
Abstract
Infection and inflammation are known risk factors for neonatal brain injury. Mycoplasma and Gram-positive bacteria, for which Toll-like receptor 2 (TLR2) plays a key role in recognition and inflammatory response, are among the most common pathogens in the perinatal period. Here, we report that systemic activation of TLR2 by Pam3CSK4 (P3C) increases neural tissue loss and demyelination induced by subsequent hypoxia–ischemia (HI) in neonatal mice. High-resolution respirometry of brain isolated mitochondria revealed that P3C suppresses ADP-induced oxidative phosphorylation, the main pathway of cellular energy production. The results suggest that infection and inflammation might contribute to HI-induced energy failure.
Introduction
Neonatal brain injury remains one of the leading causes of neurological morbidity worldwide, affecting 1–3/1000 of newborns. 1 Hypoxia/ischemia (HI) is one important underlying factor behind neurological disability in the newborn. 2 Infection and inflammation are well-described risk factors for HI brain injury.4,5 Toll-like receptors (TLRs) are a major family of pattern recognition receptors, sensing and responding to different stimuli originating from microbial pathogens or injured cells. TLR2 has a pivotal role in inflammatory responses to Gram-positive bacteria and mycoplasma. 7 Although a large proportion of perinatal infections are caused by these pathogens, 8 the role of TLR2 in neonatal brain injury and the underlying mechanisms have not been fully elucidated. We recently determined the peripheral and central inflammatory profile induced by TLR2 ligand PAM3CSK4 (P3C) in neonatal mice. 9 Here we report for the first time that acute systemic activation of TLR2 sensitizes the developing brain to subsequent HI injury. High-resolution respirometry reveals that TLR2 activation suppresses phosphorylating respiration of brain mitochondria that potentially can contribute to HI-induced energy failure.
Material and methods
Animals
C57Bl/6J mice were purchased from Charles River Laboratory. TLR2−/− (B6.129-Tlr2tm1Kir/J) mice were purchased from the Jackson Laboratory and were bred with C57BL/6J mice to obtain TLR2 heterozygote mice (TLR2+/−). From these TLR2+/− mice, TLR2+/+ and TLR2−/− pups, of both gender, were obtained and used in this study. Genotyping was performed according to the Jackson Laboratory protocol. Animal experimental procedures conformed to guidelines established by the Swedish Board of Agriculture (SJVFS 2015: 38), were approved by the Gothenburg Animal Ethics Committee (18–2015), and are reported in a manner consistent with the ARRIVE (Animal Research: Reporting in Vivo Experiments) guidelines.
P3C administration and HI
Postnatal-day 8 (P8), mice were injected intraperitoneally (i.p.) with 5 mg/kg P3C (Invivogen, France) or saline (0.9% NaCl) 14 h before HI. P3C dosage and sample sizes were based on our previous study. 9 The HI procedure has been described previously. 10 Briefly, pups were anesthetized by isoflurane, the left carotid artery was ligated using a non-absorbable prolene suture. Following vessel ligation pups were returned to the cage to recover for 1 h, and were then placed in a hypoxic chamber (10% oxygen, 36°C) for 50 min.
Immunohistochemistry
Immunohistochemistry (IHC) procedures were performed as described previously.
10
Five days after HI, mice were sacrificed with an overdose of thiopental and perfused with saline and 5% paraformaldehyde (Histofix; Histolab, Sweden). Brains were further fixed in Histofix overnight at 4°C, dehydrated, and embedded in paraffin;10 µm thick coronal sections were cut at 5 levels, 40 sections apart (Figure 1). After antigen retrieval and endogenous peroxidase activity blocking (3% H2O2 in phosphate-buffered saline (PBS), sections were incubated in 4% horse serum and 3% bovine serum albumin in PBS (1 h at room temperature (RT)) to block unspecific binding. Sections were then incubated with primary antibodies microtubule-associated protein-2 (MAP-2; clone HM-2, 1:1000; Sigma-Aldrich) or mouse monoclonal antibody myelin basic protein (MBP, 1:10,000, SMI-94R; Covance) at 4°C overnight, and then with corresponding biotinylated secondary antibodies (Vector Laboratories, 1 h, RT). To enhance peroxidase activity, Vectastain ABC Elite kit was used following manufacture’s protocol. Enzyme substrate DAB was used to visualize the target proteins.
Systemic activation of TLR2 by P3C exacerbates hypoxic-ischemic (HI) brain injury in neonatal mice. (a) The percentage loss of MAP2 stained area at five levels (represented in lower panels) of brain of mice injected i.p. with saline (n = 14) or P3C (n = 15) 14 h prior to HI. (b) The percentage loss of MAP2 stained area at five levels of brain of TLR2+/+ (n = 13) and TLR2−/− (n = 20) mice injected with P3C and exposed to HI. (c) The percentage loss of MBP stained area in subcortical white matter at three levels of brain of mice injected with saline (n = 14) or P3C (n = 15) and exposed to HI. (d) The percentage loss of MBP stained area at three levels of brain of TLR2+/+ (n = 13) and TLR2−/− (n = 20) mice treated with P3C and exposed to HI. (e,f) Total MAP2 percentage loss in mice described in (a) and (b). (g,h) Total MBP percentage loss in mice described in (c) and (d). The data are presented as Mean ± SEM. The asterisks on the bars in (a) to (d) mark significant difference obtained by two-way ANOVA analysis followed by Bonferroni post hoc test. The numbers on the bars in (e) to (h) show the p values obtained by student t-test.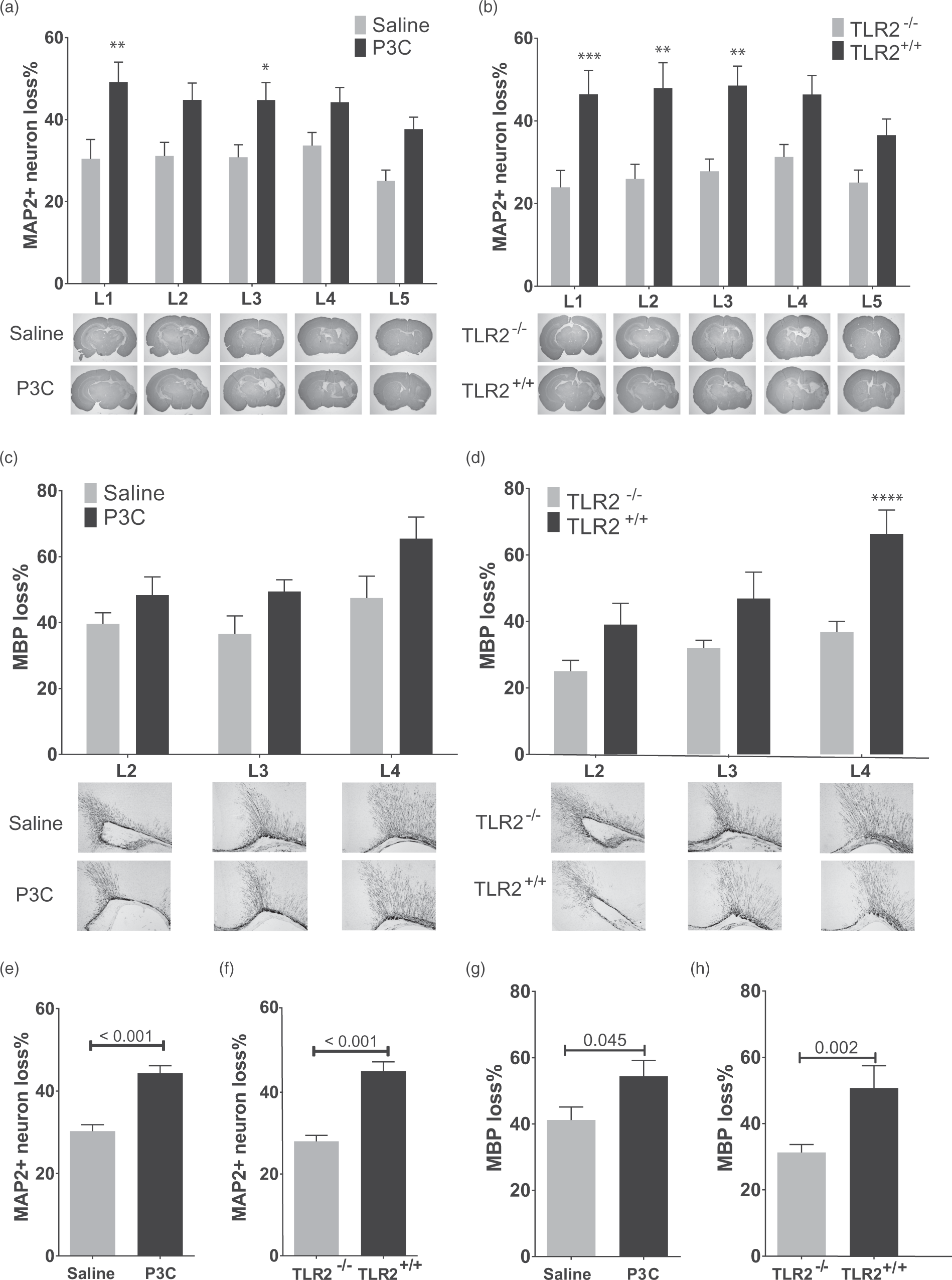
Assessment of brain injury
A Nikon Optiphot-2 microscope equipped with AVT dolphin F145B camera (Allied Vision Technologies) was used for capturing images. Positively stained areas were measured using Olympus Micro Image analysis software system (V4.0, Olympus Optical). The MAP-2 stained area in ipsilateral hemisphere was subtracted from contralateral hemisphere and the result was divided by MAP-2 area in contralateral hemisphere to obtain MAP-2 loss ratio. MBP stained area was measured in subcortical region and the loss ratio was calculated as for MAP-2. One section at each brain level was analyzed. All image analysis was performed by an investigator blind to sample treatment and genotype. Data were analyzed by two-way ANOVA followed by Bonferroni post hoc test when analyzing brain injury at different levels, and by two-tailed unpaired t-test when comparing total brain injury. Graphpad Prism V 6.01 was used for data analysis, and data are presented as Mean ± SEM.
Mitochondrial isolation
Brain mitochondria was isolated as described in detail previously (method A). 11 Briefly, mice were sacrificed by decapitation, and the cerebrum was immediately collected in ice-cold isolation buffer (IB) and homogenized in a Dounce homogenizer. The brain homogenate was centrifuged at 1100g for 2 min at 4°C, the supernatant was mixed with 75 µl of 80% Percoll solution (GE Healthcare) in Percoll diluent buffer, and added on top of 750 µl of 10% Percoll in IB, and centrifuged at 18,500 g for 10 min. The supernatant was discarded and the pellet was washed in 1 ml washing buffer twice, and the final pellet was resuspended in 100 µl of MIR05 buffer.
Mitochondria respiration analysis
Mitochondrial respiration was measured as described previously 11 using a high-resolution respirometer, Oroboros Oxygraph-2 k (Oroboros Instruments, Innsbruck, Austria). Samples were run in pairs of saline/P3C in two chambers of O2K devices. All measurements were performed at 25°C in 2.1 mL MIR05 buffer. The O2 flux was monitored in real-time using DatLab V 6.0 software. After injecting 50 µL of isolated mitochondria into chambers, saturating dosages of substrates were added to obtain final concentration of 4.8, 2.5, 0.12 mM for pyruvate, malate, and ADP respectively and 1 µmol/L of carbonyl cyanide 4-trifluoromethoxy phenylhydrazone (FCCP). Pyruvate and malate initiate leak state respiration, ADP induces complex I-mediated oxidative phosphorylation (OXPHOS capacity), and FCCP induces uncoupled respiration that corresponds to electron transport system (ETS) capacity. Respiratory control ratio (RCR) was calculated by dividing OXPHOS capacity by leak state respiration. OXPHOS capacity was corrected for leak state respiration by subtracting leak state value from max OXPHOS to obtain Free OXPHOS capacity. Free ETS capacity was also calculated by subtracting leak state value from ETS capacity. All obtained values were normalized to total protein content, and two-tailed paired t-test was performed to analyze the data.
Results
P3C exacerbates neural tissue loss after HI in neonatal mice in a TLR2-dependent manner
To investigate the effect of TLR2 activation on neonatal HI, TLR1/2 ligand P3C or saline was injected 14 h before HI. The measurement of neural tissue loss was performed 5 d after HI at PND 14 by MAP-2 staining of neurons on coronal sections at five levels (L1-L5) of cerebrum (Figure 1(a) and (b)). P3C administration significantly increased the HI-induced neural loss at two levels (Figure 1(a)) and the average loss of neural tissue of all five levels was increased by 14.0% ± 2.4 (p < 0.001, Figure 1(e)). No difference in sensitization was observed between female and male pups (data not shown). P3C did not change rectal temperature at the time of HI induction (saline: 35.2 ± 0.2°C; P3C: 35.4 ±0.1°C, n = 8/group, p = 0.51). To investigate whether the sensitization effect of P3C on HI brain injury is TLR2-dependent, TLR2−/− and TLR2+/+ littermates were exposed to HI 14 h after administration of P3C. TLR2+/+ mice (n = 13) had higher loss of MAP2+ neurons than TLR2−/− mice (n = 20) (Figure 1(b)) and overall, TLR2−/− mice were significantly protected against injury compared to TLR2+/+ mice by 17.1% ± 2.5 (p < 0.001, Figure 1(f)).
P3C augments HI-induced myelin loss in neonatal mice in a TLR2-dependent manner
The effect of TLR2 activation on myelin loss after HI was explored by MBP staining of coronal section of cerebrum at three levels. P3C administration caused a 13.2% ± 6.2 overall increase in HI-induced loss of MBP (Figure 1(g)). However, at individual levels of the brain, the difference was not statistically significant (Figure 1(c)). No difference was observed between female and male pups (data not shown). TLR2−/− mice were significantly protected against myelin loss particularly at the most anterior level (Figure 1(d)), and overall by 14.0% compared to TLR2+/+ mice (Figure 1(h)).
Systemic activation of TLR2 suppresses brain mitochondria respiration
Next, we asked whether inflammation induced by TLR2 activation affects mitochondrial respiration. We used a high-resolution respirometry system with high sensitivity and low noise to assess mitochondrial respiration in real-time with various substrates for complex I activity (Figure 2(a)). Respiratory control ratio was not significantly different between the P3C (13.53 ± 0.56, n = 13) and saline control mice (13.81 ± 0.51, n = 13, p = 0.544) at 14 h after P3C administration. ADP-induced phosphorylating respiration (OXPHOS capacity) was suppressed in P3C-treated mice by 23% compared to control (p = 0.040). OXPHOS corrected for leak state (Free OXPHOS capacity) was also decreased by 23.9% (Figure 2(b)). P3C diminished the electron transport system (ETS) capacity by 21%, although the difference did not reach statistical significance (p = 0.054). Free ETS capacity showed a 22% suppression by P3C treatment (Figure 2(c)).
Systemic activation of TLR2 suppresses brain mitochondrial respiration. (a) A representative respirogram of brain mitochondria isolated from mice injected i.p. with saline (red line) or P3C (green line) at 14 h-time point. L: leak state; D = ADP-induced OXPHOS and U = uncoupled respiration. (b) Free OXPHOS capacity (max OXPHOS capacity mediated by complex I activity and corrected for leak respiration) as calculated by subtracting L from D, and normalized to protein concentration. (n = 13 per group) (c) Free ETS capacity (uncoupled respiration corrected for leak respiration) as calculated by subtracting L from U, and normalized to protein concentration (n = 13 per group). The data are presented as Mean ± SEM. The number above the bars in (b) and (c) shows the p value obtained by student t-test.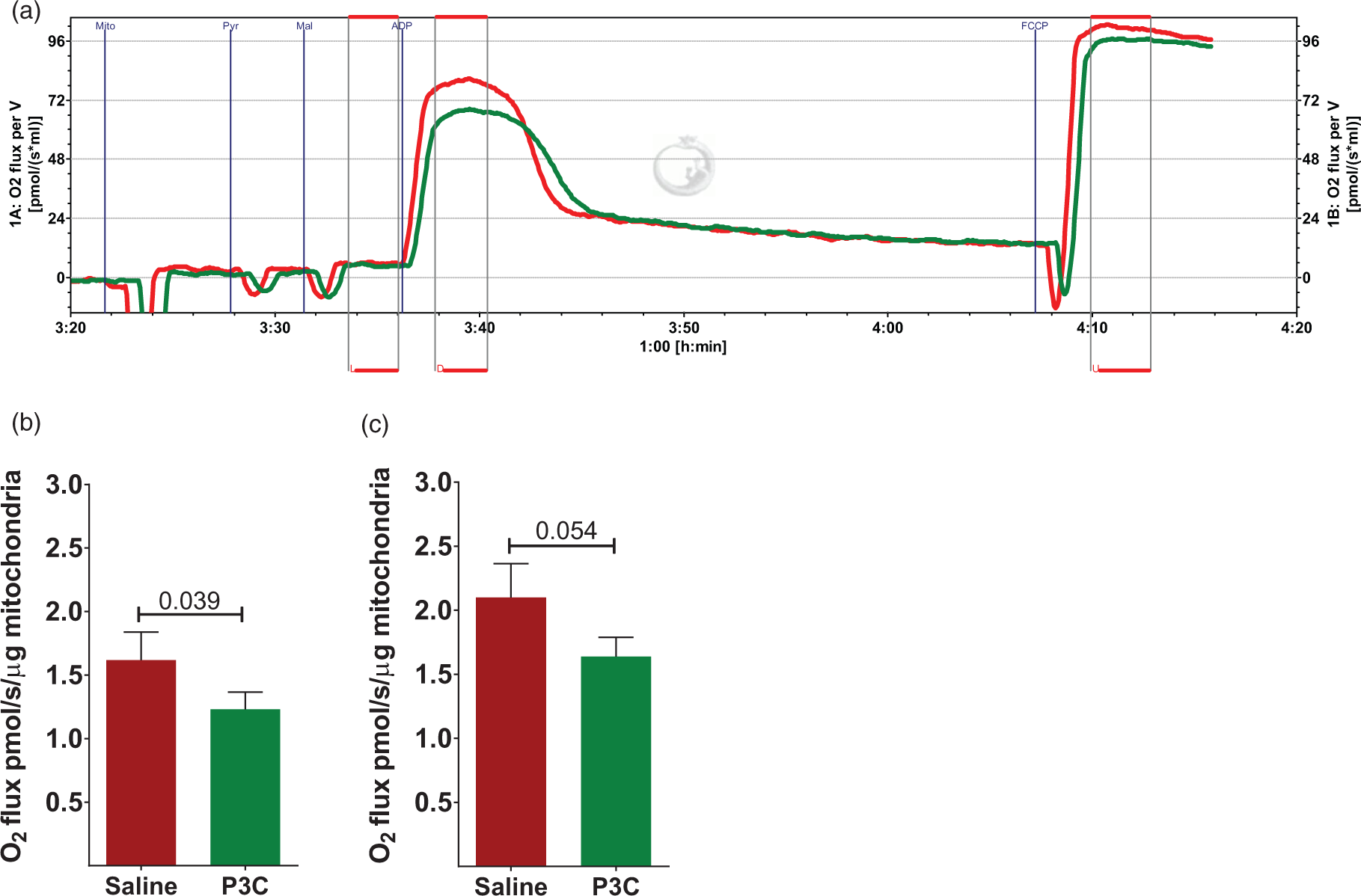
Discussion
Perinatal HI is a major cause of neurodevelopmental disorders such as cerebral palsy. 12 Infection and inflammation are associated with increased risk of brain injuries in both term and pre-term infants.4,5 Perinatal infections, such as choroioamnionitis and neonatal sepsis, are caused to a large extent by genital mycoplasmas and Gram-positive bacteria for which TLR2 plays a key role in initiating an inflammatory response.5,13 We have previously shown that repeated injections of P3C to neonatal mice impair brain development 14 and that HI-induced brain injury was ameliorated in TLR2−/− mice. 15 Here we report, for the first time, that TLR2 activation by a single dose of P3C exacerbates neonatal HI brain injury by augmenting neural tissue loss and demyelination. This is in line with previously reported inflammation-induced sensitization of neonatal brain by other TLR ligands.10,16 However, the results are in contrast with ischemia/reperfusion injury in adult mice where administration of P3C before the ischemic insult protects the brain.17,18 This discrepancy might be due to different immune response of the newborn to the inflammatory stimuli compared to the adult, 19 or different interval between P3C administration and cerebral ischemia in the adult studies. We have previously shown that the time interval between LPS and neonatal HI is important for neuronal loss. 20
The mechanisms by which infection/inflammation increase the vulnerability of the developing brain to injuries is unknown. In this study, we found that systemic activation of TLR2 suppressed ADP-induced complex I-mediated OXPHOS by mitochondria, a primary source of ATP production and thus cellular energy. 22 It should be noted that the mitochondrial respiration analysis performed in this study provides an overall estimation of the cellular respiratory state during TLR2-induced systemic inflammation and a more in-depth analysis of mitochondria function, especially the respiratory and enzymatic activity of different mitochondrial complexes is required to elucidate the full impact of TLR2-mediated inflammation on brain mitochondrial physiological functions. Evidence of the effect of local or systemic inflammation on brain mitochondrial function is limited, but a detrimental impact has been reported in an LPS-induced systemic inflammation model 23 and in a sepsis model. 24
How suppressed mitochondrial respiration might contribute to HI brain injury warrants further investigation. One potential mechanism could be that suppressed respiration has a cumulative effect on ATP depletion during the HI insult leading to initiation of necrosis pathways. 25 Another speculation is that neural cells with impaired respiration become more susceptible to glutamate excitotoxicity, an early event in neonatal HI, 25 as it has been shown that subcritical hypoxia makes hippocampal neurons vulnerable to glutamate toxicity. 26 In addition, glutamate itself might inhibit neural OXPHOS, 27 that in combination with TLR2-mediated detrimental effects on cellular respiration may result in an aggravated secondary energy failure. 25 Our results may also explain the detrimental effect of long-term TLR2 activation on brain development. 14
In summary, we show for the first time that systemic activation of TLR2 exacerbates neonatal HI brain injury. TLR2 is an important sensor of Gram-positive bacteria and the findings suggest a potential synergistic link between sub-clinical infections and other events, such as cerebral hypoxia-ischemia in the neonatal period. Our data suggest that inflammation-induced suppression of mitochondrial respiration may contribute to increased susceptibility to HI injury. Hence, mitochondrial dysfunction may be a novel therapeutic target in inflammation-related neonatal brain injury.
Funding
This work was supported by the Swedish Research Council (VR2012–2992 CM; VR 2015-02493 HH; EU; VR 529-2014-7551 HH), Government grant in Public Health Service at the Sahlgrenska University Hospital (ALFGBG-142881 CM; ALFGBG-426401 HH), the Leducq foundation (DSRR_P34404), Åhlén Foundation the Swedish Brain Foundation (FO2013–095, CM; 2015-0004, HH), Torsten Söderberg (M98/15, CM).
Footnotes
Acknowledgments
We would like to thank Anna-Lena Leverin for excellent technical assistance, and Mona Svedman at University of Gothenburg animal facility.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
AM, XW, HH, JE, and CM designed the experiments and analyzed the data. AM performed the surgical operations. PS, AM, and CJM did the immunohistochemistry and brain injury measurements. AM and SN performed the respirometry experiments. AM drafted the manuscript. All authors contributed to the writing of final version of the manuscript.