
Abstract
The blood–brain barrier is a microvascular network that (1) provides neuroprotection from metabolic and environmental toxins and (2) limits the delivery of therapeutics to the central nervous system (CNS). The ATP-binding cassette transporter P-glycoprotein contributes to the latter by actively pumping clinical substrates back into circulation before they can reach the brain parenchyma. Targeting P-glycoprotein has proven effective in increasing the delivery of therapeutics to their cerebral targets. We provide a novel mechanism to achieve this end in functioning, intact rat brain capillaries, whereby the bioactive phospholipid lysophosphatidic acid (LPA) and tricyclic antidepressant (TCA) amitriptyline reduce basal P-glycoprotein transport activity through a distinct lysophosphatidic acid 1 receptor-mediated signaling cascade that requires G-protein coupling, Src kinase, and ERK 1/2. Furthermore, we demonstrate the ability of LPA and TCA amitriptyline to decrease induced P-glycoprotein transport activity in a human SOD1 transgenic rat model of amyotrophic lateral sclerosis. This work may translate to new clinical strategies for increasing the cerebral penetration of therapeutics in patients suffering from CNS diseases marked by exacerbated pharmacoresistance.
Keywords
Introduction
Two microvascular components maintain the dynamic and selective permeability of the blood–brain barrier. Mechanical tight junctions anchor neighboring endothelial cells to one another and ATP-binding cassette (ABC) transporters that line both sides of the capillary endothelium actively pump substrates between brain parenchyma and circulation. Together, they maintain a homeostatic balance of both large and small solutes between each and provide neuroprotection from environmental and pathogenic toxins. 1 However, therapeutics developed to treat diseases of the CNS must cross this barrier in order to reach full clinical potential. The ABC transporter P-glycoprotein is a major obstacle for the targeted delivery of many of these drugs, including the amyotrophic lateral sclerosis (ALS) drug riluzole. 2 Its high expression relative to other transporters at the brain microvasculature and a directionality that results in the net efflux of its clinical substrates from brain to blood directly oppose efforts made by physicians to treat their patients with CNS therapeutics. For CNS diseases like epilepsy and ALS, bypassing P-glycoprotein-driven pharmacoresistance is especially challenging because (1) therapeutics developed to treat them include P-glycoprotein substrates and (2) their pathogeneses are marked by transporter induction.3–5
Attempts to rapidly and reversibly decrease P-glycoprotein-mediated drug efflux in order to enhance blood-to-brain therapeutic delivery, although numerous, have not been clinically adopted. For the first time, we demonstrate the ability of the endogenous phospholipid lysophosphatidic acid (LPA) and tricyclic antidepressant (TCA) amitriptyline to achieve this end ex vivo. Once considered to be a quiescent metabolic intermediate, LPA has been shown to elicit a range of cell- and tissue-specific responses through a six-membered family of G-protein-coupled receptors (GPCRs), LPA 1-6 receptors, and has been implicated in both the development and treatment of different pathologies.6–9 Here, we show that the treatment of freshly isolated rat brain microvessels to nanomolar concentrations of LPA reduces P-glycoprotein transport activity through one of its six GPCRs: lysophosphatidic acid 1 receptor (LPA1R). We exploited these findings to determine that the TCA amitriptyline also reduces P-glycoprotein transport activity through LPA1R at the rat brain microvasculature at equally low concentrations. We translated these findings to a human SOD1 transgenic ALS rat model and demonstrate the ability of both LPA and the TCA amitriptyline to reduce P-glycoprotein-mediated substrate transport when its expression is upregulated with disease progression. Given both the established presence of amitriptyline and other antidepressant drugs in the clinic and limited therapies available for treating ALS, these findings may contribute to the development of new methods by which to increase therapeutic delivery to the CNS, especially in disease states characterized by exacerbated P-glycoprotein-driven pharmacoresistance.
Materials and methods
Materials
1-Oleoyl-LPA was purchased from Santa Cruz Biotechnology. Mouse monoclonal P-glycoprotein antibody was obtained from Covance. Secondary goat anti-rabbit IgG (H+L) Alexa Fluor 488 antibody was purchased from Invitrogen. The P-glycoprotein-specific inhibitor PSC833, multidrug resistance-associated protein-2 (Mrp2)-specific inhibitor MK571, breast cancer resistance protein (Bcrp)-specific inhibitor KO143, ERK 1/2 inhibitor FR 180204, Src kinase inhibitor PP2, PI3K inhibitor LY294002, and Rho/SRF inhibitor CCG1423 were all purchased from Tocris Bioscience. The dual LPA1R-LPA3R antagonist Ki16425, LPA3R-specific agonist (2S)-OMPT, rabbit polyclonal LPA1R antibody, and rabbit polyclonal LPA3R antibody were obtained from Cayman Chemical. The LPA1R-specific antagonist AM095 was purchased from ApexBio. The fluorescent P-glycoprotein substrate N-ɛ(4-nitrobenzofurazan-7-yl)-D-Lys8 cyclosporin A (NBD-CSA) was custom-synthesized, the fluorescent Bcrp substrate BODIPY® FL prazosin was purchased from Invitrogen, and the fluorescent Mrp2 substrate sulforhodamine 101 free acid (Texas Red) was purchased from Sigma-Aldrich. 10 Mouse monoclonal β-actin antibody, LPA1R-specific agonist NAEPA, amitriptyline hydrochloride, actinomycin D, cycloheximide, Ficoll, and all other chemicals were also purchased from Sigma-Aldrich.
Animals
All experiments were approved by the Animal Care and Use Committee at the National Institute of Environmental Health Sciences (NIEHS), executed in accordance with the National Institutes of Health animal care and use guidelines, and presented in agreement with ARRIVE reporting guidelines. Adult male Sprague Dawley rats between the ages of six and nine months and human SOD1 transgenic ALS model rats were obtained from Taconic. All animals were housed in temperature- and humidity-controlled rooms with a 12-h light/dark cycle and ad libitum access to food and water. Animals were killed by carbon dioxide inhalation and immediate decapitation. Human SOD1 transgenic ALS model rats were considered symptomatic and sacrificed after disease presentation, indicated by the development of limb paralysis with simultaneous weight loss indicative of muscle wasting.11,12 Brain capillaries were isolated and either immediately used for transport assay and immunohistochemistry studies or prepared and frozen for additional Western blotting analysis.
Capillary isolation
Procedures for capillary isolation were outlined previously.13,14 The step involving syringe column passage has been replaced with one utilizing 30 µm cell strainers (pluriSelect). After decapitation, rat brains were transferred to cold PBS (in mM: 2.7 KCl, 1.5 KH2PO4, 136.9 NaCl, 8.1 Na2HPO4, 1 MgCl2, 1 CaCl2, supplemented with 5 D-glucose, 1 sodium pyruvate, pH 7.4). Tissue was kept in cold PBS for the remainder of the capillary isolation process. White matter, meninges, midbrain, choroid plexus, blood vessels, and olfactory lobes were dissected away and remaining brain tissue was homogenized. An aliquot of 30% Ficoll was added to an equivalent volume of brain homogenate and capillaries were separated from remaining brain parenchyma by centrifugation at 5800 g for 20 min. Capillary pellets were washed with 1% BSA in PBS and passed through a series of 30 µm cell strainers (pluriSelect), washed sequentially with PBS, and used immediately.
Transport assay
Confocal microscopy- and imaging-based transport assays with isolated rat brain capillaries were characterized previously. 15 All transport assay studies were conducted at room temperature within coverslip-bottomed imaging chamber slides filled with PBS. The fluorescent substrates, NBD-CSA for P-glycoprotein, Texas Red for Mrp2, and BODIPY® FL prazosin for Bcrp, were added with or without either LPA or amitriptyline and luminal substrate accumulation, indicated by luminal fluorescence intensity, was measured at varying time-points in the presence or absence of different antagonists, agonists, and inhibitors. 16 In every study, a specific inhibitor, PSC833 for P-glycoprotein, MK571 for Mrp2, and KO143 for Bcrp, was also added in order to determine the component of luminal substrate accumulation that was transport mediated. Capillaries were imaged with a Zeiss 510-inverted confocal laser-scanning microscope through a 40 × water-immersion objective (numeric aperture of 1.2) using a 488 nm laser line for both NBD-CSA and BODIPY® FL prazosin and a 543 nm laser line for Texas Red. Images were saved to a thumb drive, transferred, and luminal florescence was quantitated using NIH Image J software as characterized previously. 10 Data reported are for a single study that is representative of three to five experimental replicates.
Western blotting
Endothelial membranes were isolated from control and LPA-exposed brain-derived microvessels as outlined previously.17,18 Membrane protein was assayed by the Bradford method and an aliquot of the membrane protein determined from the Bradford-derived standard curve was mixed with NuPAGE 4 × sample buffer obtained from Invitrogen. The mixture was loaded onto a 4–12% Bis-Tris NuPAGE gel, electrophoresed, and subsequently transferred to an Immobilon-FL membrane obtained from Millipore. Odyssey Blocking Buffer from Li-Cor Biosciences was added to the membrane at room temperature for 30 min, after which the membrane was immunoblotted with mouse monoclonal P-glycoprotein antibody (170 kDa, 1:200). The membrane was then stained with goat anti-mouse fluorescent dye IRDye 800CW (1:1000) in PBS at room temperature for 3 h and sequentially washed in 0.05% Tween in PBS. The membrane was imaged using an Odyssey Infrared Imaging System from Li-Cor Biosciences. β-actin (42 kDa, 1:10,000) was used as a loading control.
Immunostaining for LPA1R and LPA3R
Isolated brain capillaries were fixed, permeabilized, and blocked in PBS as outlined previously. 16 Capillaries were then incubated at 4℃ overnight in either the primary rabbit polyclonal LPA1R antibody or primary rabbit polyclonal LPA3R antibody, and then at room temperature for 90 min in secondary goat anti-rabbit IgG (H+L) Alexa Fluor 488 antibody. The primary antibodies used recognized either LPA1R or LPA3R, which are both membrane-localized G-protein-coupled receptors. Immunostained capillaries were imaged with a Zeiss 510-inverted confocal laser-scanning microscope through a 40X water-immersion objective (numeric aperture of 1.2) using a 488 nm laser line. Images were saved to a thumb drive, transferred, and the fluorescence at the luminal and abluminal membranes was determined using NIH ImageJ software.
In situ brain perfusion
Brain perfusion was executed as described previously. 19 Rats were anesthetized with a 1 mL/kg ketamine mixture (79 mg/mL ketamine, 3 mg/mL xylazine, 0.6 mg/mL acepromazine) and administered heparin (10 kU/kg) via intraperitoneal injection. After exposing the common carotid arteries by midline incision at the neck, the common carotid arteries were perfused with oxygenated Ringer’s solution at 37℃ (in mM,117 NaCl, 4.7 KCl, 0.8 MgSO4, 24.8 NaHCO3, 1.2 KH2PO4, 2.5 CaCl2, 10 D-glucose; in g/L, 39 dextran, 1 BSA and 0.055 Evans blue) at 3 mL/min. [3H]-verapamil (0.1 μCi/mL) was infused into the circuit via syringe pump at 0.5 mL/min for 20 min. Samples of perfusate were collected from the cannulae at the end of each experiment. Brains were removed, separated by hemisphere, stripped of meninges, midbrain, and choroid plexuses, and minced. Tissue and 100 µL perfusate samples were solubilized and counted. The results were expressed as the ratio of disintegrations per minute in the brain to disintegrations per minute in the perfusate (Rbr µL/g).
Statistical analyses
Quantitative data are expressed as mean ± SEM. Statistical analyses of differences between experimental groups were performed by one-way ANOVA (Tukey multiple comparison test) or t-test using Prism software. Differences between experimental group means were considered significant when p < 0.05.
Results
LPA and amitriptyline exposure rapidly and reversibly reduce specific P-glycoprotein transport activity ex vivo
We used an established confocal imaging-based assay to determine the transport activity of P-glycoprotein in freshly isolated rat brain microvessels, which constitute the blood–brain barrier ex vivo.
10
Our assay and resulting analyses rely on the luminal accumulation of the fluorescent P-glycoprotein substrate N-ɛ(4-nitrobenzofurazan-7-yl)-D-Lys8 cyclosporine A (NBD-CSA). Figure 1(a) includes the representative confocal micrographs of freshly isolated rat brain capillaries incubated for 30 min to steady-state, followed by one of the four conditions: (1) medium containing only 2 µM NBD-CSA to represent control or baseline levels of luminal substrate accumulation, (2) medium containing both 2 µM NBD-CSA and 10 µM PSC833, a P-glycoprotein-specific inhibitor, (3) medium containing both 2 µM NBD-CSA and 10 nM LPA, an endogenous phospholipid, and (4) medium containing both 2 µM NBD-CSA and 10 nM amitriptyline, a TCA. Luminal NBD-CSA accumulation, indicated by luminal fluorescence, is visibly reduced in confocal micrographs of cerebral microvessels exposed to PSC833, LPA, or amitriptyline, when compared to control. It is important to note that LPA and amitriptyline exposure resulted in a loss of luminal NBD-CSA accumulation comparable to that elicited by PSC833, which is considered to be both maximal and specific. To this end, we only considered the PSC833-sensitive component of luminal NBD-CSA accumulation to be P-glycoprotein-mediated and used that component to measure specific P-glycoprotein transport activity, as done previously.19–21
LPA- and amitriptyline-dependent changes in P-glycoprotein-mediated transport at the rat brain microvasculature (a) Representative confocal micrographs of rat brain microvessels after incubation with 2 µm NBD-CSA for thirty minutes. Treatment with PSC833, LPA, or AMT results in visibly reduced luminal fluorescence (scale bars, 10 µm). (b) LPA- and AMT-elicited decreases in P-glycoprotein transport activity are concentration dependent. Actions of LPA and AMT on P-glycoprotein transport activity are rapid and reversible. Each point depicts the mean value for 10 to 16 microvessels from a single isolation, with tissue pooled from 5 to 10 rats. SEM bars represent experimental variability, and units are arbitrary fluorescence. Statistical comparisons: ***, significantly different than control, p < 0.001; *, significantly different than control, p < 0.05.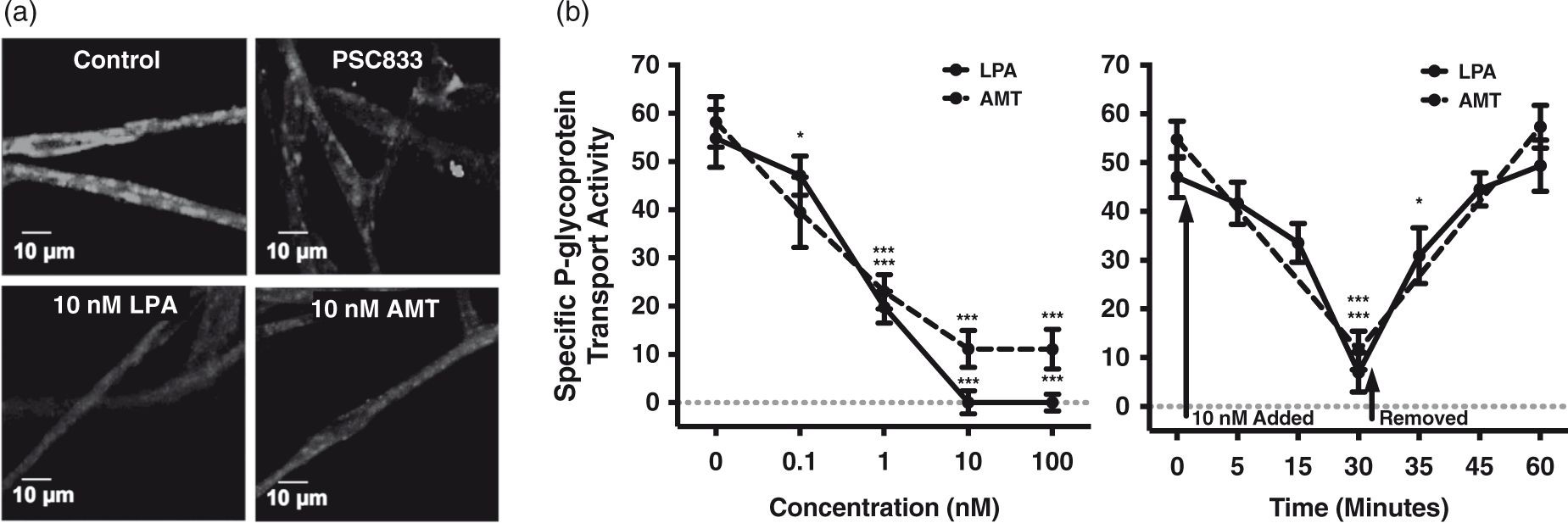
Exposing brain-derived microvessels to 0.1–100 nM LPA or amitriptyline for 30 min reduced P-glycoprotein transport activity in a concentration-dependent manner (Figure 1(b)). To determine whether the reduction in transport activity was reversible post-exposure, we performed a combined time-course and reversibility assay. After incubation for 30 min to steady-state, brain capillaries were incubated in media containing either 10 nM LPA for 5, 15, and 30 min or amitriptyline for 30 min, at which point these solutions were replaced with media containing only 2 µM NBD-CSA and incubated for a resulting 5, 15, and/or 30 min. Confocal micrographs were obtained for each of these time points, and the results are shown in Figure 1(b). Exposing brain capillaries to 10 nM LPA or 10 nM amitriptyline for 30 min resulted in rapid and maximal reduction in P-glycoprotein transport. However, transport activity returned to control levels within 30 min of removing LPA or amitriptyline, indicating the LPA- and amitriptyline-elicited decreases in specific P-glycoprotein transport activity are also reversible.
Signaling through LPA1R
LPA potentially signals through a family of six GPCRs shown to have tissue-specific expression profiles.
22
Exposing rat brain capillaries to 0.1–100 µM Ki16425, a dual LPAIR-LPA3R antagonist, blocked the LPA-elicited decrease in specific P-glycoprotein transport activity in a concentration-dependent manner (Figure 2(a)). To determine whether the LPA-dependent decrease in P-glycoprotein transport activity is mediated by LPAIR, LPA3R, or a combination of both, we used a complement of LPA1R- and LPA3R-specific agonists and antagonists. Treatment with 10 nM (2 S)-OMPT, an LPA3R-specific agonist, did not reduce luminal NBD-CSA accumulation, while treatment with 10 nM NAEPA, an LPA1R-specific agonist, did so maximally. Exposing rat brain capillaries to either 10 µM AM095 or 100 µM Ki16425, an LPA1R-specific antagonist and dual LPA1R-LPA3R antagonist respectively, completely blocked the LPA-elicited decrease in P-glycoprotein transport activity. Treatment with 10 µM AM095 also blocked the action of amitriptyline on P-glycoprotein transport activity (Figure 2(b)). To visually illustrate this point, representative confocal micrographs are included in Figure 2(c). Freshly isolated rat brain capillaries were incubated for 30 min to steady-state and exposed to one of the three conditions: (1) medium containing only 2 µM NBD-CSA to represent control or baseline levels of luminal substrate accumulation (2) medium containing both 2 µM NBD-CSA and 10 nM amitriptyline, and (3) medium containing 2 µM NBD-CSA, 10 nM amitriptyline, and 10 µM AM095. Luminal fluorescence is visibly reduced in microvessels exposed to 10 nM amitriptyline. Importantly, this reduction was not observed in the presence of an LPA1R-specific antagonist. Taken together, these data indicate the reduction in P-glycoprotein transport activity downstream of LPA exposure is an LPA1R-dependent event.
LPAR function and expression in rat brain microvessels. (a) A dual LPA1R-LPA3R antagonist blocks the LPA-elicited decrease in specific P-glycoprotein transport activity in a concentration-dependent manner. (b) An LPA1R-specific agonist decreases specific P-glycoprotein transport activity maximally, while an LPA3R-specific agonist does not. Both an LPA1R-specific and dual LPA1R-LPA3R antagonist block LPA’s action on P-glycoprotein transport activity. An LPA1R-specific antagonist blocks AMT’s action of P-glycoprotein transport activity. Each bar depicts the mean value for 8 to 14 microvessels from a single isolation, with tissue pooled from three to six rats. SEM bars represent experimental variability, and units are arbitrary fluorescence. (c) Representative confocal micrographs of brain microvessels after incubation with 2 µm NBD-CSA for thirty minutes. Treatment with AMT results in visibly reduced luminal fluorescence, but not in the presence of the LPA1R-specific antagonist AM095 (scale bars, 10 µm). (d) Representative confocal micrographs of rat brain microvessels immunostained for LPA1R and LPA3R (scale bars, 10 µm). (e) Luminal AMT exposure by perfusion increases the brain accumulation of [3H]-verapamil in vivo. Each point represents the mean value from a cerebral hemisphere, with four rats in each treatment group. SEM bars represent experimental variability, and units are the ratio of disintegrations per minute in the brain to disintegrations per minute in the perfusate (Rbr µL/g). Statistical comparisons: ***, significantly different than control, p < 0.001; *, significantly different than control, p < 0.05.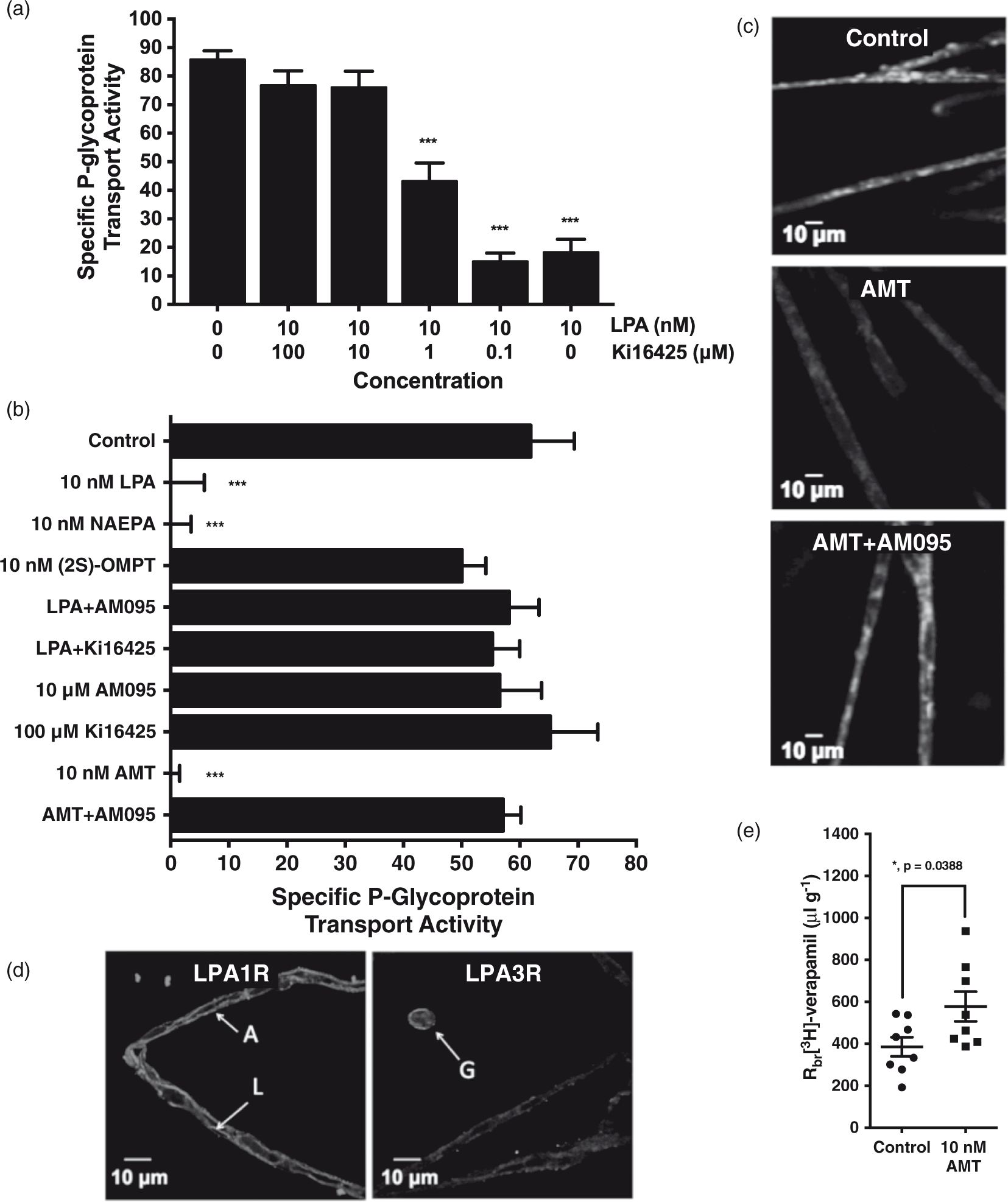
Using receptor-specific antibodies and immunofluorescence technologies, we detected the presence of LPA1R, but not LPA3R, in rat brain capillaries (Figure 2(d)). LPA1R was detected at both the luminal and abluminal membranes of cerebral microvessels, while LPA3R was detected in extravascular cells, supporting previous studies that report its expression in astrocytes and microglia.23,24 Ex vivo exposure does not allow for the luminal- or abluminal-specific receptor activation; therefore, we utilized in situ brain perfusion as an in vivo model to observe changes in P-glycoprotein transport activity given luminal amitriptyline exposure. As shown in Figure 2(e), perfusion with 10 nM amitriptyline for 20 min resulted in an increase in the cerebral penetration of the P-glycoprotein substrate [3H]-verapamil, indicating a decrease in P-glycoprotein transport activity. Thus, luminal amitriptyline exposure increases the brain deposition of a therapeutic P-glycoprotein substrate in vivo.
Intracellular events downstream of LPA1R activation
Because the reduction in P-glycoprotein transport activity caused by LPA exposure is reversible and effectively blocked by antagonizing LPA1R, we set out to elucidate the intracellular signaling events downstream of LPA1R activation by its endogenous ligand. Treatment of brain-derived microvessels with either 1 µM actinomycin D or 200 µM cycloheximide, disruptors of transcriptional and translational machinery respectively, did not block the LPA-elicited decrease in P-glycoprotein transport activity (Figure 3(a)). Western blots indicated no change in P-glycoprotein expression at capillary membranes after exposing whole capillaries to 10 nM LPA for thirty minutes (Figure 3(b)). These results indicate that the rapid reduction in P-glycoprotein transport activity is independent of changes in expression and protein degradation. We then assessed the effect of 10 nM LPA exposure on the transport activities of two additional ABC transporters present at the blood–brain barrier. As shown in Figure 3(c) and (d), exposing brain-derived microvessels to 10 nM LPA did not alter the transport activities of either Mrp2 or Bcrp. Representative confocal micrographs of capillaries incubated for 30 min to steady-state and exposed to either control conditions or 10 nM LPA are included for visual illustration. Treatment with sodium cyanide reduced the activity of both Mrp2 and Bcrp by depleting ATP stores. Disruption of tight junctional complex integrity would have resulted in a decrease in the luminal accumulation of the Mrp2 substrate sulforhodamine 101 free acid (Texas Red), as seen in rat brain capillaries incubated in hyperosmotic media containing 100 mM sucrose.
14
Taken together, these data indicate the LPA-elicited decrease in luminal NBD-CSA accumulation is specific to the transport activity of P-glycoprotein and does not involve the disruption of either tight junctional complex integrity or ATP availability, P-glycoprotein degradation, or changes in transcription or translation.
LPA-elicited decrease in P-glycoprotein transport activity is G-protein-, Src kinase-, and ERK1/2-dependent. (a) Inhibitors of transcription and translation do not block the LPA-elicited decrease in P-glycoprotein transport activity. (b) Western blot of rat brain capillary membranes; 30-min exposure to 10 nM LPA does not change P-glycoprotein expression or protein levels when normalized to β-actin. (c) LPA does not affect the transport activity of Mrp2. Representative confocal micrographs of capillaries incubated with 2 µm Texas Red illustrate LPA exposure has no effect on luminal Mrp2 substrate accumulation (scale bars, 10 µm). (d) LPA does not affect the transport activity of Bcrp. Representative confocal micrographs of capillaries incubated in 2 µm BODIPY® FL prazosin illustrate LPA has no effect on luminal Bcrp substrate accumulation (scale bars, 10 µm). (e) A G-protein antagonist, Src kinase inhibitor, and ERK1/2 inhibitor all block the action of LPA on P-glycoprotein-mediated transport, while the inhibitors of P13K and the Rho/SRF pathway do not. (f) Abbreviated working model. Each bar depicts the mean value for 8 to 14 microvessels from a single isolation, with tissue pooled from three to six rats. SEM bars represent experimental variability, and units are arbitrary fluorescence. Statistical comparisons: ***, significantly different than control, p < 0.001.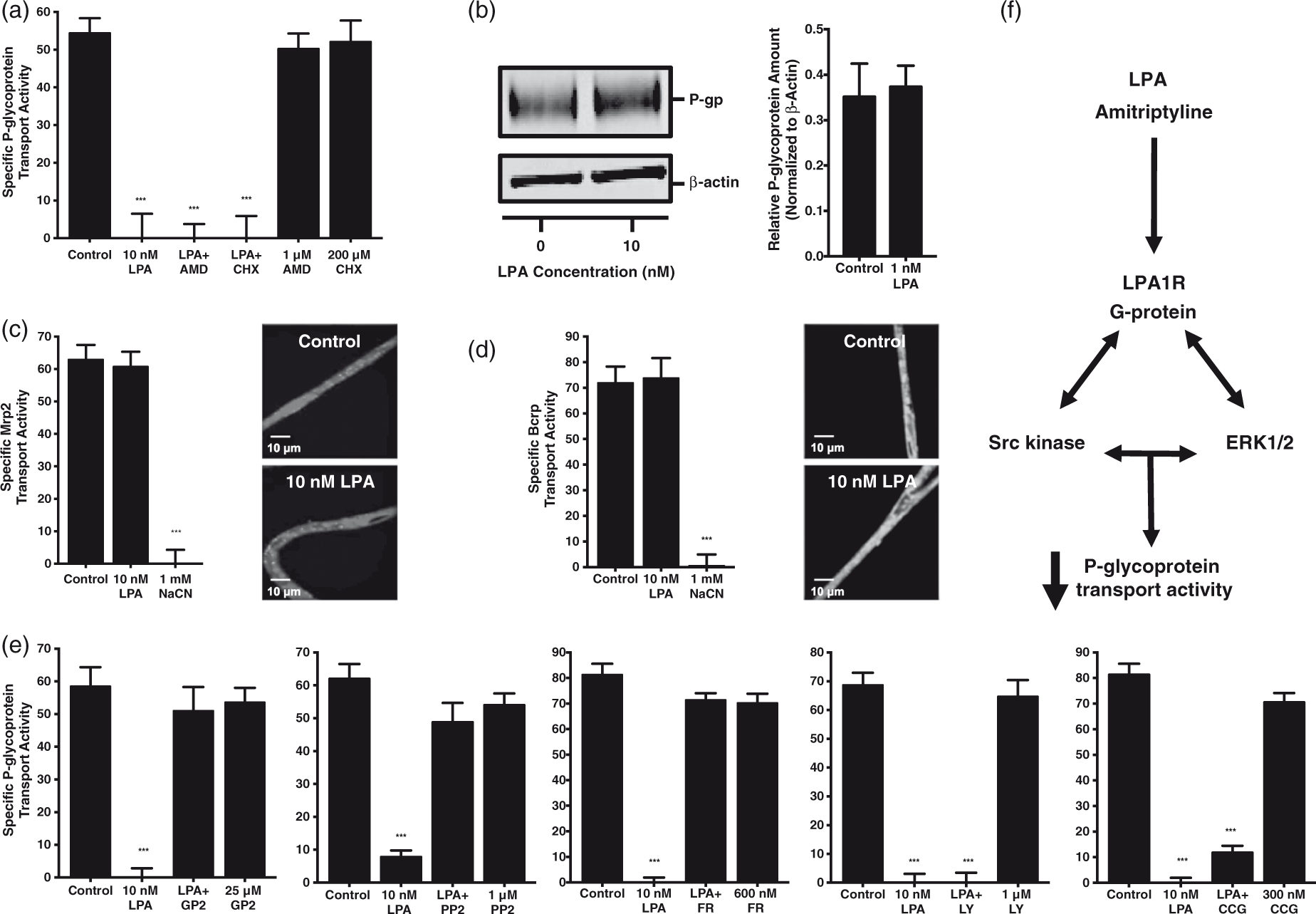
Treatment of brain-derived microvessels with 25 µM GPAnt-2, an agent that disrupts the interaction between G-proteins and their receptors, blocked LPA’s reduction in P-glycoprotein transport activity maximally. Treatment with either 1 µM PP2 or 600 nM FR180204, inhibitors of Src kinase and ERK1/2 activity respectively, blocked the reduction in P-glycoprotein transport activity caused by LPA exposure whereas treatment with either 1 µM LY294002 or 300 nM CCG1423, inhibitors of PI3K activity and the Rho/SRF pathway respectively, did not (Figure 3(e)). These results indicate the activities of G-proteins, Src kinase, and ERK 1/2, but not of PI3K or Rho/SRF, are required for the decrease in P-glycoprotein transport downstream of LPA1R at the rat brain microvasculature. We have included an abbreviated model outlining the receptor activation and intracellular signaling effectors required for the LPA-elicited decrease in specific P-glycoprotein transport activity (Figure 3(f)).
Mitigating ALS-driven P-glycoprotein transport induction
Increases in P-glycoprotein expression have been reported in human SOD1 transgenic animals, implicating P-glycoprotein in the pharmacoresistance associated with ALS progression.4,25–27 Western blots indicated that P-glycoprotein protein levels were increased in brain capillary membranes isolated from symptomatic human SOD1 transgenic ALS model rats compared to those derived from wildtype controls (Figure 4(a)). To investigate the relationship between increased P-glycoprotein expression and P-glycoprotein transport activity at the brain microvasculature, we measured luminal NBD-CSA accumulation in microvessels derived from both human SOD1 transgenic ALS model rats and wildtype controls. Figure 4(b) includes representative confocal micrographs of brain capillaries isolated from both symptomatic human SOD1 transgenic ALS model and wildtype control rats incubated for 30 min to steady-state in media containing only 2 µM NBD-CSA. Luminal substrate accumulation is clearly increased in capillaries isolated from symptomatic human SOD1 transgenic ALS model rats compared to age-matched wildtype controls.
LPA and AMT decrease ALS-induced P-glycoprotein transport activity. (a) Western blot of rat brain capillary membranes; P-glycoprotein expression is upregulated in human SOD1 transgenic ALS model rats compared to wildtype controls when normalized to β-actin. (b) Representative confocal micrographs of brain microvessels from either wildtype or human SOD1 transgenic ALS model rats. Note that the increase in luminal fluorescence seen in microvessels isolated from human SOD1 transgenic ALS model rats compared to wildtype controls, and the decrease in luminal fluorescence that occurs with AMT treatment (scale bars, 10 µm). (c) P-glycoprotein transport activity is significantly increased in SOD1 rats compared to wildtype. AMT and LPA treatments mitigate the induced P-glycoprotein-mediated transport observed in human SOD1 transgenic ALS model rats, and this effect is completely blocked by treatment with an LPA1R-specific antagonist. Each point depicts a single microvessel, with tissue pooled from five to six rats. SEM bars represent experimental variability, and units are arbitrary fluorescence. Statistical comparisons: ***, significantly different than control, p < 0.001; *, significantly different than control, p < 0.05.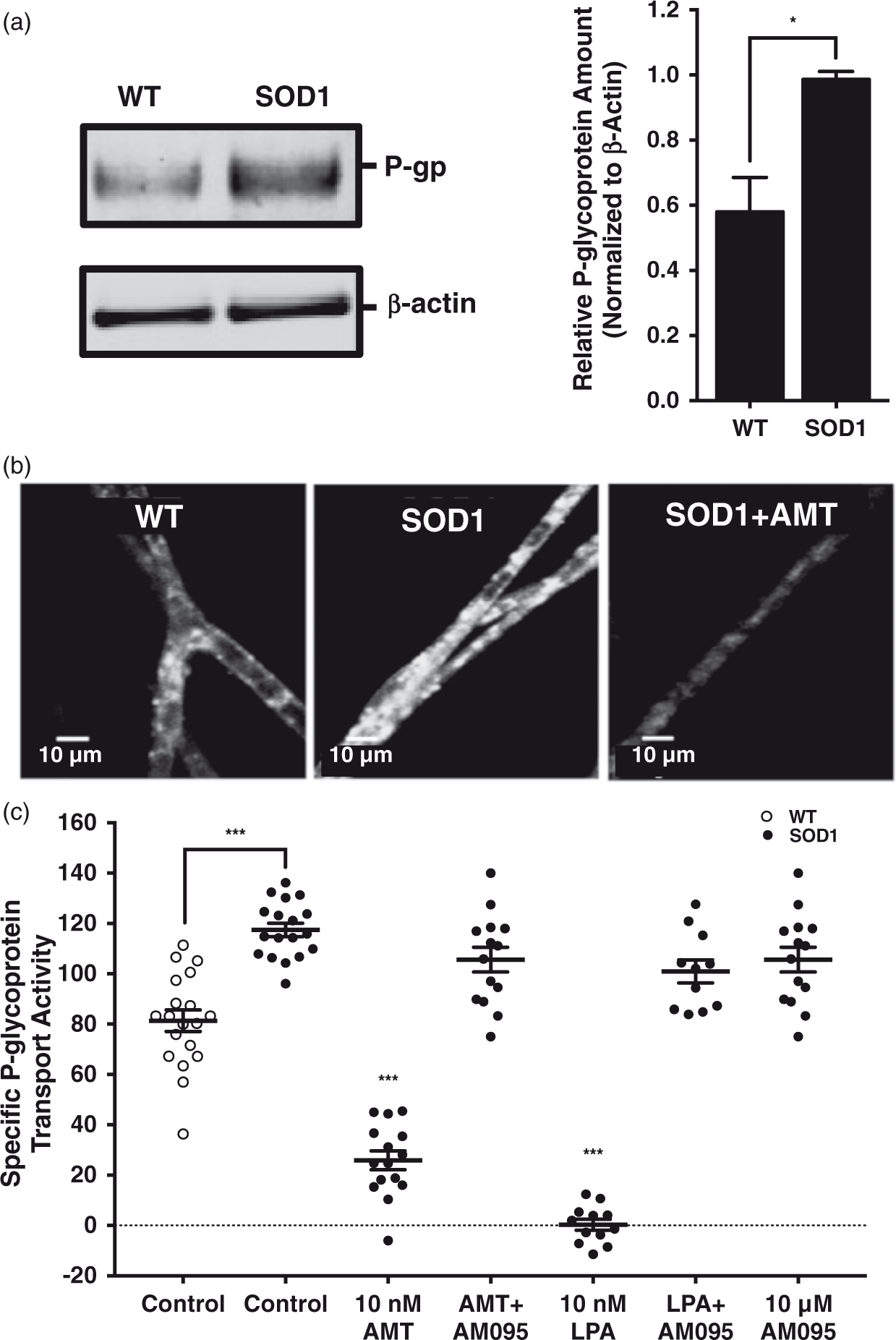
To determine if disease-associated increases in P-glycoprotein transport are sensitive to reduction through LPA1R, capillaries isolated from human SOD1 transgenic ALS model rats were exposed to either 10 nM LPA or amitriptyline in the presence or absence of the LPA1R-specific antagonist AM095. Note the decrease in luminal fluorescence that occurs with treatment of brain capillaries isolated from SOD1 transgenic ALS model rats with 10 nM amitriptyline (Figure 4(b)). Treatment of cerebral microvessels derived from symptomatic human SOD1 transgenic ALS model rats with either 10 nM LPA or amitriptyline reduced P-glycoprotein transport activity maximally. Importantly, both reductions were blocked by treatment with 10 µM AM095, an LPA1R-specific antagonist (Figure 4(c)). Taken together, these data indicate that LPA and amitriptyline reduce the specific transport activity of P-glycoprotein in symptomatic human SOD1 transgenic ALS model rat cerebral microvessels in an LPAIR-mediated manner, even when induced as a component of disease pathology.
Discussion
In this report, we demonstrate that exposure of intact rat brain capillaries to nanomolar concentrations of either LPA or amitriptyline specifically reduces the transport activity of P-glycoprotein through LPA1R at the cerebral microvasculature. Intracellular events downstream of LPA1R involve the activities of G-protein coupling, Src kinase, and ERK 1/2, but occur independently of P13K and Rho/SRF activity, expression, P-glycoprotein degradation, compensatory efflux by other ABC transporters, disruption of microvascular integrity, and metabolic poisoning. We exploited these findings in human SOD1 transgenic ALS model rat brain microvessels as a model of P-glycoprotein induction and exacerbated pharmacoresistance.
Several groups have reported on the effects of LPA on blood–brain barrier permeability previously. Their mechanistic interpretations include non-specific disruption of vascular integrity, proteolytic cleavage, and large-scale structural reorganization of the neurovascular unit.28,29 We identify a receptor-specific mode of action for LPA at the blood–brain barrier through modulation of the transport activity of the ABC transporter P-glycoprotein. The varied findings could result from different experimental parameters. First, our model utilizes freshly isolated, brain-derived microvessels that are metabolically active and maintain their in vivo membrane polarity. Second, in previous studies, the concentrations of LPA used to elicit changes in vascular permeability were in the micromolar range, whereas we observed maximal changes in transporter-mediated permeability in the nanomolar range. The specificity of LPA’s action and downstream signaling may be linked to these differences in concentration. Additionally, because of the range of phospholipid bioactivity at the vasculature, our results may not represent events outside of P-glycoprotein transport. We do, however, provide evidence that LPA exposure does not affect the activities of two other ABC transporters (Bcrp and Mrp2) or cause capillary leakage.
A number of signaling cascades that regulate ABC transport activity and expression at the capillary endothelium have been identified.14,18,20 We show the LPA-elicited decrease in P-glycoprotein transport activity requires LPA1R, G-protein coupling and the activities of both Src kinase and ERK1/2. These findings are consistent with reports showing the involvement of Src kinase in the decrease in P-glycoprotein transport activity downstream of flk-1 activation by vascular endothelial growth factor (VEGF). 19 After establishing that LPA1R-mediated signaling reduced P-glycoprotein transport activity at the cerebral microvasculature, we investigated the ability of the TCA amitriptyline to act as an LPA1R ligand and elicit the same effect. While ERK1/2 has not been implicated as a modulator of P-glycoprotein transport activity, ERK1/2 phosphorylation has been observed downstream of LPA1R activation by certain antidepressants, including amitriptyline, in CHO-K1 fibroblasts and glial cells.30,31 We determined that amitriptyline also reduces P-glycoprotein-driven efflux at the blood–brain barrier in an LPAIR-dependent manner, revealing a novel and direct receptor-mediated effect on P-glycoprotein by amitriptyline. Previous reports have characterized the cerebral penetration of amitriptyline as a P-glycoprotein substrate, and found little difference between P-glycoprotein knockout and wildtype mice at certain time points after dosing. 32 Our in situ brain perfusion results indicate the cerebral penetration of amitriptyline in P-glycoprotein knockout and wildtype mice may converge due to an LPA1R-mediated inhibition of P-glycoprotein transport activity at the brain microvasculature, whereby amitriptyline increases its own cerebral penetration independently of its primary therapeutic mechanism as an antidepressant.
The role of ABC transporters in limiting the penetration of CNS drugs into the brain is well documented. P-glycoprotein is a major contributor to this clinical challenge, so much so that variations in its expression can forecast treatment outcomes for certain cancers, including neuroblastoma. 33 Rapidly and reversibly decreasing its transport activity is a means by which to increase the delivery of CNS therapeutics while maintaining the neuroprotective function of the blood–brain barrier long term. Modulating its transport potential may be an especially important consideration for clinicians whose patients suffer from diseases marked by P-glycoprotein upregulation, including epilepsy and ALS. Riluzole is currently the only drug approved by the Federal Drug Administration for the treatment of ALS; hence, determining tissue-level changes that may occur with disease is clinically important. 34 We have previously characterized changes in ABC transporter expression in the human SOD1 transgenic ALS model rat. 27 For this reason, we investigated the potential of LPA1R activation to mitigate induced P-glycoprotein transport activity at the blood–brain barrier in a human SOD1 transgenic ALS rat model. We determined that treating SOD1-derived microvessels with either LPA or amitriptyline reduced P-glycoprotein transport activity via LPA1R activation, even when induced with CNS pathology. We predict this LPA1R-mediated reduction may translate to other CNS pathologies (e.g. epilepsy, stroke, and certain cancers) shown to exhibit induced P-glycoprotein-driven pharmacoresistance.35–37
Amitriptyline is already used in the clinic to treat depression, somatoform pain disorders, and insomnia.38,39 The established use of amitriptyline in the clinic is based on known therapeutic modes of action that have not been shown to involve phospholipid receptor activation at the vasculature. We demonstrate an amitriptyline-elicited decrease in specific P-glycoprotein transport activity through LPA1R at the rat blood–brain barrier ex vivo. Our work cannot rule out the possibility that amitriptyline signals through LPA1R indirectly; however, this does not diminish the potential clinical benefit of using amitriptyline as an adjunct therapy to enhance drug delivery to the CNS.
The rapid and reversible nature of transport reduction and nanomolar concentrations required indicate that amitriptyline may be a candidate for adjunct therapies, but with special consideration given to dosing parameters, as well as potential drug–drug interactions and toxicity. It is important to note the time course and reversibility observed may translate to rapid circulation of a drug following dosing; however, more pharmacokinetic work should be conducted in order to determine the exact dosing parameters needed to achieve the desired reduction. The increase in delivery following in situ brain perfusion we report provides hopeful but limited insight into the timing and dosing required. Additionally, if the amitriptyline-elicited decrease in P-glycoprotein-mediated efflux translates to other excretory tissues, there is also potential for a systemic increase in plasma concentration of co-administered drugs, which may result in adverse drug reactions (ADRs). For this reason, the amitriptyline-elicited effect we report should be further investigated as it relates to possible ADRs associated with different drugs and across different tissues.
Further in vivo studies wherein adjunct therapies with amitriptyline and other antidepressant drugs are employed to determine increased brain penetration and therapeutic efficacy should be conducted. Indirect inhibition of P-glycoprotein has already been shown to increase the therapeutic efficacy of ALS drugs in murine models. 40 However, we provide an alternative, and potentially more effective, means by which to directly target P-glycoprotein through a distinct signaling cascade that reduces its efflux potential in order to increase the delivery of CNS therapeutics. Activating an endogenous phospholipid-induced signaling cascade at the capillary endothelium with nanomolar concentrations of a therapeutic already present in the clinic may provide a safe and effective means to target P-glycoprotein while avoiding side effects associated with dosing and specificity. We determine the LPA1R-transport activity relationship holds true in cases of both basal and induced P-glycoprotein-mediated substrate efflux ex vivo, and reason for clinical implementation after further in vivo validation.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Intramural Research Program at the National Institute of Environmental Health Sciences, National Institutes of Health.
Acknowledgements
We thank the members of the Intracellular Regulation Group for technical support: Emily Mesev and Joyce Blaisdell. We also thank the staff at the NIEHS animal facility for providing excellent care to animals used in this study.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: David B. Banks and Ronald E. Cannon are listed as co-inventors on a submitted provisional patent (application number 62453718) entitled: Methods for Improving Drug Delivery Across the Blood-Brain-Barrier.
Authors’ contributions
Participated in research design: Banks, Cannon and Miller. Conducted experiments: Banks, Chan and Evans. Performed data analysis: Banks, Miller and Cannon. Manuscript writing: Banks and Cannon.