5% and 10% CO2 inhalation, respectively. This response was similar in 8-month (Figures 2A and 2D, gray symbols) and 12-month-old mice (Figures 2A and 2D, dark gray symbols), indicating that the response of the whole cerebrovascular tree to CO2 was not affected by age. In contrast to the CO2-induced CBF response, dilation of pial vessels to CO2 was significantly reduced in aged mice (Figure 3A). Although in young mice pial arterioles dilated as expected by 20% and 40% on inhalation of 5% and 10% CO2, respectively (Figures 3B and 3E, white symbols), the increase in 8-month (P = 0.001 versus 6 weeks) and 12-month-old mice (P = 0.001 versus 6 weeks) was significantly smaller (Figures 3B and 3E, gray symbols). In 12-month-old mice, the response after application of 5% CO2 was almost eliminated (Figure 3B, dark gray symbols) and analysis of individual vessels revealed that arteries in some animals even displayed an inverted response (Figure 3D, dark gray symbols), i.e., vasoconstriction instead of vasodilation. By doubling the concentration of inhaled CO2 to 10%, we were able to dilate pial arterioles in 12-month-old mice (Figure 3C, dark gray symbols); however, the response was significantly attenuated (P < 0.002) and still showed an inverted response in some arteries (Figure 3E, dark gray symbols). These results show that exposure to CO2, which results in vasodilation in young mice, is dramatically reduced in pial arterioles already at an age of 8 months.
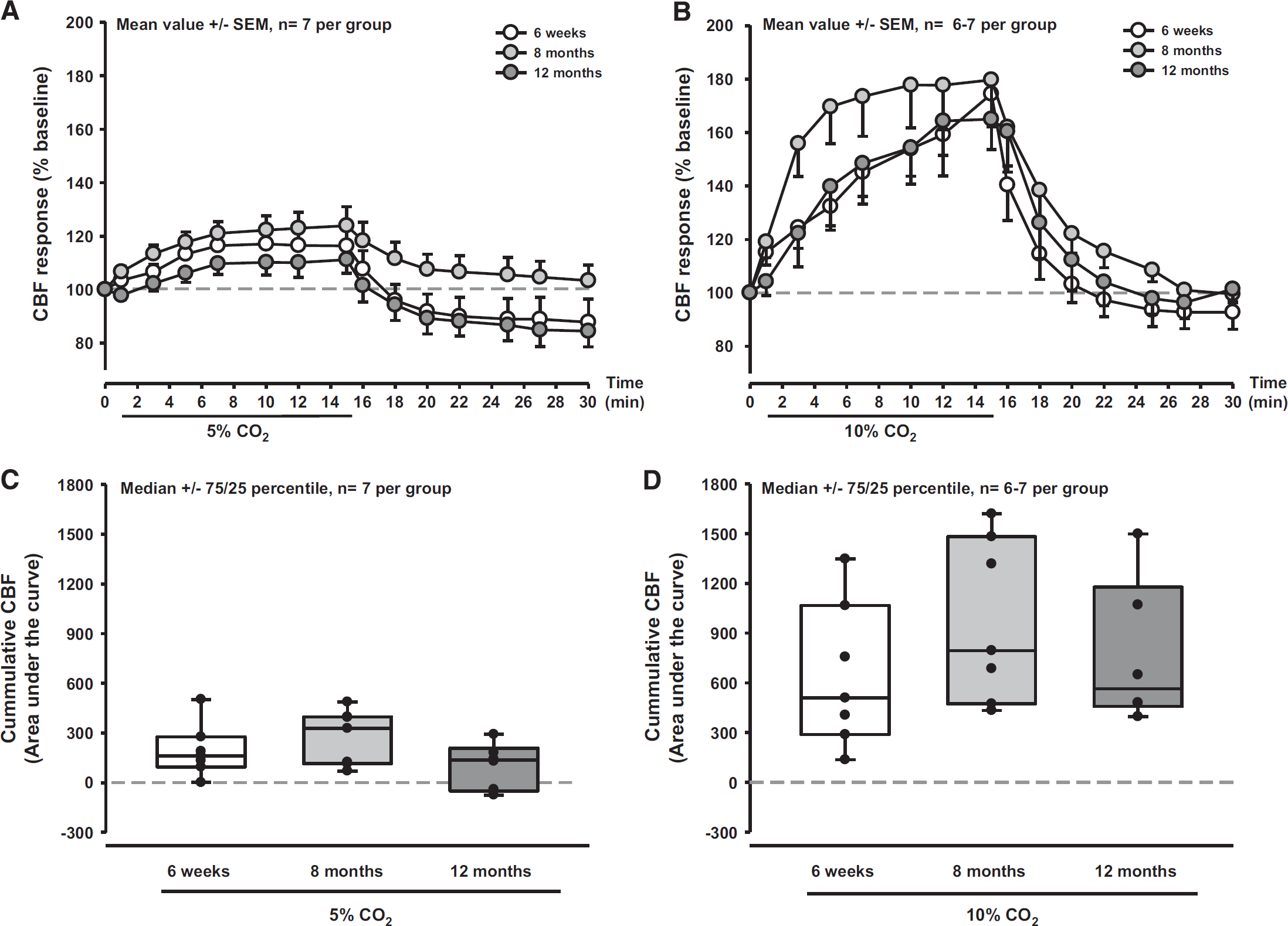
Cerebral blood flow (CBF) response to inhalation of CO2 in young and aged mice. Cerebral blood flow response was not different in 6-week, 8-month, and 12-month-old-mice after inhalation of 5% (
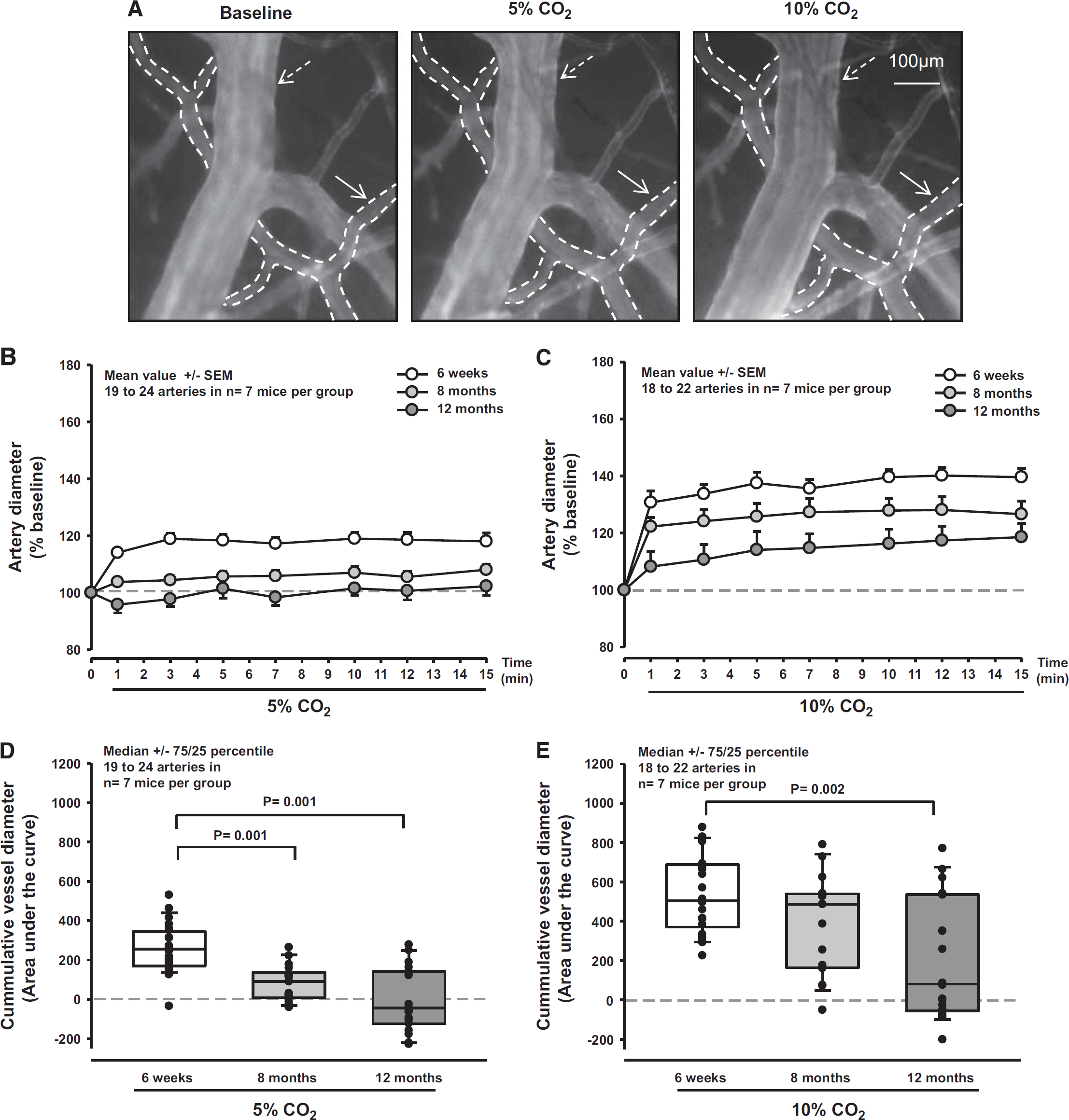
Diameter of pial arterioles in response to CO2 in young and aged mice. (
Effect of Age on Neurovascular Coupling
Sensory stimulation of the forepaw with 10 consecutive stimulation cycles resulted in an increase of CBF by up to 40% in young mice (Figure 4A, white symbols). The first four stimuli had a weaker effect (20% to 30%); the maximum effect of 40% increase was reached only after the fifth stimulus. The response to the first four stimuli was not different between young and 8- or 12-month-old mice (Figures 4A and 4B), however, thereafter aged mice showed a significantly (P < 0.04 to 0.02) decreased response (Figures 4A and 4C). After the seventh stimulus, the CBF increase toward sensory stimulation in aged animals was completely eliminated. Six animals even display a decrease in CBF (Figure 4C). To ensure that sensation of the forepaw was not lost with age thereby resulting in reduced neuronal stimulation and subsequent NVC, we tested young and 7-month-old animals using a hot plate paradigm. All animals showed the same response (Supplementary Figure S4) suggesting not a loss of sensory input but age was responsible for the decline of NVC in old mice. Accordingly, these results point to a severe age-related neurovascular dysfunction already present after one-third of the life span of a mouse. 21
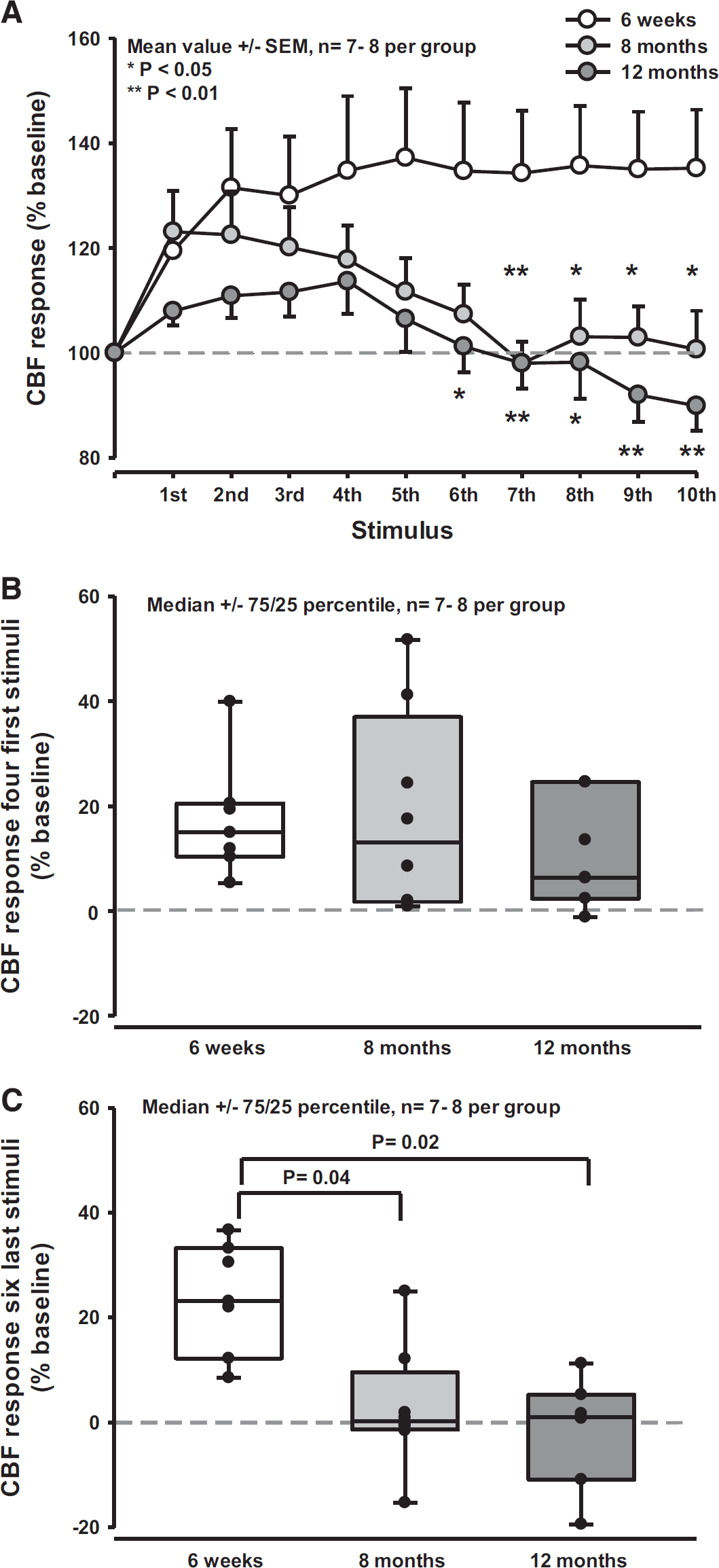
Neurovascular coupling in young and aged mice. (
Structural Changes of the Neurovascular Unit During Aging
To investigate whether structural changes of the NVU—e.g., loss of pericytes or opening of the blood–brain barrier 22 —are responsible for the observed functional deficits of aged cerebral vessels, we assessed and quantified important components of the NVU during aging by immunostaining (Figure 5A and Supplementary Figure S2B). We examined capillary density (lectin staining), coverage of arterioles with smooth muscle cells (α-sma), the coverage of capillaries with pericytes and the total number of pericytes in the microcirculation (PDGFRβ expression), the coverage of capillaries with astrocytic endfeet (GFAP expression), and leakage of the blood–brain barrier by assessing the presence of blood plasma proteins in the brain parenchyma (antialbumin staining). We did not observe any significant change in the endothelium or in the structure of the NVU up to 12 months of age (Figure 5B and Supplementary Figure S2C), suggesting that structural integrity of the NVU is not affected by aging.
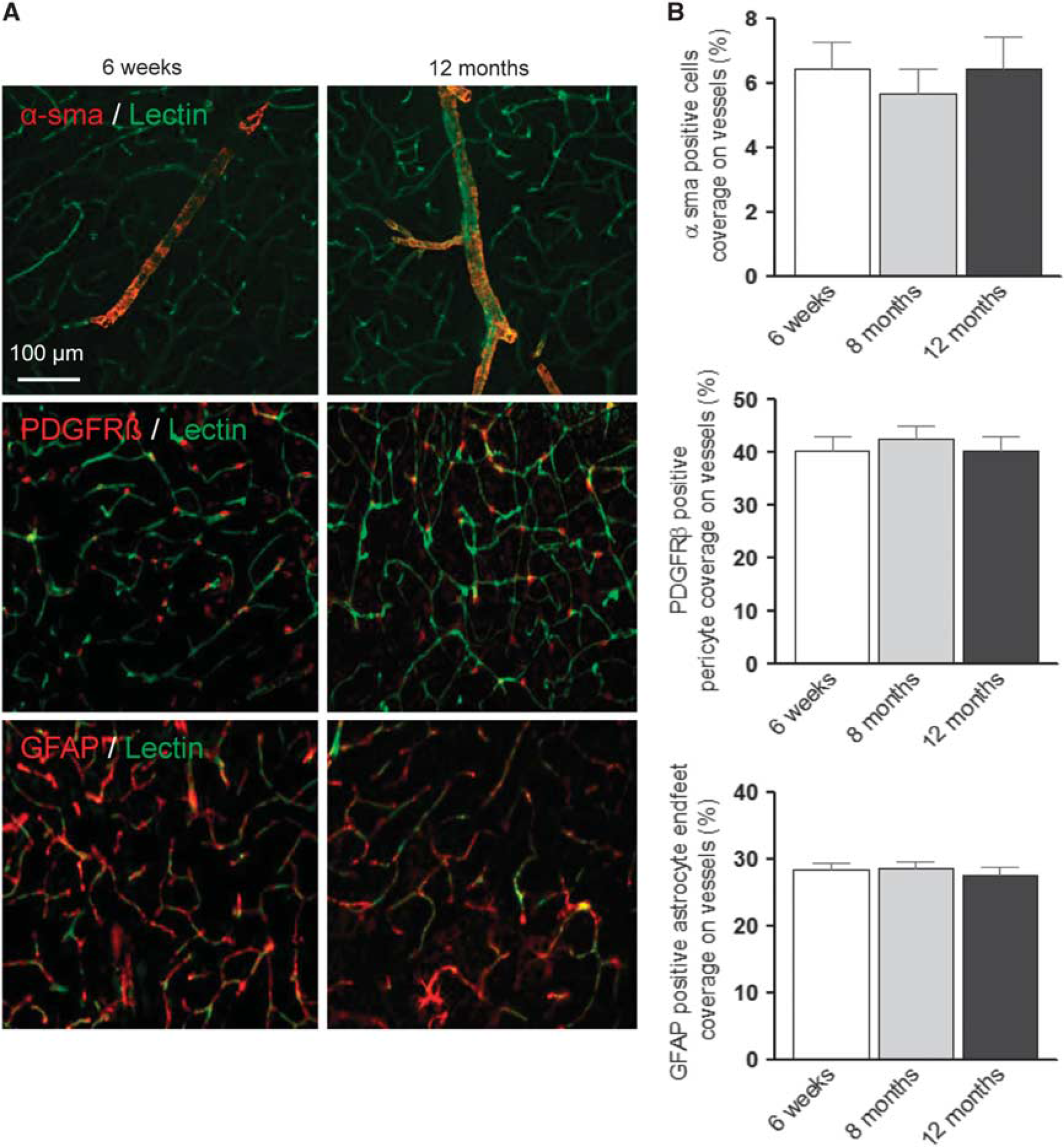
Quantification of the cellular components of the neurovascular unit in young and aged FVB/N mouse cortex. (
Neurovascular Coupling is Impaired In Situ in Aged Mice
To investigate the effects of aging on the function of penetrating arterioles, we used a brain slice model of NVC in which electrical field stimulation was used to initiate neurovascular signaling. By using this established paradigm,19,23,24 we observed that the vasodilatory response to electrical field stimulation was abrogated at 18 months (Figures 6A and 6B). In fact, the prevailing response was a slight constriction in old mice (2- to 3-month-old: 30% ± 7% dilation, n = 8 arterioles, four mice; 18 months: - 3% ± 8% constriction, n = 10 arterioles, five mice). A key step in NVC is an increase in concentration of astrocytic endfoot Ca2+, which leads to the production or release of vasoactive factors onto the underlying pericytes or smooth muscle cells. 25 We observed no difference in either resting endfoot or the level of evoked endfoot calcium between young and 18-month-old mice (control resting: 92 ± 15 nmol/L, control evoked: 435 ± 93 nmol/L, n = 6 endfeet, four mice; aged resting: 98 ± 30 nmol/L; aged evoked 371 ± 70 nmol/L, n = 6 endfeet, four mice; Figures 6C and 6D).
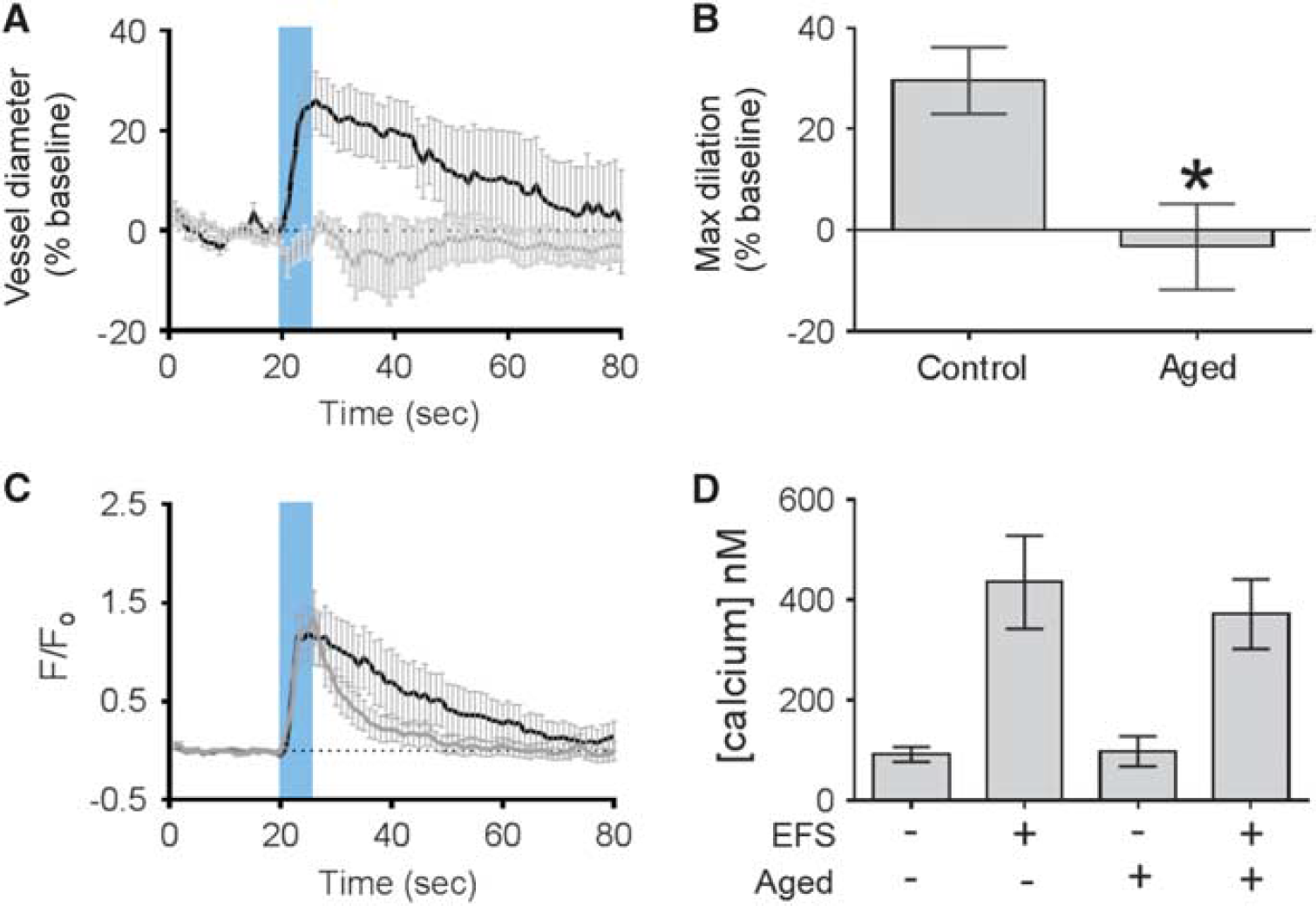
Neurovascular coupling in brain slices from young and old mice. (
DISCUSSION
The findings of the current study suggest that the function of cerebral vessels is differentially altered by aging and that cerebral vessels are structurally intact despite severe functional impairment. In addition, direct visualization of the pial microcirculation allowed us to detect neurovascular dysfunction to CO2 inhalation much earlier that with regional measurements of CBF alone. Consistent with this, rCBF in response to forepaw stimulation was substantially reduced by 8 and 12 months, showing that the cerebral vasculature in mice is already functionally impaired in early adulthood.
An increase in CBF on neuronal activation, also termed functional hyperemia, is essential for the brain's proper function. Reduced hyperemia during brain activation limits the delivery of energy substrates and oxygen to activated neurons thereby resulting in mismatch of blood flow and metabolism, metabolic stress, and eventually neuronal damage as shown in various pathologic conditions, e.g., hypertension and Alzheimer's disease.26,27 However, in addition to cerebrovascular and neurodegenerative diseases, physiologic aging was also reported to result in dysfunction of cerebral vessels. Aging is associated with a decrease of resting CBF 20 and aged vessels tend to display a reduced response to endothelium-dependent vasodilators. 28 It was suggested that the main underlying mechanism is the production of reactive oxygen species by reduced nicotinamide adenine dinucleotide phosphate oxidase in the vessel wall, where reactive oxygen species convert nitric oxide (NO) to peroxynitrite, thereby reducing the bioavailability of NO and damaging vascular structures.13,29
Despite these mechanistic insights, the spatiotemporal dynamics of ACD and its cellular and molecular basis remain poorly characterized. Functional hyperemia to whisker stimulation is reduced by ~40% in 12-month-old mice suggesting dysfunction of the neuron–astrocyte–pericyte/smooth muscle cell axis. 13 The results of the current study are in line with these findings, but detect an impaired NVC already at an age of 8 months. This was identified using trains of repetitive stimuli. By using this approach, we identified that after the first stimulus 6-week and 8-month-old mice reacted normally while 12-month-old mice showed a reduced CBF response. When repeating the stimulation up to ten times, however, it became apparent that vessels in 6-week-young mice maintained their CBF response over time, while 8-month and 12-month-old mice could not maintain their initial response level and showed almost no functional hyperemia. To the contrary, 6 of 16 of the old animals (38%) even showed inverse NVC, i.e., a reduction rather than an increase in CBF. Accordingly, NVC impairment seems to be a much earlier event than previously anticipated since it occurs already at 8 months of age, i.e., in the first third of a mouse's life.
Mechanistically, this vascular ‘fatigue effect’ could be explained by a structural damage to the NVU, by impaired neuron–astrocyte–vascular signaling, or by dysfunction within the vascular wall itself. To assess these hypotheses, we first investigated the NVU by quantifying capillary density, smooth muscle cell coverage of cerebral arterioles, pericyte and astrocytic endfeet coverage of capillaries, and leakage of the BBB by three-dimensional immunohistochemistry. None of these parameters was altered up to an age of 12 months suggesting that arterioles and capillaries are structurally completely intact while NVC is already impaired. To clarify whether neuron–astrocyte–vascular signaling might be impaired, we performed experiments on isolated brain slices from aged mice. Vessels in control slices dilated robustly to neuronal activation, while vessels in slices from aged mice showed a response very similar to that observed in vivo at 12 months, namely a complete loss of NVC with a tendency toward vasoconstriction. However, the increase of astrocytic endfoot Ca2+ on neuronal activation was very similar to the response observed in slices from young mice. Accordingly, we conclude that aging principally affects the functionality of cerebral vessels without affecting neuronal–astrocytic signaling or the structure of the NVU, as also discussed above and suggested by others.13,20,28,29
The absence of an alteration in astrocytic endfoot Ca2+ levels, coupled with impairment of the reactivity of intracerebral arterioles, suggests that mechanisms downstream of this signaling step are affected by aging. One possible mechanism underlying the disturbance of NVC in the current study might be the impairment of potassium ion (K+) signaling.14,30 The Ca2+-driven release of K+ release from astrocytic endfeet plays a critical role in generating rapid vasodilation of cerebral parenchymal arterioles in response to neural activity, 23 through activation of smooth muscle cell potassium inward rectifier channels, driving membrane potential hyperpolarization and vasodilation.25,31 We have previously shown that this mechanism is sensitive to disruption at the level of the SMC KIR channel. 19 Future studies will need to explore the role of vascular Kir channels in the ACD in more detail.
To further explore the time course and localization of vascular dysfunction during aging, we used hypercapnia by inhalation of CO2, which induces nitric-oxide dependent vasodilation.29,32,33 Examining the global increase of CBF after inhalation of 5% or 10% CO2, we were not able to find any neurovascular dysfunction in mice up to an age of 12 months as previously published by Park et al. 13 However, visualization of pial microvessels by in vivo microscopy allowed us to detect a significant lack of CO2-induced vasodilation in pial arterioles already in 8-month-old mice. These findings suggest that AVD starts in pial arteries and precipitates down the neurovascular tree during aging. This is supported by published data showing that aging affects the global increase of CBF after inhalation of CO2 only in mice older than 1.5 years 13 and by experiments showing accelerated aging of pial arteries in a mouse model small-vessel disease. 7 Furthermore, since CO2-induced vasodilation is mediated by constitutively expressed endothelial and neuronal NO synthases, the current data also suggest that AVD may be caused by disturbed NO signaling.
Taken together, our results show that age impairs neurovascular coupling at an unexpectedly young age in mice (30% of the mouse's mean life span). This dysfunction starts in pial vessels and is not accompanied by any changes on the cellular composition of the NVU or by impaired astrocytic Ca2+ reactivity. The lack of vascular CO2 reactivity suggests that defective constitutive NO signaling may represent a possible mechanism. The current study identified the onset and location of age-related neurovascular dysfunction and suggests a putative mechanism, thereby paving the way for the development of novel strategies to maintain neurovascular function during aging.
AUTHOR CONTRIBUTIONS
MB performed all in vivo experiments and wrote the manuscript. MG performed all immunohistochemical staining. TAL performed all in vitro experiments, contributed wiring, and edited the manuscript. MJV and BG analyzed data on neuronal firing. FH worked as a statistical consultant and edited the manuscript. AL worked as a statistical consultant. MTN edited the manuscript. NP designed the study, wrote, and edited the manuscript
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.