
Abstract
We characterized the relationship between the plasma concentration of the phospodiesterase (PDE)-4 inhibitor GSK356278 and occupancy of the PDE4 enzyme in the brain of healthy volunteers, using the positron emission tomography (PET) tracer [11C](R)-rolipram. To this end, PET scans were acquired in eight male volunteers before and at 3 and 8 h after a single 14 mg oral dose of GSK356278. A metabolite-corrected arterial input function was used in conjunction with the dynamic PET emission data to estimate volumes of distribution (VT) from a two-tissue compartment model. The administration of GSK356278 reduced [11C](R)-rolipram whole brain VT by 17% at 3 h post-dose (p = 0.01) and by 4% at 8 h post-dose. The mean plasma Cmax was 42.3 ng/ml, leading to a PDE4 occupancy of 48% at Tmax. The in vivo affinity of GSK356278 was estimated as EC50 = 46 ± 3.6 ng/ml. We present the first report of a direct estimation of PDE4 blockade in the living human brain. In vivo affinity of GSK356278 for the PDE4, estimated in this early phase study, was combined with GSK356278 safety and tolerability data to decide on a therapeutic dose for future clinical development.
Introduction
Rolipram is a selective inhibitor of the enzyme phospodiesterase (PDE)-4, a member of the PDE enzyme family, which hydrolyses the second messenger cyclic adenosine monophosphate (cAMP). The carbon-11-labelled (R) enantiomer of rolipram has been demonstrated to be suitable for the in vivo evaluation of PDE4 availability and activity with positron emission tomography (PET).1,2 [11C](R)-rolipram binding is sensitive to pharmacological challenges in rat 3 and porcine brain. 4 However, to this point, no human blocking studies with PDE4 modulators have been published. GSK356278 is a potent, CNS penetrant inhibitor of cAMP hydrolytic activity 5 that binds to the high-affinity rolipram binding site (HARBS) with a pIC50 of 8.6 and is nonselective for the PDE4A, B and D isoforms. 6 In a model of anxiety in common marmosets, the therapeutic index for GSK356278 was >10 versus <1 for rolipram, 6 which may be explained by the faster HARBS dissociation rate compared to rolipram. The present study was designed to evaluate the relationship between the plasma GSK356278 concentration and the occupancy of the brain PDE4 enzyme in healthy male subjects. These data were to be utilized to optimize the dose range for future Phase 1 and 2 studies.
Methods
This was an open-label study in eight healthy male volunteers with a mean (±standard deviation, SD) age of 34.4 ± 10.7. This study was approved by the Edinburgh Independent Ethics Committee for Medical Research and the Administration of Radioactive Substances Advisory Committee (ARSAC) and conducted in accordance with ICH GCP and the 2008 revision of the Declaration of Helsinki. PET scanning was performed at Imanova, Centre for Imaging Sciences (Hammersmith Hospital, London, UK). All subjects gave written informed consent, tested negative for drugs in urine, and were free from clinically significant illness or disease as determined by their medical history, a full physical examination, blood laboratory tests and electrocardiography.
Radiopharmaceutical preparation
[11C](R)-rolipram was prepared by O-[11C] methylation of its desmethyl analog (1 mg) in dimethylformamide (350 μl) using [11C]methyl iodide in the presence of tetrabutylammonium hydroxide (0.5 M, 4 μl). The reaction mixture was heated to 85℃ for 5 min, diluted with high-performance liquid chromatography (HPLC) mobile phase (1 ml) and loaded onto semi-preparative HPLC for purification (Agilent Eclipse XDB C18, 5 µm, 250 × 9.4 mm). The retention time of [11C](R)-rolipram was 5.4 min through isocratic elution with a mixture of 10 mM ammonium formate buffer/acetonitrile (60/40, v/v) at a flow rate of 6 ml/min. The desired fraction was collected, diluted with water (20 ml) and loaded onto a SepPak® Classic C18 cartridge for reformulation. Following an initial wash with sterile water (10 ml), [11C](R)-rolipram was eluted off the cartridge with ethanol (1 ml) and subsequently diluted with saline (10 ml). In a final step, the resulting formulation solution was filtered through a 0.22 µm sterile filter into a sterile, pyrogen-free vial. Typical synthesis from a 55 μA, 30 min beam provided 3.35 ± 1.4 GBq (uncorrected) of [11C](R)-rolipram in a total synthesis time of 35 min with a specific activity of 142 ± 133 GBq/µmol. Quality control showed that doses were obtained with radiochemical purities >99% and that the final product for injection was sterile and pyrogen free.
Positron emission tomography data acquisition
In total, 22 [11C](R)-rolipram PET scans were acquired on a Siemens Biograph HiRez XVI PET-CT scanner (Siemens Healthcare, Erlangen, Germany) of which eight scans were acquired at baseline (PET 1) and seven scans at approximately 3 and 8 h post-dose each (PET 2 and PET 3 respectively). A CT scan of the head was acquired for attenuation and model-based scatter correction. Subjects were injected with an intravenous bolus of the radioligand, and dynamic emission data were acquired continuously for 90 min. PET data were reconstructed using filtered backprojection with corrections for attenuation, dead time, random coincidences and scatter. Dynamic data were binned into 26 frames (8 × 15 s, 3 × 1, 5 × 2, 5 × 5 and 5 × 10 min). A continuous sampling system (ABSS Allogg, Mariefred, Sweden) was used to measure whole blood activity for the first 15 min (sampled at 5 ml/min). Discrete blood samples were acquired to measure blood and plasma radioactivity concentration at 5, 10, 15, 20, 30, 40, 50, 70 and 90 min and to determine with HPLC the fraction of radioactivity corresponding to intact parent [11C](R)-rolipram in arterial plasma. Plasmafree fraction (fP) was determined by ultrafiltration. The three discrete blood samples at 5, 10 and 15 min post-injection were used to calibrate the continuous blood data. The continuous and discrete datasets were used to form a whole-blood activity curve, covering the duration of the scan. Radioactivity concentrations in discrete plasma samples were divided by the corresponding whole-blood samples to form plasma-over-blood (POB) data, and a constant POB model was fitted (i.e. average of POB values). This POB value was then multiplied by the whole blood curve to generate a total plasma curve. Parent fraction data were fitted to a sigmoid model f = ((1−(t3/(t3 + 10a)))b + c)/(1 + c), where t is time and a, b and c are fitted parameters. The resulting fitted parent fraction profile was multiplied by the total plasma curve and then smoothed post-peak using a tri-exponential fit to derive the required parent plasma input function. For each scan, a time delay was fitted and applied to the input function to account for any temporal delay between blood sample measurement and the tomographic measurements of the tissue data.
Image analysis
T1-weighted magnetic resonance images (MRI) were acquired to aid in the definition of the anatomic regions of interest (ROIs) using a Siemens Magnetom Trio 3T MRI scanner at Imanova. Dynamic PET images were registered to each individual subject’s MRI, and corrected for motion using a frame-to-frame registration process with a mutual information cost function. Regional analysis was facilitated by using a human brain atlas containing ROIs that had been manually delineated on a T1-MR image template according to strict anatomical criteria. 7 The target ROIs included the caudate, putamen, thalamus, hippocampus, frontal cortex, parietal lobe, temporal lobe, occipital pole and cerebellum. For each subject, the template and the corresponding atlas were individually warped to the subject’s anatomical MRI which was previously linearly registered to the PET images and used to generate time activity curves (TACs).
Compartmental modelling and specific signal estimation
The individual metabolite-corrected arterial input function and the whole-blood activity curve were used in conjunction with the dynamic PET data to estimate regional volumes of distribution (VT) using a two-tissue compartment model. The blood volume component (VB) was fixed at 5%.
The VT is the sum of the tracer’s specific (Vs) and non-displaceable (VND) binding. As there is no suitable (pseudo)reference region in the human brain, devoid of PDE4, it is not straightforward to estimate VND and calculate the non-displaceable binding potential (BPND). Target occupancy and VND may be estimated from the VT data and the related plasma concentration of GSK356278 if a sufficiently large reduction in VT is observed post-block. However, we were not able to acquire data following high levels of PDE4 blockade due to the safety limitations on the maximal dose of GSK356278 we could administer. Therefore, an estimate of the average brain binding potential (
Results
The mean (±SD) injected dose for the 22 PET scans in this study was 270 ± 61 MBq with specific activity of 76 ± 56 GBq/µmol. After injection, the tracer readily entered the brain and showed widespread distribution. The images and the regional TACs for a representative subject are presented in Figure 1(a) and (b), respectively.
(a) Anatomical MRI and PET-integrated activity from 30 to 90 min post [11C](R)-rolipram injection at baseline, post-dose 1 (3 h) and post-dose 2 (8 h) for subject 2. Data for each scan have been normalized by the injected activity per liter (%ID/l). (b) Regional measured data (circles) and model fits (lines) for subject 2. For each region, the baseline (red), post-dose 1 (3 h, green) and post-dose 2 (8 h, blue) time activity curves are shown.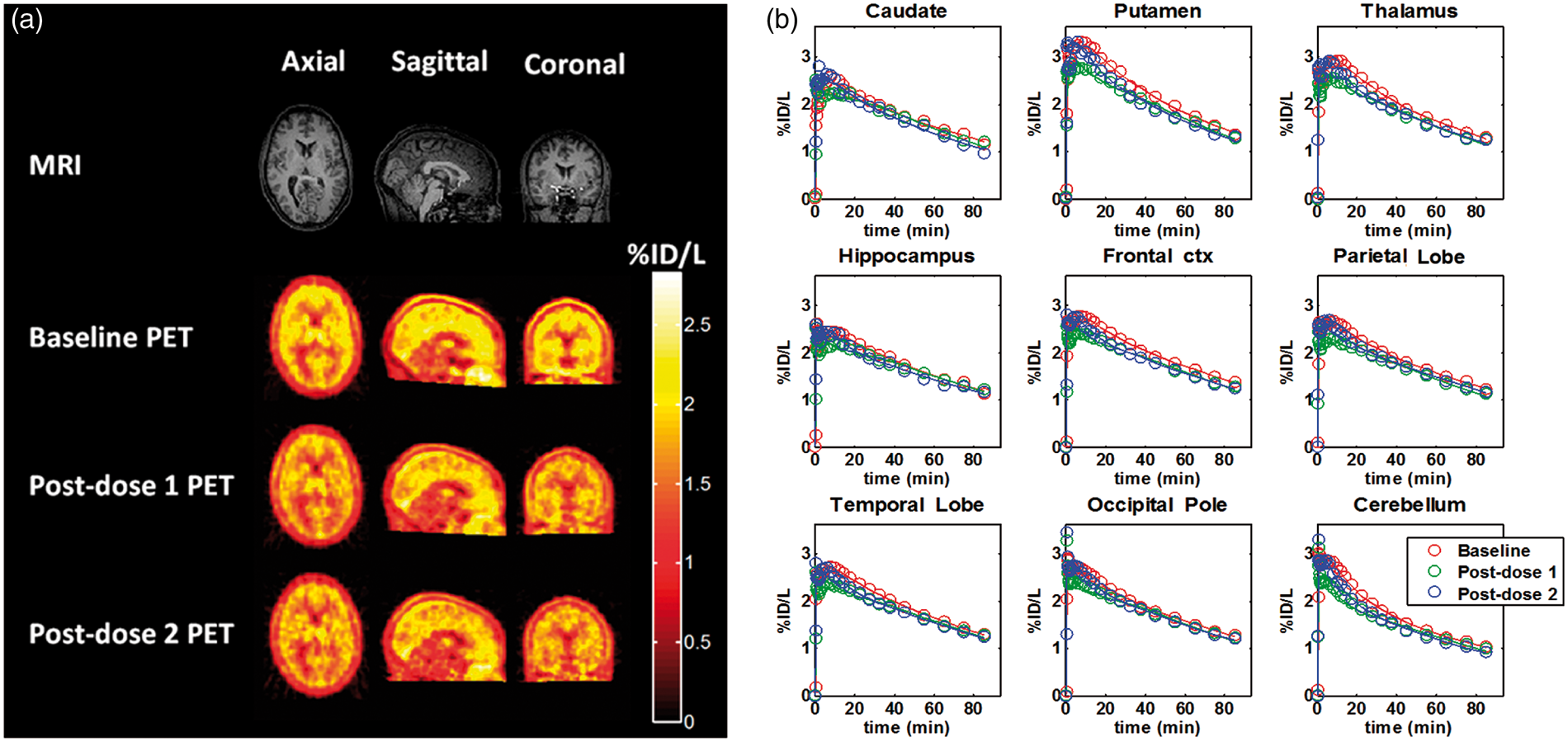
[11C](R)-rolipram volumes of distribution (VT), plasma GSK356278 concentration and PDE4 occupancy at post-dose scans.
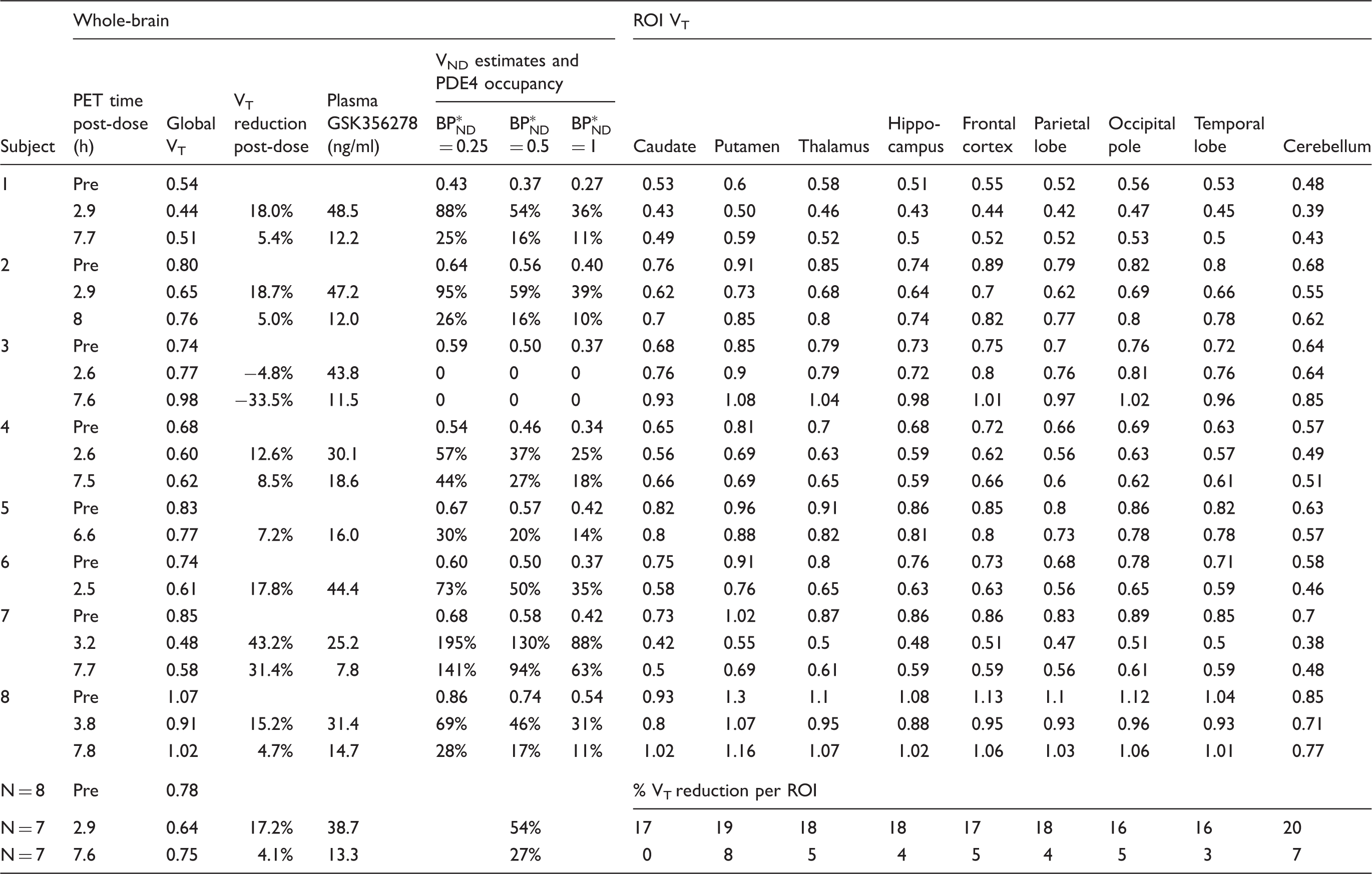
VT: volume of distribution; VND: non-displaceable volume of distribution;
[11C](R)-rolipram binding in all eight subjects expressed as VT for the whole brain (global) and nine regions of interest (ROI). Plasma GSK356278 concentration was measured at the start of PET. The bottom three rows show the means for all subjects, i.e. N = 8 for baseline scans and N = 7 for post-dose scans.
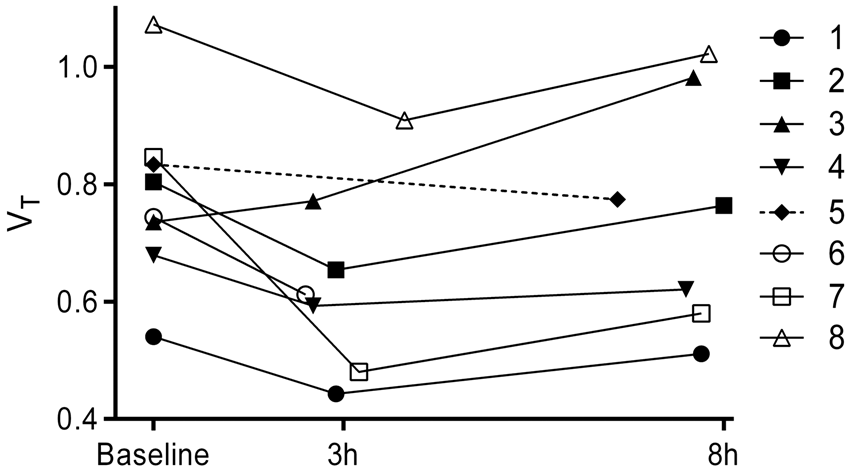
Global brain volumes of distribution (VT) for baseline, 3 and 8 h post-dose scans in all eight subjects. Subjects 5 and 6 completed one post-dose scan.
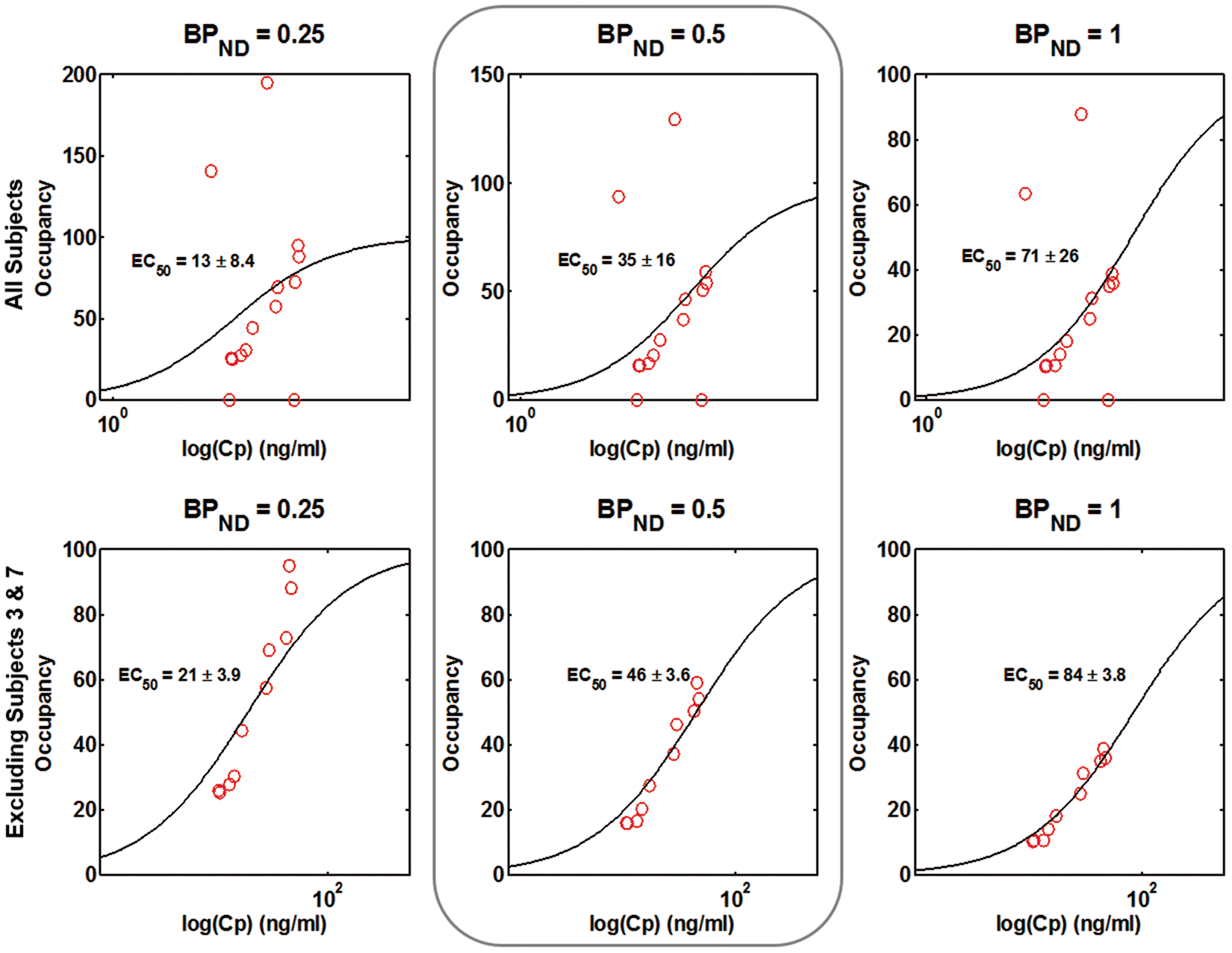
The individual plasma GSK356278 concentration–time curves are presented in Figure 3. The mean Cmax was 42.3 ng/ml. Figure 4 shows plasma GSK356278–PDE4 occupancy plots using three different estimates of Individual plasma GSK356278 concentration-time curves. The dotted lines show the average time of the post-dose scans. Model fits of the PET occupancy data and plasma GSK356278 concentration with estimated EC50 in all subjects examined (top row) and for the dataset excluding subjects 3 and 7 (bottom row). Column 2 represents the most likely value of the non-displaceable binding potential (BPND) while the other two columns are intended to show the effect of a different estimate of BPND on the EC50.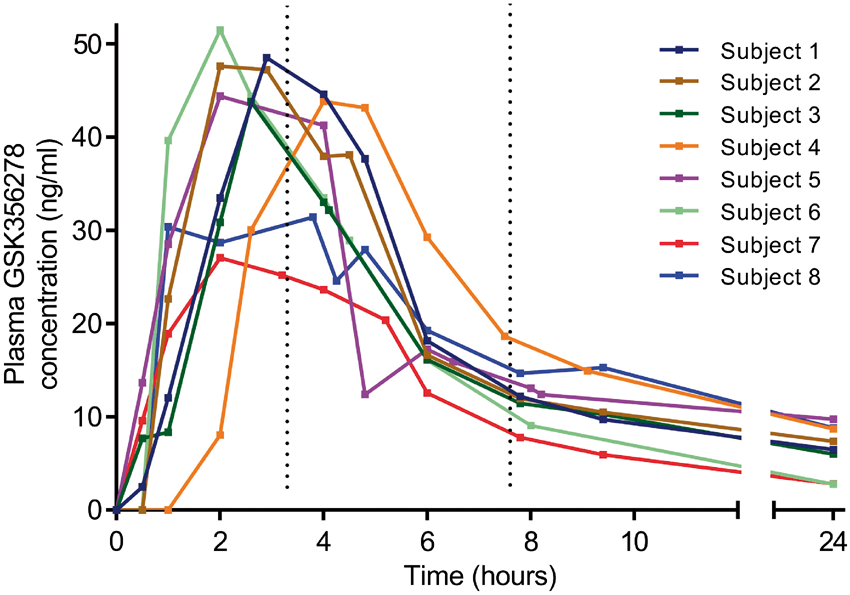
Discussion
To our knowledge, this is the first study to explore the blockade of the human PDE4 enzyme in the brain in vivo. Baseline VT values obtained in this study were consistent with those seen in previous human studies.2,8 The between-subject variability (coefficient of variation) of the baseline VT was 20%, comparable to earlier reports of 25%. 8 Oral administration of the PDE4 inhibitor GSK356278 led to a mean VT reduction of 17% around 3 h post-dose compared to the baseline VT and a reduction of 4% around 8 h post-dose, consistent with the hypothesis that GSK356278 enters the brain readily and binds to PDE4. VT change in all subjects followed the plasma concentration of GSK356278, with ΔVT PET 2 > than ΔVT PET 3. The estimated relationship between the plasma concentration of GSK356278 and PDE4 occupancy indicates that occupancies of close to 50% of PDE4 are achieved at Tmax following the administration of single oral doses of 14 mg of GSK356278. The available data provide no evidence for indirect pharmacokinetics for GSK356278 in the human brain, suggesting that the plasma EC50 (46 ng/ml) estimated in this study can be used to calculate PDE4 occupancy following repeat dose administration.
The assessment of the relationship between PDE4 occupancy and plasma GSK356278 concentration was complicated by our inability to estimate the [11C](R)-rolipram BPND directly from the study data, due to the relatively low levels of occupancy achieved. Our population estimate of BPND from the literature ignores between-subject variability in PDE4 expression. Although an error in the estimate of BPND would lead to an error in the estimated EC50, a relatively large range of BPND estimates (0.25–1) produced a modest difference in GSK356278 EC50 (21–84 ng/ml), indicating that our estimated EC50 is relatively robust to variability in assumed
Subjects 3 and 7 were identified as outliers based on abnormal post-dose VT compared to the baseline. Specifically, subject 3 showed an increase in post-dose VT, whereas subject 7 showed an exceptionally large reduction, leading to occupancy at 3 h > 100%. The metabolite-corrected arterial input function and plasmafree fraction of [11C](R)-rolipram for these subjects did not differ between scans, making it unlikely that changes in blood flow or plasma protein binding could explain these findings. Variability in estimated parameters is thus the most likely explanation for the findings in these subjects. Test–retest variability of VT in healthy humans was shown to be approximately 19% in an earlier study of 12 subjects. 8
PDE enzyme activity dysfunction has been implicated in disease states such as asthma, ischemic stroke and CNS disorders. In a study of patients with major depressive disorder, [11C](R)-rolipram binding was reduced by 18% compared to healthy control subjects and could be partially normalized with selective serotonin reuptake inhibitor treatment. 9 PDE4 selectively metabolizes cAMP in the brain to the inactive monophosphate and is therefore an important component of the cAMP cascade. The enzymatic activity of PDE4 is regulated by protein kinase A (PKA) via a feedback mechanism, with high concentrations of cAMP stimulating PKA to phosphorylate PDE4, thereby increasing its enzymatic activity and returning the concentration of cAMP to steady state. Unilateral injection of a PKA activator and inhibitor into the rat striatum was shown to significantly increase and decrease, respectively, the binding of [11C](R)-rolipram as measured with PET. 3 Upregulation of the cAMP cascade through long-term pharmacological inhibition of the PDE4 enzyme is a promising therapeutic intervention for a range of conditions. In preclinical studies, GSK356278 was shown to improve performance in an object retrieval test in cynomolgus macaques, 6 consistent with the reported effects of rolipram in various tests of cognition. 10 Despite the possible benefits of brain-penetrant PDE4 inhibitors, clinical use has been limited by mechanism-dependent adverse events such as nausea and emesis. 11 The highest dose in this study was limited to 14 mg (equivalent to ∼50% occupancy) by these adverse events in Phase 1 studies.
In conclusion, we present the first human report of PDE4 occupancy measured directly in the human brain with PET. Our data will be used in conjunction with the known plasma GSK356278 pharmacokinetics to determine optimal doses to be used in future clinical development.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by GlaxoSmithKline. Portions of this work have previously been presented in abstract form at the XXVI International Symposium on Cerebral Blood Flow, Metabolism and Function (BRAIN 2013).
Acknowledgements
We thank all participants, the PET technicians, MRI radiographers and the research nurses at Imanova for their support with the execution of the study. We also thank Prof. Dr. Lammertsma for comments on the manuscript.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: This study was funded by GlaxoSmithKline. At the time this work was conducted, John Tonkyn and Frank A Gray were employees of GlaxoSmithKline.
Authors’ contributions
Jvda, JT, FAG, JP, RNG and EAR designed the study. JvdA, RD, SPM, AAW and JP performed the experiments and acquired the data. JvdA, CS, JP, RNG and EAR analyzed the data. All authors contributed to the interpretation of the results and writing the manuscript. All authors approved it for publication.