
Abstract
The current absence of a disease-modifying treatment for Alzheimer’s disease (AD) and vascular cognitive impairment and dementia (VCID) highlights the necessity for investigating the benefits of non-pharmacological approaches such as physical exercise (PE). Although evidence exists to support an association between regular PE and higher scores on cognitive function tests, and a slower rate of cognitive decline, there is no clear consensus on the underlying molecular mechanisms of the advantages of PE. This review seeks to summarize the positive effects of PE in human and animal studies while highlighting the vascular link between these benefits. Lifestyle factors such as cardiovascular diseases, metabolic syndrome, and sleep apnea will be addressed in relation to the risk they pose in developing AD and VCID, as will molecular factors known to have an impact on either the initiation or the progression of AD and/or VCID. This will include amyloid-beta clearance, oxidative stress, inflammatory responses, neurogenesis, angiogenesis, glucose metabolism, and white matter integrity. Particularly, this review will address how engaging in PE can counter factors that contribute to disease pathogenesis, and how these alterations are linked to endothelial cell function.
Introduction
Together, Alzheimer’s disease (AD) and vascular cognitive impairment and dementia (VCID) are the two most common forms of dementia. While the neuronal pathology in AD has been extensively studied and well characterized, there is also a less well-known cerebrovascular pathology that displays substantial similarities with VCID.1,2 VCID is caused by a number of vascular and/or ischemic lesions that may be focal or diffuse in the basal ganglia, thalamus, white matter (WM) tracts, and subfrontal areas that lead to deafferentation of neuronal networks. 3 Vascular pathology in AD includes accumulation of amyloid-beta (Aβ) peptide in the vessel wall referred to as cerebral amyloid angiopathy (CAA), 4 and atherosclerosis of cerebral vessels. 5 Other pathology shared between AD and VCID that affects the vasculature includes vascular fibrosis, microvascular changes including decreased capillary density, increased amounts of degenerating capillaries (string vessel pathology), pericytes and astroglial endfeet surrounding cerebral microvessels, decreased blood brain barrier (BBB) tight junction proteins, and reduced mitochondrial content in endothelial cells.6,7 Alterations such as fragmented or collapsing microvessels, and damaged vessel endothelium can decrease cerebral perfusion. If cerebral hypoperfusion is chronic, it may be a determining factor in precipitating cognitive dysfunction. 8 Most of the literature on physical exercise (PE) interventions in dementia research has focused around AD. However, given the substantial pathological overlap between AD and VCID, it would not be unreasonable to posit that the reported positive effects of PE in the AD population can also be applied to those afflicted with VCID.
In fact, a frequently recommended preventative therapy against cognitive impairment in both AD and VCID is regular PE, a strategy that has a clear link to vascular health. In healthy, cognitively normal older adults, cardiovascular fitness has been shown to be protective against cognitive decline. 9 Regular PE has also been shown to improve cognitive function in healthy older adults in terms of processing speed, attention, and executive function.10,11 Such findings have led to randomized control studies in early to moderate AD patients using aerobic PE interventions from 3-month to 2-year long periods that have reported slower rates of cognitive decline.12–15 A similar protection has also been shown in several animal models of AD,16–21 and has been attributed to a variety of underlying molecular mechanisms. Explanations for these beneficial outcomes include decreased levels of the hyperphosphorylated form of the microtubule-associated protein tau, Aβ plaque load, inflammation and oxidative stress, improved cerebral perfusion, and increased neurogenesis and synaptic plasticity.
Due to numerous failed clinical trials 22 and the current absence of a disease-modifying treatment for AD or VCID, it is crucial, despite recent reports of declining incidence rates in North America and Europe, 23 to better understand the potential benefits of nonpharmacological approaches, namely, PE. Although the beneficial effects of PE have been well reviewed,24–29 there remains no consensus as to which molecular mechanism(s) is(are) responsible for preserving cognitive function. In this review, we will first present the risk factors of developing dementia, including vascular risk factors and sleep quality. We will then present how PE can modify molecular mechanisms by focusing on toxic protein aggregates, oxidative stress, neuroinflammation, neurorepair, as well as glucose metabolism and WM integrity. Finally, we will present a central role for endothelial cells in mediating the benefits of PE. Overall, this review seeks to summarize the reported benefits of PE in both human and animal studies in an attempt to highlight the cerebrovascular basis that may underlie its effects, and potentially provide a novel pharmacological target for future curative therapies. We have been careful to indicate studies that used forced or voluntary PE in animal studies, as there have been reported differences in their effects. Main differences being animals forced to exercise show more anxiety on behavioral tests, whereas no consensus exists regarding differences in how forced or voluntary exercise affect markers of hippocampal neurogenesis such as positively labeled BrdU cells (double-labeled with NeuN, a neuronal marker) and changes in levels of BDNF.30,31
Lifestyle risk factors of developing dementia and their modification by exercise
Vascular risk factors
Aside from age and genetics, the most common risk factors for sporadic AD are of a vascular nature,8,32 and these risk factors can predict the rate at which an individual’s cognitive abilities will decline.
33
Epidemiological studies most often identify hypertension as the most prominent vascular risk factor, followed by cardiovascular diseases (CVD) including myocardial infarction, and stroke, diabetes mellitus, hyperhomocysteinemia, hypercholesterolemia, hyperlipidemia, and tobacco smoking, all of which share the common feature of compromised endothelial cell function (Figure 1).34,35 Vascular pathology associated with hypertension includes reduced vasodilator reserve and microvascular density.
36
In diabetes, disrupted insulin signaling causes vessels to remain in a constricted state due to decreased bioavailability of nitric oxide (NO), a vasodilator produced in endothelial cells
37
that helps vessels maintain their basal tone. Hyperhomocysteinemia, often a result of low folate consumption, primarily affects endothelial cells by inhibiting NO and endothelium-derived hyperpolarizing factor, promoting oxidative stress, angiotensin II receptor activation, induction of the potent vasoconstrictor endothelin-1 (ET-1), and disruption in the regulation of prostanoids.
38
Hypercholesterolemia, which is often the leading cause of CVD, results in fatty deposits in the vessel wall. These deposits lead to endothelial dysfunction, impaired vessel reactivity by decreasing NO bioavailability, increased oxidative stress and promotion of a proinflammatory environment.39,40 Interestingly, an increase in mid-life body weight adiposity has been associated with decreased adiponectin levels, a hormone that typically has protective effects against cerebrovascular damage and inflammation through endothelial NO synthase (eNOS)-dependent and NF-κB pathways.
41
Summary of the effects of lifestyle factors on cognition and how physical exercise (PE) modifies them. Cardiovascular risk factors have the common effect of disrupting endothelial cell function, while poor sleep impacts white matter integrity and clearance of toxic proteins. PE has been shown to decrease risk of developing vascular risk factors and improve quality of sleep, both of which decrease the likelihood of cognitive decline. CVD: cardiovascular disease; NO: nitric oxide; eNOS: endothelial nitric oxide synthase.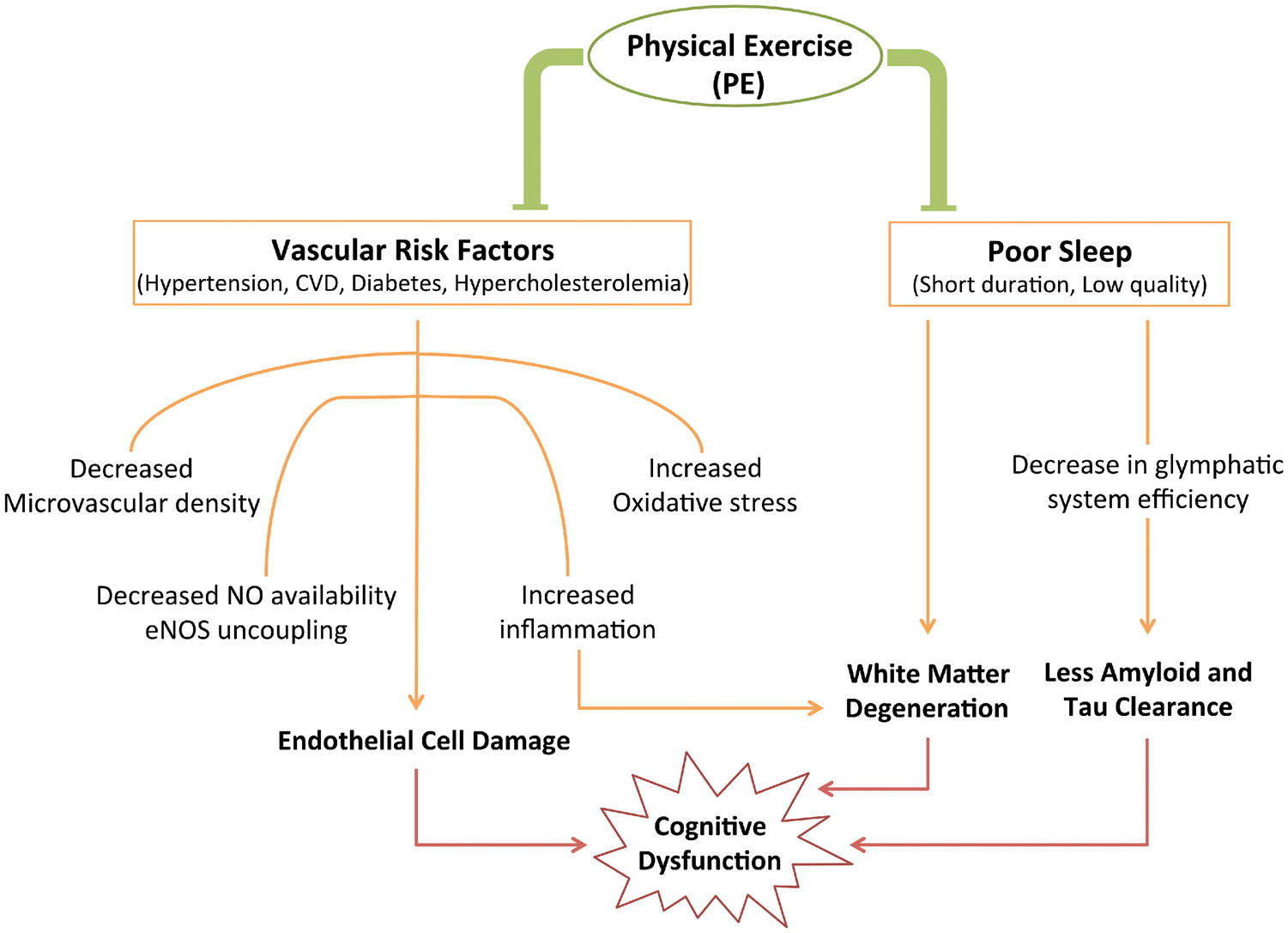
According to the Centers for Disease Control and Prevention in America (2015), 150 min/week of aerobic PE at a moderate intensity is sufficient to significantly decrease the probability of acquiring the vascular risk factors mentioned above. This decreased risk has been attributed to the ability of PE to decrease blood pressure, lower total cholesterol levels likely by increasing levels of high-density lipoprotein cholesterol, and control blood-glucose levels. In addition, PE has been shown to maintain or improve endothelial cell function by increasing eNOS activity,42,43 further highlighting endothelial cell function as a common path underlying the beneficial effects of PE in counteracting vascular risk factors.
It thus appears that CVD all have endothelial cell dysfunction in common and increase likelihood of dementia incidence, whereas PE reduces the risk of CVD, improves endothelial cell function and thus lowers the risk of developing dementia, 24 and has the potential to delay onset of dementia.
Sleep quality
Another life-style factor that can contribute to the progression of dementia is disrupted sleep (Figure 1). It has been reported that both the quantity and the quality of one’s sleep decreases with age.44,45 This is noted by changes in both macro- and micro-sleep architecture, which include decreased non-REM (rapid eye movement) slow wave sleep, and a reduction in the number of sleep spindles. 46 Although these changes occur in healthy older adults, decreased quality of sleep has been implicated in contributing to cognitive dysfunction. Both obstructive sleep apnea 47 and insomnia 48 are predictive factors for cognitive decline. A proposed explanation for poor sleep leading to cognitive decline is reduced clearance of excess fluid and toxic interstitial solutes from the brain parenchyma. A recently characterized waste clearance system in the CNS, known as the glymphatic system, is reported to be mainly functional during sleep to enable the clearance of Aβ through interstitial fluid bulk flow and cerebrospinal fluid absorption clearance. 49 A dysfunctional glymphatic system has also been reported to exacerbate tau pathology, 50 another hallmark of AD. However, it is important to note that clearance may also occur via perivascular drainage, 51 and that the physiological relevance of the glymphatic system has recently been questioned.52,53
An additional mechanism through which sleep can impact cognition is through changes in WM that occur with increased episodes of obstructive sleep apnea. 54 Potential mechanisms proposed by Kim et al. 54 include increased cardiac output and increased vasoconstriction. These rapid changes in flow can create a shear stress in the cerebrovasculature leading to damage of small vessels within the brain. This damage is particularly prominent in the WM where blood flow is typically lower than in grey matter, making it more susceptible to injury. 55 Another mechanism linked to cerebrovascular dysfunction is increased vascular oxidative stress, as shown in mice subjected to chronic intermittent hypoxia, a common feature of obstructive sleep apnea. Levels of reactive oxygen species (ROS) were significantly increased in cerebral arterioles of these mice. However, when mice lacked a subunit of the ROS producing enzyme NADPH oxidase, or when NADPH oxidase was inhibited, vascular oxidative stress was not significantly different from control mice. 56 Interestingly, PE has been shown to improve quality and duration of sleep, as well as decrease the number of obstructive sleep apnea episodes.57–60 PE could thus indirectly improve Aβ and hyperphosphorylated tau clearance through the glymphatic system, 52 help maintain WM integrity, and reduce oxidative stress, the latter two of which are discussed in more detail in sections to follow.
Low quality of sleep can perpetuate the accumulation of toxic protein aggregates in the parenchyma and around blood vessels, rendering clearance via the glymphatic system or other mechanisms less efficient. PE has been shown to improve quality and quantity of sleep and could thus indirectly help with clearance efficiency.
Modification of molecular mechanisms by exercise
Exercise and toxic protein aggregates
The two key neuropathological features of AD are neurofibrillary tangles of hyperphosphorylated tau, and increased levels of soluble and insoluble Aβ peptides in the brain parenchyma and vessel walls. While Aβ peptides lead to retraction of neuronal branches and impaired synaptic transmission and long-term potentiation (LTP), 61 a biological process thought to be important for learning and memory, they also compromise vascular reactivity in the brain. 62
There is a tight equilibrium between Aβ efflux and influx across the BBB that is disrupted in AD and leads to accumulation of toxic protein aggregates. This equilibrium requires degradation of Aβ by insulin degrading enzyme and neprilysin, Aβ transport across the BBB into the brain by receptor for advanced glycation end products (RAGE), and transport out of the brain by low-density lipoprotein receptor-related protein 1 (LRP-1). LRP-1 is found on endothelial cells, vascular smooth muscle cells, glia, and neurons. 63 Lower levels of LRP-1 expression in endothelial cells at the BBB have been reported in both normal aging and in AD patients, and have been shown to be upregulated by PE. 64 Inversely, RAGE levels in endothelial cells and neurons are increased in normal aging and AD, 65 causing more Aβ to accumulate in the brain parenchyma. Interestingly, RAGE levels have been shown to decrease following PE. 64 Hypoperfusion has also been linked to increased levels of Aβ and hyperphosphorylated tau in the 3xTg-AD mouse (homozygous for the presenilin1, Psen1, mutation and homozygous for the Swedish APP and tauP301L transgenes) following bilateral common carotid artery occlusion. 66
Aside from receptors and enzymes, another factor that may contribute to clearance efficiency is CBF. It is not yet clear whether global CBF increases during PE. This is in part due to differences in methodology and timing of CBF measurements, length of interventions, and it also appears to vary by species. Despite a number of studies reporting no change in CBF following PE, or only region-specific increases in CBF, 67 there is a general consensus that PE of a moderate intensity (approximately 60% maximal oxygen uptake) improves global CBF,68,69 particularly in humans. This improvement in CBF could diminish the effects of hypoperfusion, which may assist in Aβ and tau clearance. Very recently, lack of eNOS in APP/PS1 transgenic mice (Tg2576 mouse crossed with mice that overexpress human Psen-1 with the M146L mutation) led to increased levels of hyperphosphorylated tau, 70 and PE has been demonstrated to increase eNOS levels, 43 which could therefore assist in maintaining tau homeostasis.
Several studies using mouse models of AD have reported cognitive benefits following PE interventions and concluded that it was due to a reduction in Aβ plaque load, hyperphosphorylated tau, or both (Figure 2). After 1 h of daily voluntary PE for 4 months, Tg2576 mice that contain the APPSw mutation (K670N, M671L) showed improved recognition memory using the short-term novel object recognition task. The explanation for the improved cognition was a reduction in the number of mature dense-core plaques in the hippocampus and overlying cortex.
71
Similarly, 5 months of forced treadmill PE in the APP/PS1 mouse model revealed a drastic and consistent decrease in Aβ deposition and reduced tau hyperphosphorylation.
72
Long-term voluntary PE (9 months) in THY-Tau22 mice (mutations at G272V and P301S in the 412 amino acid isoform of human 4-repeat tau) resulted in more spontaneous alternation in a 2-trial Y-maze task. This was attributed to both preserved levels of the acetylcholine-synthesizing enzyme (choline acetyltransferase, ChAT) in the medial septum, and reduced tau hyperphosphorylation identified by AT100 immunohistochemistry.
19
Another group found that 3 months of forced treadmill PE in aged P301S mice, a model of taupathology with the P301S mutation in the microtubule-associated protein tau gene (MAPT), significantly reduced phosphorylation of tau at sites serine 202 and threonine 205 in the hippocampus.
73
Although the studies above attribute the cognitive benefits to reduced Aβ and tau pathology, it is possible that there are other explanations for the reported improvements. One possibility is that less amyloid can enter astrocytes (due to more amyloid being cleared by PE) and, therefore, less proinflammatory mediators being released near vessels by astrocytic perivascular endfeet.
74
Another viable explanation comes from PE’s ability to increase global CBF and vascular integrity. Improved CBF has been correlated with reduced CAA in hAPP J20 mice (hAPP model that carries both the Swedish, K670N, M671L, and Indiana, V717F, familial mutations) treated chronically with rapamycin,
75
which made it easier to clear the brain of toxic protein aggregates.
Effects of physical exercise (PE) on various molecular phenomena including neurorepair, clearance, glucose metabolism, oxidative stress, and inflammation. ERK: extracellular signal-regulated kinases; Akt: protein kinase B; PI3K: phosphatidylinositol 3-kinase; GSK-3β: glycogen synthase-3β; SIRT3: sirtuin 3; SOD: superoxide dismutase; HIF-1α: hypoxia-inducible factor 1α; INF-γ: interferon gamma; IL-1β: interleukin 1β; TNF-α: tumor necrosis factor alpha; IL-6: interleukin 6; COX-2: cycloxygenase 2; VCAM-1: vascular cell adhesion molecule 1; ICAM-1: intracellular adhesion molecule 1.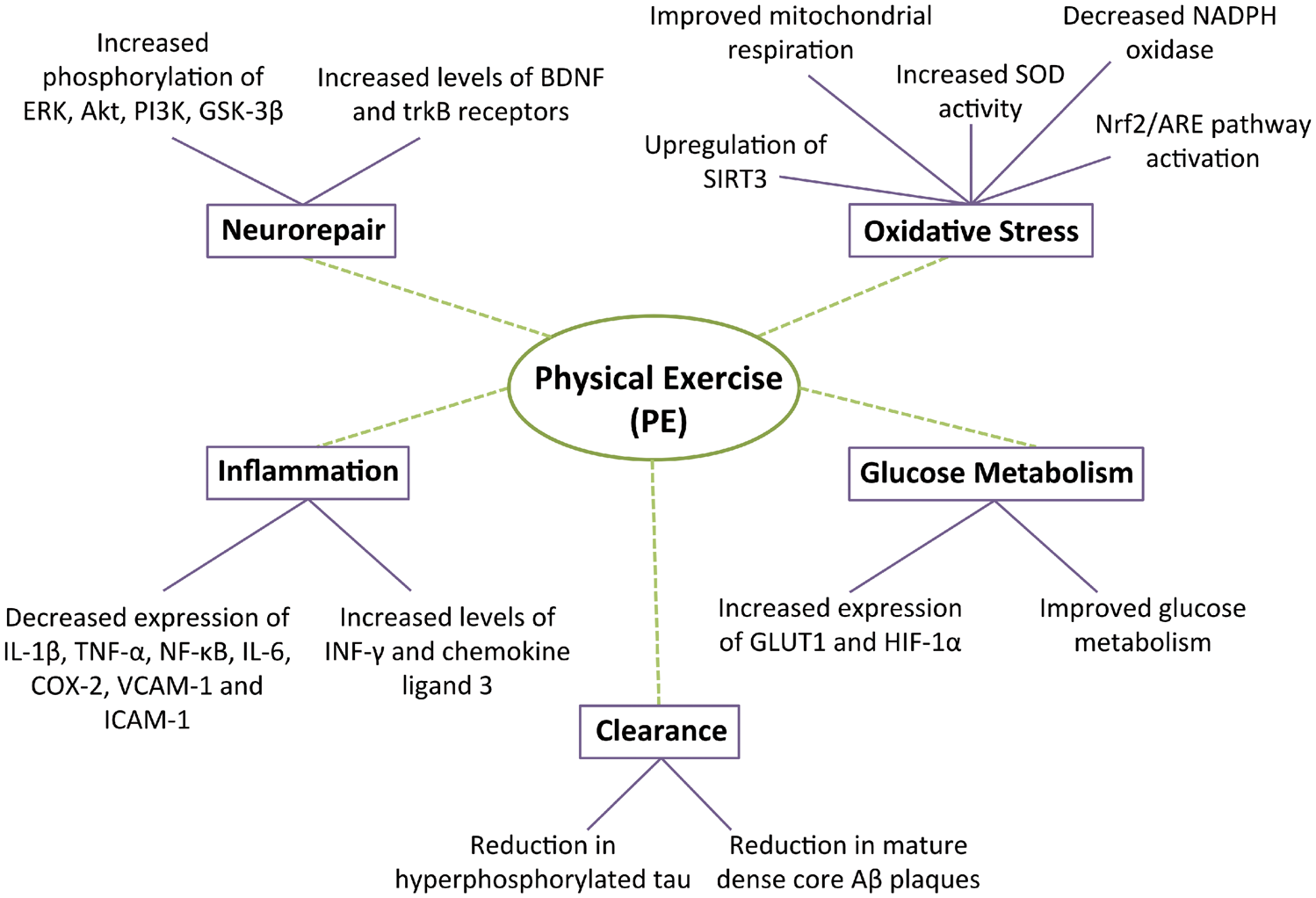
Toxic protein aggregates are thought to start accumulating before dementia onset and are considered key to the neurodegenerative process. PE has been shown to significantly reduce Aβ plaque load and levels of hyperphosphorylated tau through unclear mechanisms. We suggest that as a result of less amyloid, there is less vascular damage induced by the release of proinflammatory/oxidant mediators.
Exercise and oxidative stress
Endothelial cells have a precarious relationship with oxidative stress, as they are very sensitive to ROS and can exacerbate an already toxic environment by producing NO. NO can interact with superoxide ions (O2.-) to form peroxynitrite, which can then decompose into highly toxic hydroxyl radicals and nitrogen dioxide that can lead to mitochondrial dysfunction and interfere with transport across the BBB. 76 In environments that are high in oxidative stress, O2.- limits the amount of NO available and can result in eNOS uncoupling, 77 where eNOS produces O2.- ions instead of NO, further reducing the amount of NO required to maintain basal vascular tone. Transgenic J20 APP mice aged 4–18 months show increased levels of manganese superoxide dismutase (or SOD2) in pial vessels and show complete recovery of endothelial cell-dependent vasodilation to ACh and basal NO production or bioavailability after incubating cerebral arteries in vitro with antioxidants like superoxide dismutase (SOD) or catalase. 78
Cerebral endothelial cells contain multiple defense mechanisms against oxidative stress including glutathione (GSH), GSH peroxidase, GSH reductase, catalase, and SOD. 79 One response to oxidative stress is activation of the Nrf2/ARE pathway, which regulates the transcription of several antioxidant genes. Interestingly, this antioxidant pathway can be activated in the brain through the increase of peripheral insulin like growth factor 1 (IGF-1) that occurs during PE (Figure 2).80,81 In vitro experiments have shown that increased expression of Nrf2 increases intracellular GSH levels and can suppress tumor necrosis factor alpha (TNF-α)-induced leukocyte adherence to peripheral endothelial cells, preventing leukocyte migration into inflamed tissue. 82
Several studies report an inverted U-shape relationship between PE and oxidative stress, where both low and high levels of PE result in high levels of oxidative damage whereas a moderate amount of PE seems to be protective. Using a mouse model that mimics a key component (cerebral hypoperfusion) of VCID, elevated ROS levels were found in damaged WM following 1 month of bilateral common carotid artery stenosis, which was accompanied by working memory deficits on a spontaneous alternation Y-maze. 83 As shown in Tg2576 and J20 AD mouse models, the main source of ROS in cerebral vessels is NADPH oxidase, which is responsible for Aβ-induced cerebrovascular dysfunction as measured by neurovascular coupling and cerebrovascular reactivity experiments.78,84 The antioxidant properties of PE consists of decreased activity of the ROS-generating enzyme NADPH oxidase and increased activity of antioxidant enzymes Cu-Zn superoxide dismutase (or SOD1), located in cell cytoplasm and SOD3 located extracellularly, both of which function to increase conversion of ROS into less toxic compounds (Figure 2). 85 It has been reported that long-term treadmill exercise (5 months) in APP/PS1 double transgenic mice improved mitochondrial respiration via increased activity of complexes 1, 4, and 5, as well as decreased ROS production and oxidation of mitochondrial DNA in the hippocampus (Figure 2). 20 Moreover, sirtuin 3 (SIRT3), a mitochondrial matrix protein that regulates acetylation of metabolic enzymes, was upregulated by PE. This increase in SIRT3 could decrease acetylation of mitochondrial 8-oxoguanine DNA glycosylase-1 and SOD2 (Figure 2), and thus PE can positively influence mitochondrial antioxidant enzyme activity. Oxidative stress may be an early phenomenon in AD pathology and is certainly present throughout the disease progression, as well as in VCID. The above findings point to PE improving mitochondrial function and decreasing ROS production as a viable neuroprotective mechanism, indicating that PE can act as a form of antioxidant therapy on the brain’s vasculature.
Exercise and neuroinflammation
Although inflammation is fundamentally a protective response, when unmanaged it has the potential to exacerbate pathology. 86 The innate immune system naturally shifts toward a proinflammatory status with aging, making it most relevant for sporadic AD and VCID. In AD, Aβ deposits and soluble Aβ42 oligomers are able to activate astrocytes, which leads to the release of proinflammatory mediators including TNF-α, and interleukin IL-1β.87,88 In early stages of AD, microglia are thought to slow disease progression due to their ability to phagocytose Aβ; however, constant microglial production of proinflammatory cytokines TNF-α, IL-1β, IL-6, and IL-8 in later stages is detrimental. 89 As part of a sustained inflammatory response, TNF-α and IL-1 bind on the extracellular surface of endothelial cells to their respective receptors, which results in the activation of nuclear transcription factors NF-κB and activator protein 1. These factors are then capable of initiating transcription of cyclooxygenase-2 (COX2) and proinflammatory adhesion proteins that can bind leukocytes. Transcription and subsequent activation of COX2 in endothelial cells increase the conversion of arachidonic acid into prostacyclin (PGI2), which serves mainly to reduce inflammation. 90 Additionally, PGI2 binds to its receptors in smooth muscle cells and circulating platelets, which results in the synthesis of cAMP that activates protein kinase A, which relaxes smooth muscle cells, causing the vessel to dilate. 91 Therefore, any damage incurred upon endothelial cells will greatly influence inflammatory and vasomotor responses.
Other contributors to neuroinflammation are perivascular macrophages located adjacent to endothelial cells 92 that have recently been implicated in neurovascular dysfunction and cognitive impairments in a mouse model of chronic hypertension. 93 When perivascular macrophages were pharmacologically depleted in an AD mouse model with the Swedish and Indiana mutations (TgCRND8 mouse), CAA was significantly increased. Conversely, when perivascular macrophages were activated using chitin, Aβ clearance increased, 94 thus implicating them in Aβ clearance. Systemic inflammation can influence central inflammation through breakdown of tight junction proteins at the BBB. 95 Inflammation from the periphery can also cause leukocytes and cytokines to bind to BBB endothelium due to an upregulation of cell adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and P-selectin. Both of these signals increase leukocyte and platelet adhesion and promote a proinflammatory state in the brain’s parenchyma, hence allowing for potential entry of blood-derived monocytes into the brain.96,97 Conversely, endothelial NO has been implicated in inhibiting leukocyte activation and preventing the fusion of vesicles containing chemokines on the endothelial cell surface.98,99
PE reportedly reduces and even inhibits inflammatory processes, which when disinhibited negatively influence growth factor signaling and precipitate inflammation in the CNS. 81 When investigating the effects of PE in the Tg2576 AD mouse model, it was found that sedentary mice had higher hippocampal expression of proinflammatory interleukins IL-1β and TNF-α, and reduced levels of interferon-gamma (IFN-γ) and chemokine ligand 3. In contrast, when given the opportunity to run on a spinning wheel, IL-1β and TNF-α levels were reduced while INF-γ and chemokine ligand 3 levels were increased to WT levels (Figure 2). In a rat model of VCID, Lee et al. 100 fed aged rats a high fat diet to induce atherosclerosis. Although cognitive function was not assessed, 8 weeks of swimming exercise (1 h/day; 5 days/week) decreased peripheral inflammatory markers including NF-kB, TNF-α, IL-6, COX-2, vascular cell adhesion molecule (VCAM-1), and ICAM-1.
Neuroinflammation has gained attention in dementia research as it is thought to play an important role not only throughout the disease but also as a possible trigger in a number of deleterious cascades in early stages. PE at moderate levels is well established to reduce peripheral inflammation and neuroinflammatory responses, all of which reduce vascular inflammation and damage, and can help maintain BBB integrity.
Exercise and neurorepair
In the previous sections, we summarized how PE can reduce toxic protein aggregates, oxidative stress, and inflammation, we will now describe how it can also enhance biological processes to improve overall neuronal and vascular function.
Neurogenesis
Neurogenesis occurs in the highly vascularized subventricular zone in lateral ventricles of the forebrain and in the subgranular zone of the dentate gyrus in the hippocampus. 101 This process is thought to be relevant for learning and memory as new neurons can be integrated into existing circuits and provides the potential for new connections to form. An important mechanism involved in the proliferation of neural stem cells in adults is the Wnt/β-catenin pathway, which is modulated by the multifactorial serine/threonine kinase glycogen synthase-3β (GSK-3β). Patients with AD have more active GSK-3β, 102 which phosphorylates β-catenin and promotes its degradation. 103 This results in lower levels of β-catenin, 104 which is important for endothelial cells’ tight junction structures comprising β-catenin and vascular endothelial (VE)-cadherin. 105 β-catenin also binds to neural (N)-cadherin expressed on endothelial cell membranes, and it has been shown that N-cadherin influences the expression of VE-cadherin. 106
Another important contributor to neuron proliferation is brain-derived neurotrophic factor (BDNF) that can be synthesized and secreted by endothelial cells. 107 Unsurprisingly, brains of AD patients have lower levels of BDNF in temporal cortex and patients that are APOEɛ4 positive (a genetic risk factor reported to significantly increase likelihood of acquiring sporadic AD) display polymorphisms at BDNF loci.108,109 BDNF activates tropomyosin-related kinase B (trkB) leading to activation of mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways that are relevant for synaptic plasticity and LTP. 110 Binding of BDNF to trkB receptors on oligodendrocyte precursor cells has been implicated in the myelination process by promoting the formation of new oligodendrocytes, 111 which can affect signal transmission efficiency.
When improved cognitive performance is observed following a PE intervention in mouse models of AD, a commonly used explanation is increased neurogenesis in the hippocampus, along with angiogenesis and synaptic plasticity. 112 One study investigating the effects of voluntary PE on 10–12 month-old APOEɛ4 transgenic mice attributed improved performance on a radial arm maze task to increased levels of BDNF and upregulation of trkB receptors (Figure 2). 113 Strong positive correlations were found between BDNF levels and cognitive function following 3 months of voluntary PE in ovariectomized 3xTg-AD mice. 114 Several markers of neuronal cell death were investigated in another study using 24-month NSE/PS2m old transgenic mice, which express a human presenilin-2 mutation (PS2m) under the regulation of the rat neuron-specific enolase (NSE) promoter. Results showed increased phosphorylation of extracellular signal-regulated kinases (ERK), PI3K, protein kinase B (Akt), and GSK-3β, and increased levels of BDNF after exercising for 1 h/day on a treadmill for 3 months (Figure 2). 115 Another study examined synaptic transmission in the perforant path in a rat model of AD (intracerebroventricular infusion of Aβ1-42) and reported normalized levels of calmodulin-dependent protein kinase II (CAMKII) and BDNF, molecules important in LTP and memory formation. 17 However, in order to reap the benefits of these neuronal effects on PE, an adequate vascular niche is required to sustain the metabolic demands of pathways involved in neurogenesis.
Angiogenesis
Due to an increase in metabolic demand that occurs in early stages of neurogenesis, there is a lot of crosstalk between neural stem cells and blood vessels through direct physical contact and diffusible signals from vascular and perivascular cells.101,116 Thus, neurogenesis has been tightly linked to the concept of angiogenesis. To further support this link, BDNF and vascular endothelial growth factor (VEGF) regulate each other, 117 and proteins produced by the Wnt/β-catenin pathway are involved in angiogenesis. 118 Angiogenesis is the process of formation of new capillaries from existing vessels. Briefly, pericytes must first detach from the location in which the new vessel will form. This allows proteases to degrade the endothelial cell basement membrane and extracellular matrix in order for a new matrix to be synthesized by growth factors that promote endothelial cell division and stabilization. 119 Angiogenesis also depends on production and availability of a number of growth factors. However, one driving step in the formation of new capillaries is VEGF, which can be secreted by macrophages, pericytes, vascular smooth muscles cells, and astroglial cells, but whose receptors are almost exclusively found on endothelial cells. 120
In relation to dementia, studies have found altered serum levels of progenitor endothelial cells121,122 and angiogenic factors such as transforming growth factor β1 (TGF-β1) and VEGF in individuals with AD when compared to participants with amnestic mild cognitive impairment (MCI). 123 Lower levels of angiogenic mediators were associated with worse cognitive scores, suggesting that AD progression may be associated with a decreased capacity to regenerate endothelium. Interestingly, a form of small vessel disease associated with cognitive impairment called cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) is associated with increased TGF-β1 signaling in the brain. 124
A small group of cognitively normal, elderly individuals identified as high (3 h/week over the course of 10 years) or low (no record of regular PE in past 10 years or less than 3 h/week) aerobic activity levels were assessed for vessel number, radius, and tortuosity using MR angiography. A significantly higher number of small vessels (<1 mm diameter) were found in the anterior cerebral circulation in the high activity group. 125 PE-induced angiogenesis through production of endothelial progenitor cells 122 not only improves peripheral organ blood flow, but has been associated with increased cerebral blood volume in motor cortex 126 and improved CBF, 127 as measured in a rat model of stroke. However, it should be noted that increased metabolic demand induced by PE may partly account for the improved brain perfusion that allows for rapid transport of necessary substrates such as oxygen and glucose, in order for angiogenesis to occur. 128 Improved CBF could be indicative of improved neurovascular coupling, a tightly regulated phenomenon disrupted in AD and VCID, which ensures adequate supply of oxygen and energy to active neurons. 129
In normal aging neurogenesis and angiogenesis are decreased, and are often even lower in cases of dementia. PE has been shown to increase both phenomena as well as brain levels of BDNF and VEGF 130 that can be synthesized by endothelial cells. Together, these findings highlight the importance of endothelial cells in neurogenic and angiogenic mechanisms upon which PE can act.
Exercise and glucose transport
Activated neurons rely on a supply of glucose from the blood in order to function properly. Brain imaging studies have shown that subjects afflicted with AD, and even those with prodromal AD, show decreased glucose uptake and CBF throughout the brain. Findings reporting decreased glucose metabolism and high levels of oxidative stress in AD brains has lead to the hypothesis of AD being a form of diabetes, namely type 3 diabetes. 131 Sanabria-Dia et al. 132 analyzed local cerebral metabolic rate for glucose at rest in healthy elderly controls, MCI, and AD patients. Analyses revealed significant hypometabolism in AD patients compared to controls in temporal, parietal, and occipital lobes as well as structures in the limbic system. A study conducted in a 5xFAD mouse model that co-expresses five mutations of familial AD (APP KM670/671NL, APP I716V, APP V717I, PSEN1 M146L, PSEN1 L286V) making it a model of rapid amyloidosis, showed regional changes in glucose uptake at 2 months of age using FDG-PET that made them distinguishable from WT controls. 133
The main transporter of glucose at the BBB is the glucose transporter 1 (GLUT1), which is located in the endothelium of the brain capillaries. A deficiency in this transporter has been implicated in the vascular hypothesis of AD that places breakdown of the BBB early in the disease process. In an AD mouse model with the APPSw mutation, it was shown that when crossed with GLUT1-deficient mice, cerebral amyloid accumulation increased more rapidly. 134 This increase was attributed to decreases in LRP-1, CBF, and in BBB breakdown occurring earlier compared to the APPSw mouse.
PE has been recommended as a preventative measure for both diabetes mellitus and dementia, and studies report PE-induced changes in glucose metabolism (Figure 2). While examining the effects of PE on insulin signaling in a rat model of mild diabetes mellitus it was found that after 2 months of forced treadmill exercise, glycogen storage and glycogen synthase activity in the cortex and hypothalamus were improved in comparison to sedentary controls. 135 Similarly, a study conducted in elderly MCI adults investigated the effects of a 6-month high-intensity aerobic PE intervention. The authors reported improvements on a battery of executive function tasks that was accompanied by improved glucose metabolism as measured by an increase in insulin sensitivity. 136
A recent study reported that even a brief amount of PE (18 h) is sufficient to upregulate the number of endothelial GLUT1 transporters in the rat cerebral cortex (Figure 2), and increase the levels of lactate, an important component required for myelin synthesis. 137 A mere 5–10 h of PE increased the expression of hypoxia-inducible factor-1α (HIF-1α), a key mediator of glucose metabolism in endothelial cells 138 (Figure 2). Another testament to the importance of endothelial cells in glucose metabolism is their abundant expression of insulin and IGF-1 receptors. When insulin binds to its receptors on endothelial cells, they can release the vasodilator NO through the PI3K-Akt/eNOS pathway and vasoconstrictor ET-1 through MAPK pathways. 139 These pathways are involved in angiogenesis, along with the regulation of cellular growth, proliferation, and cell survival of vascular smooth muscle cells. 140
Together these observations of well-documented glucose hypometabolism in AD, and of PE improving cortical glycogen synthase activity and overall glucose metabolism, increasing glucose transport via endothelial GLUT1, and upregulating HIF-1α, underscore the importance of brain endothelial cells in the benefits of PE.
Exercise and WM integrity
A key feature seen in VCID is WM damage, as depicted by WM hyperintensities using conventional MRI and by microstructural WM changes using diffusion tensor imaging.141,142 In humans, WM comprises approximately half of the brain’s volume and contains myelinated axons that are responsible for transmitting signals over long distances both between and within hemispheres. 143 Alterations in WM can affect information processing by altering transmission speed and coherence within and between functional networks. 144 A recent multimodal imaging study investigated correlations between CBF in parietotemporal and frontal cortices, and microstructural WM changes in tracts connecting these areas with areas in the medial temporal lobes. 145 Prodromal AD patients revealed a strong correlation between lower CBF in parietal lobes and increased microstructural damage in WM fibers in the default mode network that is typically active during resting brain states and inactive during cognitive tasks. Blood flow in WM is naturally low 55 and the WM is threefold less vascularized than grey matter, 146 and thus has less metabolic reserve, making it more vulnerable to damage relative to grey matter.
Recent studies have examined the effect of aerobic PE on WM integrity and all report the same essential finding: higher cardiorespiratory fitness levels or aerobic PE interventions increase WM connectivity across all ages.147–150 Moderate PE, which has been shown to increase cerebral perfusion68,151 and has been linked to improved performance in executive function using a Stroop test, 152 can thus counteract loss of WM integrity and potentially influence cognition. Major arteries including the posterior cerebral artery, and superior and inferior branches of the middle cerebral artery irrigate most fiber tracts. It is not surprising then that chronic cerebral hypoperfusion, which has been noted as a potential marker for prodromal AD8,153 affects WM integrity, where low CBF worsens the efficiency of the tracts. The vascular laminar shear stress provided by blood flow is an important stimulus to the endothelium, and it is implicated in initiation of NO production, and in the remodeling and formation of vessels. 154 During PE, when blood flow is elevated, the increase in shear stress can lead to higher NO levels through eNOS upregulation.
A structural component of WM that can be indirectly affected by PE is the myelin sheath provided by oligodendrocytes, whose function relies heavily on the delivery of lactate from astrocytes.155,156 Engaging in PE increases peripheral levels of lactate, which is capable of crossing the BBB through the monocarboxylate transporter 1 (MCT1) found on endothelial cells.
155
Another way the brain can synthesize lactate is through the transport of glucose to astrocytes and oligodendrocytes by GLUT1 at the BBB. Once glucose enters oligodendrocytes, it is converted to pyruvate, and then to acetyl CoA, which is further processed to ATP and lipids that contribute to the formation of the myelin sheath. In astrocytes, glucose is converted to pyruvate and then lactate, which exits the astrocytes by monocarboxylate transporter 4 (MCT4) and enters oligodendrocytes by MCT1, where it can be used to synthesize myelin (Figure 3). One in vitro study showed that when myelin extracted from rats that had access to a spinning wheel was placed on cultured cortical neurons, cdk5 activity was significantly increased compared to myelin extracted from sedentary rats.
157
Active cdk5 is known to play an important role in neuronal differentiation during the elongation process. Cultured neurons were found to have longer processes and stronger GAP43 (a protein associated with axonal growth) immunoreactivity when incubated with myelin from rats that exercised compared to the sedentary rats in this study.
Benefits of physical exercise (PE) act through or require a healthy, functional endothelium (beige cells). Items written in green are upregulated or improved by PE, while those written in red are decreased or downregulated. PE increases the bioavailability of NO, which is synthesized by eNOS in endothelial cells. PE improves quality and increases quantity of sleep, which can facilitate clearance of toxic protein aggregates including amyloid and hyperphosphorylated tau by the glymphatic system. This clearance prevents amyloid from activating astrocytes (green cell), and thus prevents proinflammatory mediators from entering blood vessels and damaging the endothelium. PE can increase LRP-1 and decrease RAGE found on endothelial cells. PE improves CBF, which can in turn decrease the amount of cerebral amyloid angiopathy (CAA) around vessels and thus improve endothelial cell health. PE can decrease TNF-α and IL-1 levels in the periphery, as well as intracellular adhesion molecule (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), which could prevent the transcription of proinflammatory proteins and entry of leukocytes across the BBB. PE increases the activity of superoxide dismutase (SOD) enzymes and SIRT3 in mitochondria (peach cell), keeping ROS at low levels that will not negatively impact endothelial cell function. PE has been shown to upregulate GULT1 receptors found on endothelial cells, allowing more glucose into the brain. When glucose enters oligodendrocytes (orange cell) it is converted into lipids that are used to synthesize myelin. Another key component to myelin synthesis is lactate, which is upregulated by PE and enters the brain through MCT1 receptors found on endothelial cells. PE increases both brain derived neurotrophic factor (BDNF) and vascular endothelial growth factor (VEGF) transcription in endothelial cells, as well as activation of the Wnt/β-catenin pathway, which synthesizes β-catenin that is part of the structure of tight junctions between endothelial cells. These mechanisms may be important for neurogenesis and angiogenesis. Akt: protein kinase B; PI3K: phosphatidylinositol 3-kinase; SIRT3: sirtuin 3; HIF-1α: hypoxia-inducible factor 1α; IL-1β: interleukin 1β; TNF-α: tumor necrosis factor alpha; COX-2: cycloxygenase 2.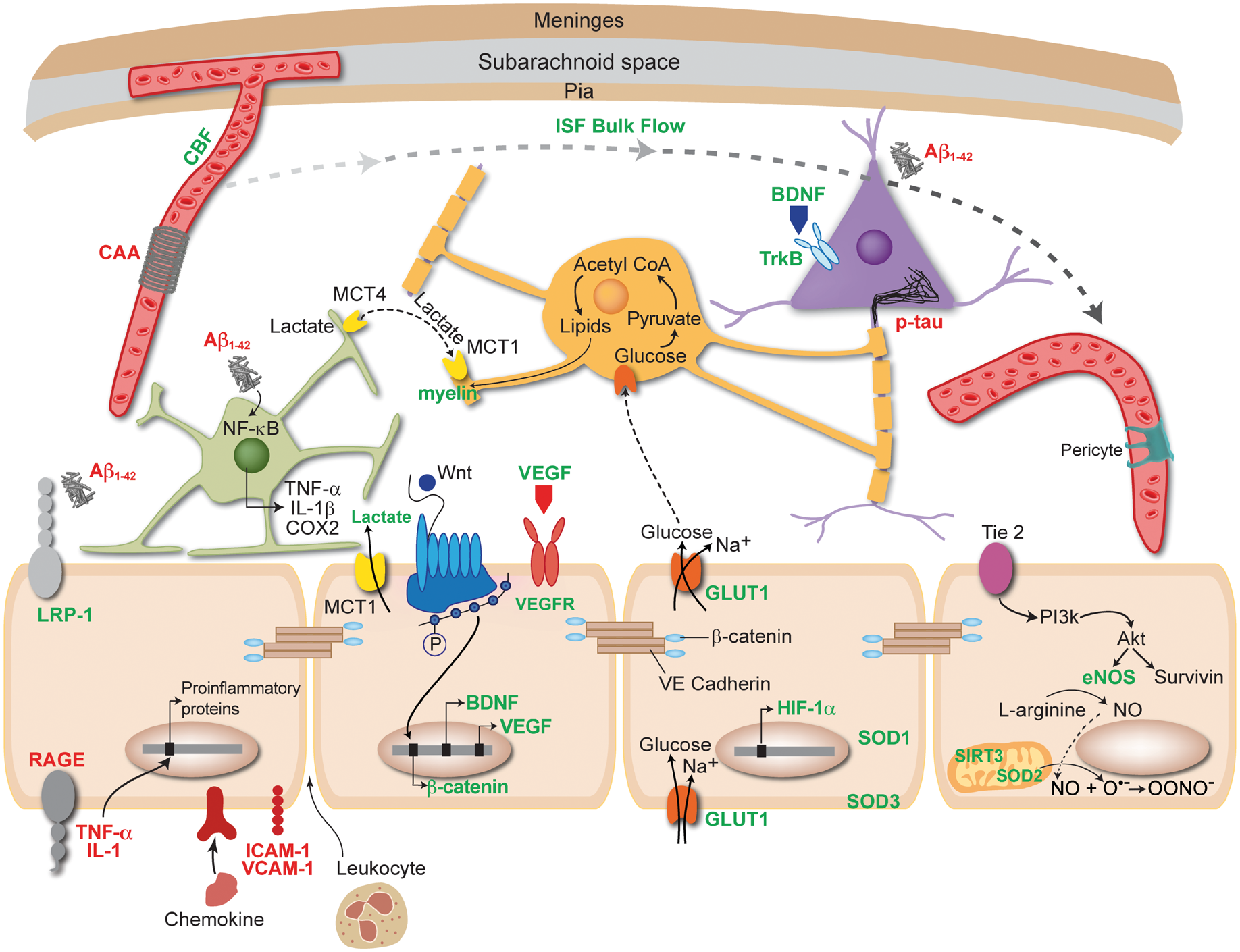
Although more indicative of VCID, WM hyperintensities are also present in cases of AD. PE has been shown to improve WM integrity, which could be explained by improved perfusion to watershed areas but also to increases in glucose and lactate transport through endothelial cells that are integral to the synthesis of myelin.
A central role for endothelial cells in mediating the benefits of exercise
Although PE is well accepted as a protective, nonpharmacological approach in modifying the likelihood or progression of AD and VCID, the mechanisms through which it exerts its benefits are not agreed upon. We propose that a possible common link between most PE-induced effects resides in the endothelium (Figure 3), whose dysfunction can have widespread ramifications as it secretes a variety of biologically active mediators, is largely responsible for maintaining vascular homeostasis, and has been implicated in key phenomena involved in the onset and progression of AD.29,158
The main vascular risk factors for developing sporadic AD and VCID all share the common feature of endothelial cell dysfunction. Disrupted insulin signaling seen in diabetes mellitus, fatty streaks from hypercholesterolemia, and increased blood pressure from hypertension all reduce the bioavailability of NO, stimulate ROS production, and promote a proinflammatory environment. 34 All of these disrupt endothelial cell function. In the presence of oxidative stress, elevated levels of O2.- react with NO to form peroxynitrite which can lead to oxidation of tetrahydrobiopterin and, consequently, eNOS uncoupling that generates O2.- rather than NO. 159 With a decreased production and availability of NO, more vesicles containing chemokines can fuse to endothelial cell surfaces and potentially permit leukocytes to enter the brain. 96 Endothelial cells are also sensitive to cytokines TNF-α and IL-1, which when bound, are capable of initiating transcription of proinflammatory proteins. 81 PE has been well established to reduce expression of proinflammatory cytokines, decrease NADPH oxidase activity, increase antioxidant enzymes SOD1, SOD2, and SOD3, upregulate SIRT3,20,85 and decrease the presence of cell adhesion molecules VCAM-1 and ICAM-1 on endothelial cells. 100
The processes of neurogenesis and angiogenesis, which are stimulated during PE, have a close interplay with endothelial cells, and neurogenesis requires a vascular niche to occur. 101 Although not the main source of BDNF, endothelial cells synthesize and secrete this growth factor, 107 which is well known to be upregulated with PE. The Wnt pathway activated by PE produces β-catenin, which is integral to endothelial cell tight junctions as part of their structural integrity. PE can increase the production of endothelial progenitor cells to promote the formation of new blood vessels, and endothelial cells also express VEGF receptors that are key to the process of angiogenesis, which can increase CBF and thus improve neurovascular coupling, a phenomenon reliant on endothelial pathways. 160 Other receptors found on endothelial cells involved in Aβ transport include RAGE and LRP-1, which may be down- and upregulated, respectively, by PE. 64 However, the precise role of PE on the clearance mechanism for Aβ has not been established to our knowledge. Where PE can play a role in the clearance of toxic proteins is through improving the quality and increasing the duration of sleep, during which Aβ and hyperphosphorylated tau can possibly be cleared via the glymphatic system, at the level of the brain vasculature. Another key transporter found in endothelial cells that is rapidly upregulated by PE is GLUT1, 137 which serves to bring glucose into the brain, and deliver it to neurons, astrocytes, and oligodendrocytes, which require glucose to support their main functions. Also, PE has been shown to increase insulin sensitivity and endothelial cells contain insulin and insulin growth factor receptors that stimulate the release of NO and ET-1 that regulate vascular tone. 139 Lastly, PE has been shown to improve WM connectivity, which may be related to a number of factors, including improved global CBF, increases in proteins involved in neuronal differentiation and axonal growth, and increased number of endothelial GLUT1 receptors that deliver glucose directly to oligodendrocytes that produce the myelin sheath.
The vascular endothelium can act as a cellular hub that plays a role in mediating the several benefits reaped by PE. Although it may not be sufficient to target the endothelium to improve cognitive function in patients with AD and VCID, it appears to be necessary. Moderate PE acts as a multi-target drug and its effects need to be studied further to know the frequency and intensity at which it is required, and to gain a deeper understanding of PE at a cellular level if an effective pharmacological therapy is to be found to prevent and potentially reverse dementia.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Canadian Institute of Health research (MOP-126001), and the Canadian Consortium on Neurodegeneration and Aging (CIHR-CCNA), and a CIHR studentship (LT). The authors gratefully thank Drs Clotilde Lecrux and María Lacalle-Aurioles, and Jessika Royea for their insightful comments and revisions, and Susan Kaupp for her work on ![]() .
.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.