
Abstract
Nucleoside diphosphate kinase B (NDPK-B) is an enzyme required for nucleoside triphosphate homeostasis, which has been shown to interact with caveolin-1 (Cav-1). In endothelial cells (ECs), NDPK-B contributes to the regulation of angiogenesis and adherens junction (AJ) integrity. We therefore investigated whether an interaction of NDPK-B with Cav-1 in ECs is required for this regulation and the involvement of VEGF signaling herein. We report that simultaneous depletion of NDPK-B/Cav-1 in HUVECs synergistically impaired sprouting angiogenesis. NDPK-B depletion alone impaired caveolae formation, VEGF-induced phosphorylation of c-Src/Cav-1 but not of ERK1/2/AKT/eNOS. In vivo, Cav-1−/− mice showed impaired retinal vascularization at postnatal-day five, whereas NDPK-B−/− mice did not. Primary mouse brain ECs (MBMECs) from NDPK-B−/− mice showed no change in caveolae content and transendothelial-electrical resistance upon VEGF stimulation. Interestingly, NDPK-B−/− MBMECs displayed an accumulation of intracellular vesicles and increased Cav-1 levels. Dextran tracer analysis showed increased vascular permeability in the brain of NDPK-B−/− mice compared to wild type. In conclusion, our data indicate that NDPK-B is required for the correct localization of Cav-1 at the plasma membrane and the formation of caveolae. The genetic ablation of NDPK-B could partially be compensated by an increased Cav-1 content, which restored caveolae formation and some endothelial functions.
Introduction
The regulation of permeability and angiogenesis in the endothelium is essential for vascular homeostasis. Angiogenesis, the formation of new vessels from pre-existing ones, contributes to physiological vessel formation and also occurs as part of the pathology of numerous diseases such as proliferative diabetic retinopathy and cancer. 1 The vascular endothelial growth factor (VEGF) – VEGF receptor type 2 (VEGFR-2) signaling cascade has been extensively investigated and its importance for physiological as well as pathological angiogenesis has been verified. Therefore, interference with VEGF-VEGFR2 activity and its downstream signaling is an already used and further sought principal in anti-angiogenic therapy. 2 Recently, we were able to demonstrate that the enzyme nucleoside diphosphate kinase (NDPK)-B contributes to the regulation endothelial permeability and angiogenesis. 3 It is required for the VEGF-induced opening of endothelial adherens junctions (AJs) and sprouting vessel growth. NDPK-B belongs to a family of proteins with 10 putative isoforms. The members of group I, consisting of NDPK-A, -B, -C and -D, exhibit the classical NTP/NDP transphosphorylase activity and thus catalyze the transfer of a phosphate group from nucleoside triphosphate (NTP) to nucleoside diphosphate (NDP). These isoforms show a high degree of sequence homology (58–88%), while most of the members of group II are catalytically inactive and share only lower degrees of homology (25–45%). NDPK-A and -B are ubiquitously expressed products of the nme1 and nme2 gene, respectively, which are located on the same gene locus. Although these isoforms are highly homologous and are able to form heterohexamers, differential biological functions have been described. Whereas NDPK-A is confirmed to act as a metastasis suppressor in vitro and in vivo, NDPK-B has been identified as a specific protein histidine kinase for heterotrimeric G protein β subunits and at least two ion channels (see Hsu et al. 4 for recent review). Interestingly, NDPK-B was also found to be important for caveolae formation. 5 Caveolae are small cholesterol-enriched invaginations at the plasma membrane, which for example cluster and compartmentalize signaling components. 6 Two classes of scaffold proteins, caveolins and cavins, form caveolae. 7 The depletion of NDPK-B, but not of NDPK-A, was associated with a loss in caveolin-1 (Cav-1) and caveolin-3 (Cav-3) expression in zebrafish. 8 Likewise, the depletion of Cav-1 or Cav-3 reduced the expression of NDPK-B in zebrafish and the absence of NDPK-B in mouse embryonic fibroblasts obtained from NDPK A/B−/− mice was associated with a largely decreased number of caveolae and a reduced Cav-1 content at the plasma membrane. 5 Also in endothelial cells (ECs), a large variety of signaling molecules, including the VEGFR-2 localize to caveolae. 9 Although ECs express the Cav-2 isoform as well, only the Cav-1 variant was reported to be indispensable for caveolae formation and has been implicated in the regulation of AJ permeability 10 and the VEGF-induced angiogenic response. 11 Moreover, the importance of Cav-1 for endothelial permeability and pathological angiogenesis in vivo has been demonstrated in Cav-1−/− mice. 12 As both NDPK-B and Cav-1 appear to be indispensable for the regulation of endothelial permeability and angiogenesis, we investigated the interaction of NDPK-B with Cav-1 in ECs and their contribution to angiogenesis in well-characterized in vitro and in vivo models. We report here that NDPK-B and Cav-1 cooperate in angiogenic sprouting and caveolae formation in EC. The VEGFR-2 induced phosphorylation of Cav-1 by c-Src, which is required for the opening of AJs, 10 is largely suppressed upon NDPK-B depletion.
Materials and methods
Animal care and handling
Animal care and experiments were performed in accordance with the Association for Research in Vision and Ophthalmology statement, the ARRIVE guidelines and were approved by the local government (35-9185.81/G-203/10 and /G124/12 Regierungspräsidium Karlsruhe, Germany). Homozygous Cav-1−/− mice 13 and wild type (WT) littermates on a C57BL/6JOIaHsd background, as well as homozygous NDPK-B−/− mice 14 with its WT littermates on a C57BL/6 background were used in this study. Genotyping was performed as described previously. 14
Whole mount retinal immunofluorescence
To evaluate the vascular morphology, eyes were fixed in 4% formalin at room temperature for 2 h. Thereafter, the retinas were dissected and washed with phosphate-buffered saline (PBS). The retinas were permeabilized and blocked in a solution containing 1% bovine serum albumin (BSA) and 0.5% Triton-X-100. Subsequently, the retinas were incubated with lectin conjugated with fluorescein isothiocyanate (FITC) or tetramethylrhodamine (TRITC) (1:50, Sigma, Germany) to visualize retinal vessels. Central avascular zones and de novo vessel formation in the retina were quantified by using an analysis program (IX81, Olympus, Hamburg, Germany) after confocal microscopy (Leica, Germany). Cav-1 was stained with anti-Cav-1 (1:50, 610059, BD Transduction Laboratories).
Isolation and culture of mouse brain microvascular endothelial cells
The mice (six weeks old) were sacrificed using isoflurane followed by cervical dislocation. The brains were placed in falcon tubes containing buffer A (153 mM NaCl, 5.6 mM KCl, 1.7 mM CaCl2, 1.2 mM MgCl2, 15 mM HEPES, 10 g/l BSA, pH 7.4) on ice. The superficial blood vessels present on the brains were carefully removed along with the meninges. The tissue with respective genotypes was homogenized with 10 strokes and then centrifuged for 10 min at 400 g, 4℃. The pellet incubated in 0.75% collagenase II for 60 min at 37℃ on a shaker. After incubation, the homogenate was centrifuged at 400 g for 10 min at 20℃ and the pellet re-suspended in Buffer B, i.e. PBS(140 mM NaCl, 0.2 mM CaCl2, 0.2 mM MgCl2, 10 mM NaHPO4, 10 mM KH2PO4) containing 25% BSA. The homogenates were centrifuged at 2000 g for 30 min at 4℃. The pellets were incubated in Buffer A with 1 mg/ml collagenase/dispase and 1 µg/ml DNase I at 37℃ for 15 min. The cells were washed with MCDB131 medium (MCDB131 Life technologies, containing 0.05 mg/ml endothelial cell growth supplement (home-made from porcine brain), 2 mM penicillin/streptomycin, 2 mM L-glutamine, 0.01 mg/ml heparin, 0.01 g/l NaHCO3, 4 mg/ml puromycin) and re-suspended in MCDB-131 medium with 20% (by vol.) fetal calf serum (FCS). Cells were seeded on collagen I-coated culture flask and used at passage (p) 0 or 1.
Isolation and culture of human umbilical vein endothelial cells
Human umbilical vein endothelial cells (HUVECs) were extracted from the human umbilical vein by dispase digestion and suspended in 10% FCS (Promocell, C 37350) to stop the enzyme activity. Cell suspensions were seeded in endothelial cell growth complete medium (ECGM, Promocell) with 10% (by vol.) FCS as described before. 3 HUVECs were grown on gelatin-coated culture flasks. At passage (p) 0, cells were split after they exhibited a cobble stone morphology and reached confluence. Cells were maintained in culture until p3.
Transfection with siRNA
HUVECs were seeded and allowed to grow in ECGM containing 10% FCS at 37℃ overnight to 70% confluence in a 12-well dish. Thereafter, the cells were transfected using lipofectamine (Invitrogen) according to the manufacturer’s protocol. An amount 100 pmol of a scrambled siRNA (AACUGGUUGACUACAAGUCUU, Eurofins MWG operon), an NDPK-B specific-(AGGUAGUGUAAUCGCCUUG)/Cav-1 specific-(AAGCAUCAACUUGCAGAAAGA) siRNA was used. Four hours after transfection, the cells were supplemented with ECGM containing 10% FCS. Analyses were conducted at 48 and 96 h post transfection.
Sprouting assay
HUVECs were suspended in 5 ml ECGM containing 1 ml methocel and 20% FCS. In vitro sprouting angiogenesis assays were performed as described previously. 3 Briefly, spheroids generated for 24 h with 400 cells were embedded into a 3D collagen-based gel placed in a 48-well plate. They were stimulated without and with 50 ng/ml VEGF for 24 h, and the cumulative sprout length was measured.
Measurement of transendothelial-electrical resistance
Transendothelial-electrical resistance (TEER) of mouse brain microvascular endothelial cell (MBMEC) monolayers was exactly performed as described 15 using a cellZscope® device.
Immunohistochemistry
Cells were seeded on gelatin-coated glass cover slips and transfected with siRNA as described. Thereafter, the cells were grown overnight in ECGM, washed with PBS, treated without and with VEGF and fixed with 4% formaldehyde for 15 min at room temperature. The cells were then permeabilized with 0.1% Triton X-100 for 3 min and incubated with primary antibodies overnight at 37℃. After washing three times for 5 min with PBS, cells were incubated with the secondary antibody for 1 h at 37℃. The cells were washed again with PBS and mounted using mowiol (Calbiochem, Germany). Used primary antibodies: anti-Cav-1 (610059), anti-Claudin-5 (35-2500, Thermofisher Scientific) and anti-vascular endothelial (VE)-Cadherin (SC-6458, Santa Cruz Biotechnology). MBMECs were stained by polyclonal secondary antibodies swine anti-rabbit FITC (F0205, DAKO). Staining intensity was quantified using the Image J software. Pixel densities were measured by drawing a line across the cell, such that the cell membrane and cytoplasmic regions were marked.
Electron microscopy
HUVEC and MBMEC monolayers were fixed with 3% glutaraldehyde and 0.1% picric acid in 0.1 M sodium cacodylate. The respective cells were scraped from the culture dishes, pelleted and post-fixed in 1% OsO4. Samples were embedded in Epon (Serva) according to standard procedure. Ultrathin sections were analyzed using an electron microscope (Zeiss EM 10).
WT or NDBKB−/− mice (six weeks old, n = 4 WT, n = 3 KO) were anesthetized and transcardially perfused with PBS for 1 min as described earlier followed by 4 min with 4% PFA/2% glutaraldehyde in PBS. The isolated brains were post-fixed with the same fixative ON at 4℃. The tissue (cut into small pieces) was additionally post-fixed with 1% OsO4 for 2.5 h at RT and stained with 2% uranyl acetate solution at 4℃ ON prior to dehydration in graded acetone and embedded in Durcupan. The polymerization was performed at 60℃ for 72 h. For the quantification of vesicles, 20 cortical capillaries of comparable size (diameter ∼ 4 µm) were analyzed for each animal. Data were analyzed by Student’s unpaired t-test.
Immunoblotting
Cells were scraped off in RIPA buffer (50 mM Tris-HCl, pH7.4, 150 mM NaCl, 1 mM dithiothreitol, 1% Triton X-100, 1% sodium deoxycholate) and centrifuged for 15 min at 4℃. The supernatant was analyzed by standard SDS-PAGE. Proteins were electrically transferred onto nitrocellulose membranes, blocked with Rotiblock (Roth) and incubated with the indicated antibodies, followed by detection with an ECL detection reagent (Thermo Scientific). The following antibodies were used: anti-NDPK-A (C-20, Santa Cruz Biotechnology), anti-NDPK-B (MC-412, Kamiya), anti-β Actin (A2228, Sigma), anti-Cav-1 (sc-894, Santa Cruz Biotechnology), anti-c-Src (sc-18, Santa Cruz Biotechnology), anti-ERK 1/2 (sc-153, Santa Cruz Biotechnology), anti-AKT (9272, cell signalling), anti-eNOS (610296, BD Transduction Laboratories) and anti-Tubulin (T5168, Sigma).
Detection of phosphorylated proteins
To measure the phosphorylation of specific proteins, HUVECs were plated at 70% confluence and transfected with the respective siRNA. At 24 h, FCS was reduced to 2% (by vol.). At 48 h, HUVECs were stimulated with or without VEGF for 30 min. Thereafter, cells were washed two times with PBS and lysed using GST-FISH buffer (50 mM Tris-HCl, pH 7.4, 10% glycerol, 1% NP-40, 100 mM NaCl, 2 mM MgCl2). Cell lysates were then analyzed by Western Blot using antibodies directed against the phosphorylated form of the protein under study: anti-P-Cav-1 (611338, BD Transduction Laboratories), anti-P-c-Src (44660G, Invitrogen), anti-P-ERK 1/2 (sc-7383, Santa Cruz Biotechnology), anti P-AKT (Ser473, cell signalling) and anti-P-eNOS (9572, cell signaling).
In vivo permeability assay
Mice (six weeks old) were anaesthetized by intraperitoneal injection of 200–250 µl of prepared anesthetic solution per mouse (4.5 ml NaCl, 1 ml Ketavet®, 0.32 ml Rompun®). The tracer, namely, 3 kDa tetramethylrhodamine (TMR/D3308, Life Technologies) diluted in PBS (1 mM), was injected intravenously (110 µl per mouse). Around 100 µl of the blood was collected for normalization to circulating tracer and the mice were then perfused with 1–2 ml of PBS. A sham mouse was perfused without tracer injection and used for background subtraction. Once the mice were sacrificed, the brain was isolated, and the hemispheres were weighed and homogenized in 300 µl PBS. The homogenate was centrifuged at 10,000 g for 15 min at 4℃. The supernatant was collected and 100 µl was transferred into a 96-well plate. The fluorescence was measured using a spectrophotometer with an excitation/emission wavelength of 550/580 nm. After background values obtained from the sham mouse were subtracted, the fluorescence intensity measured in the brain homogenate was normalized to the tissue weight (mg) and serum fluorescence. The results are presented in relative fluorescence units (RFU) per mg tissue/RFU serum with values obtained from WT mice set as 100%.
Statistical analysis
Data are presented as means ± SEM, unless otherwise stated. The data were analyzed using the GraphPad Prism software (GraphPad Software, La Jolla). The unpaired Student’s t-test without and with Welch’s correction or analysis of variance with the indicated post-tests were used for statistical analysis. A p value < 0.05 was considered statistically significant.
Results
Both, NDPK-B and Cav-1 are required for VEGF-induced sprouting angiogenesis in ECs
We previously reported that the siRNA-induced depletion of NDPK-B inhibits VEGF-sprouting angiogenesis in ECs.
3
In accordance with data reported previously,
16
the suppression of Cav-1 expression by transfection with 100 pmol of a specific siRNA completely abolished VEGF-induced sprouting angiogenesis (not shown). In order to get a first hint whether NDPK-B and Cav-1 cooperatively regulate sprouting angiogenesis, we performed single knockdown experiments with lower siRNA amounts (50 pmol) and subsequently, performed also a double knockdown in HUVEC (Figure 1), resulting in 40–50% reduction of either NDPK-B or Cav-1 expression (Figure 1(c)). The knockdown of NDPK-B had no influence on Cav-1 expression and vice versa, Cav-1 depletion did not alter NDPK-B expression. Nevertheless, even at 40–50% knockdown efficiency, the single depletion of NDPK-B or Cav-1 significantly impaired VEGF-induced endothelial sprouting. Interestingly, the simultaneous depletion of both proteins induced a further significant impairment (Figure 1(b)), resulting in the strongest inhibition. These results thus indicate that NDPK-B and Cav-1 act additively or may even act synergistically to allow for VEGF-induced sprouting of ECs.
NDPK-B and Cav-1 depletion synergistically impair VEGF-induced endothelial sprouting angiogenesis. HUVECs were transfected with control (Ctrl) siRNA, NDPK-B siRNA, Cav-1 siRNA and NDPK-B plus Cav-1 siRNA. At 72 h, the cells were transferred into the respective gels and spheroids were cultured with or without 50 ng/ml VEGF for 24 h (a). Representative images are shown (scale bar 60 µm). (b) The cumulative sprout length was measured for each condition. Data shown were analyzed by one way ANOVA with Bonferroni’s multiple comparisons (*p < 0.05, n = 3). (c) Representative immunoblots (lower panel) and quantifications of NDPK-B and Cav-1 content (upper panels) are shown. β-Actin served as loading control.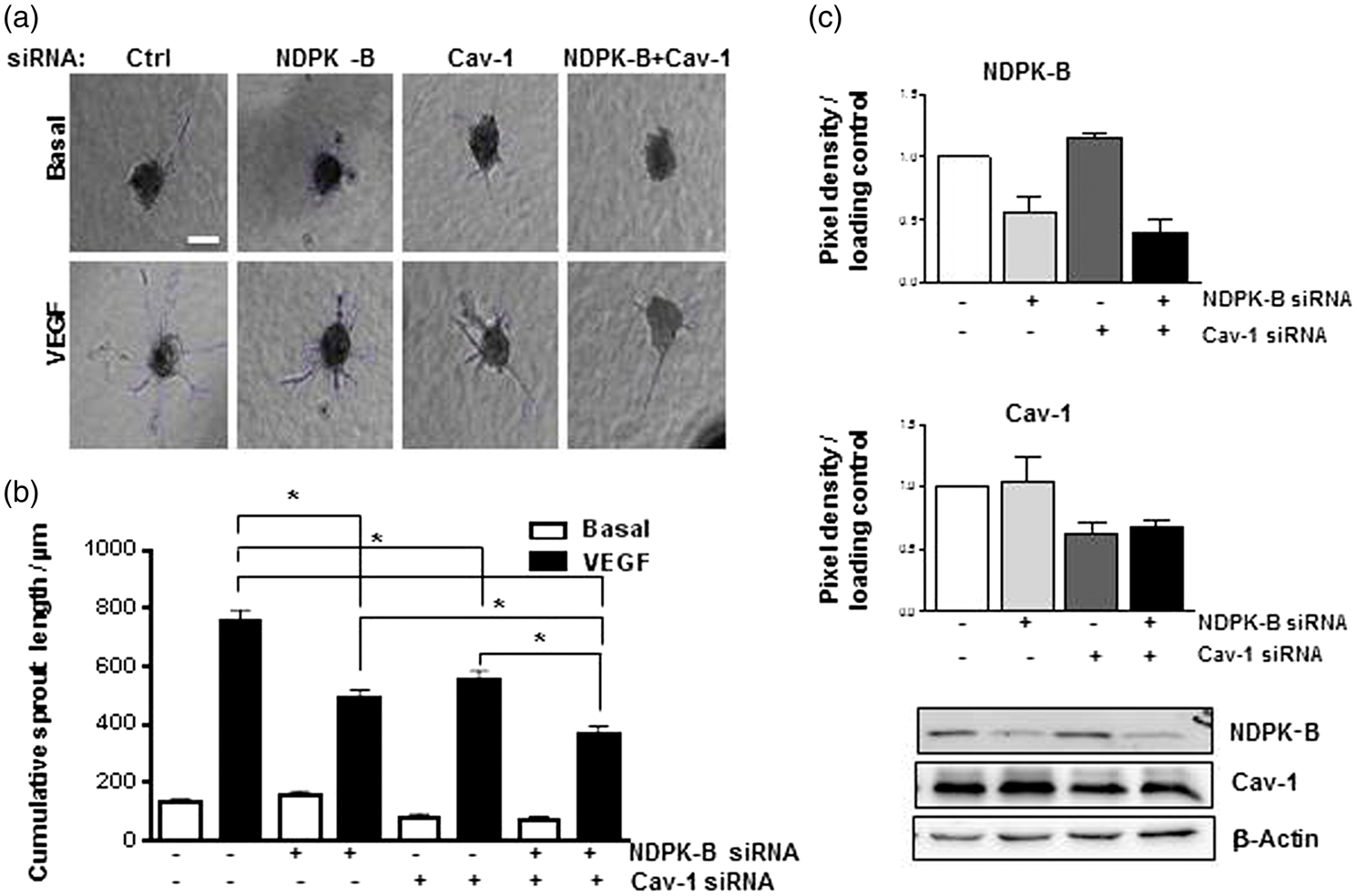
NDPK-B is required for the correct localization of Cav-1 and sustains caveolae formation at the plasma membrane
Caveolae are micro-lipid invaginations, which are abundantly present in ECs. They harbor for example the VEGFR-26 and thus contribute to the regulation of permeability and angiogenesis.9,11 As data obtained from embryonic fibroblasts of NDPK-A/-B-deficient mice suggested that especially NDPK-B is involved in caveolae formation,
5
we studied the effect of NDPK-B depletion on the caveolae content in HUVECs. Electron microscopic analysis of NDPK-B siRNA- and control-treated HUVECs revealed a significant reduction (∼60%) of the number of caveolae at the membrane in the NDPK-B-depleted cells when compared to the scrambled control (Figure 2(a) and (b)). The formation of caveolae requires the stepwise assembly of Cav-1 homo-oligomer complexes at the endoplasmic reticulum (ER) and a vesicular transport through the Golgi apparatus to the plasma membrane.
17
Therefore, we analyzed Cav-1 localization by confocal microscopy. In control siRNA-treated cells, Cav-1 showed a prominent presence at the plasma membrane but was also detected in perinuclear and vesicular structures (Figure 2(d)). Using the Image J software, we quantified the pixel density distribution of Cav-1 (Figure 2(e)) as previously described.
3
The loss of NDPK-B in HUVECs (Figure 2(c)) caused a significant decrease of Cav-1 at the plasma membrane (Figure 2(d) and (e)) and led to a more scattered localization within the cell. The data therefore indicate that NDPK-B is required for the correct localization of Cav-1 at the plasma membrane, which in turn is indispensable for caveolae formation in ECs.
NDPK-B depletion decreased caveolae and Cav-1 content at the endothelial cell membrane. (a) Respective electron microscopy images of control and NDPK-B siRNA treated HUVECs at low (left panels) and high magnifications (right panels). (b) Quantitative analysis of the caveolae content at the cell membrane. The number of flask-shaped invaginations of 50–100 nm in size on the cell membrane were counted and to the plasma membrane length in µm. Data were statistically analyzed by unpaired student’s t-test with Welch’s correction (*p < 0.05, n = 35-45, scale bar 2 µm). (c) Representative immunoblots showing the efficacy of the NDPK-B depletion. β-Actin served again as loading control. (d) Representative confocal images of HUVECs treated with control (Ctrl) and NDPK-B siRNA and stained with anti-Cav-1 antibody. The lower panels show the same images at higher magnification. (e) Representative plot profiles of line scan analysis using Image J. The representative plot profile shows a single cell pixel density distribution (Gray value), where “M” denotes the membrane peak and “P.N” the peri-nuclear peaks. (f) Quantitation of pixel density measured from the highest peak in the first 15% length of the line scan. Data were statistically analyzed by unpaired student’s t-test (*p < 0.05, n = 69, scale bar 2.5 µm).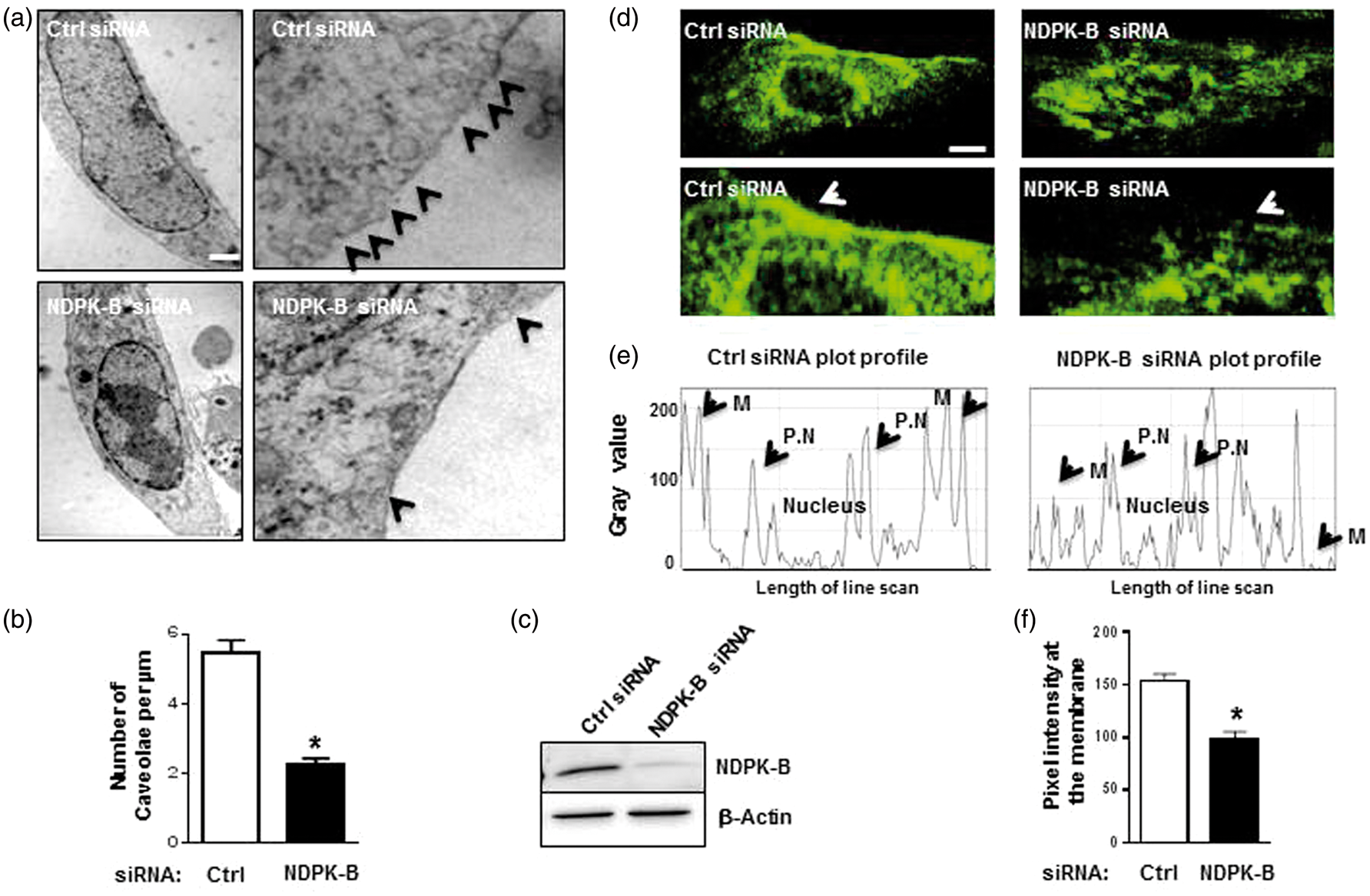
NDPK-B depletion does not alter VEGF-induced phosphorylation of ERK 1/2, AKT, and eNOS in HUVEC
VEGF binding to VEGFR-2 induces the autophosphorylation of the receptor, which triggers VEGFR-2 signaling in EC.
18
Among others extracellular-signal regulated kinase (ERK), protein kinase B (AKT) and endothelial nitric oxide synthase (eNOS) are activated by VEGFR-2 stimulation which have been attributed to differential cellular processes, namely endothelial proliferation,
19
survival
20
and permeability regulation.
21
Therefore, we monitored the activation of these pathways by analyzing the phosphorylation of these proteins with and without depletion of NDPK-B. As shown in Figure 3, we did not detect any alterations in the VEGF-induced phosphorylation of ERK 1/2, AKT or eNOS after NDPK-B depletion. Thus, these results indicate that NDPK-B does not participate in the regulation of these pathways by VEGFR-2 in ECs.
The VEGF-induced phosphorylation of ERK 1/2, AKT and eNOS is not altered in NDPK-B-depleted endothelial cells. Representative immunoblots showing the phosphorylation and total protein content of ERK 1/2 (a), AKT (b) and eNOS (c) in control-siRNA and NDPK-B-siRNA-treated HUVEC lysates obtained with or without VEGF stimulation (50 ng/ml) for 10 min at 96 h post transfection. (d) The efficacy of the NDPK-B depletion in HUVECs at 96 h post transfection is shown.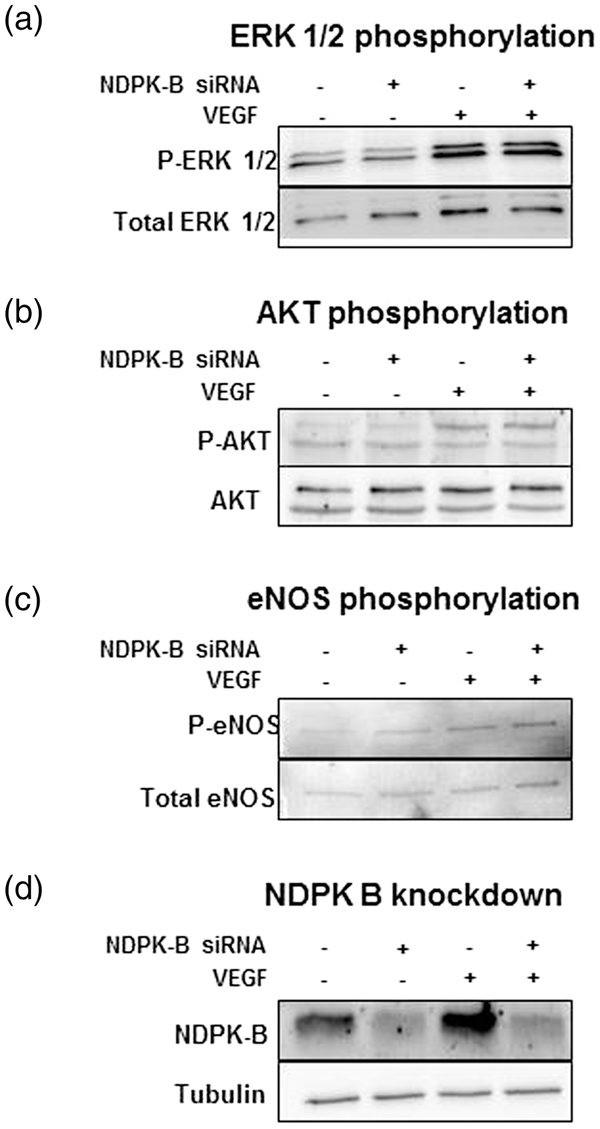
NDPK-B is required for VEGF-induced, c-Src-mediated Cav-1 phosphorylation in ECs
In our previous work, we demonstrated that NDPK-B is required for the VEGF-induced opening of AJ.
3
Recent evidence has been presented that Cav-1 is interacting with the AJ-associated proteins α-, β- and γ-catenin. Within this complex, the transient phosphorylation of Cav-1 on Tyr14 by c-Src is critical for the thrombin-induced breakdown of the paracellular barrier function in HUVEC.
10
We therefore studied whether VEGF induces an analogous phosphorylation of Cav-1 and whether such a phosphorylation is dependent on NDPK-B. As judged by its autophosphorylation, VEGF treatment induced the well-known activation of c-Src in control-transfected cells (Figure 4(a)). As shown in Figure 4(b), a significant VEGF-induced phosphorylation of Cav-1 on Tyr14 occurred concomitantly. Interestingly, both the VEGF-induced c-Src as well as the Cav-1 phosphorylation was abolished by NDPK-B depletion (Figure 4(a) and (b)). To verify that the activation of c-Src is caveolae dependent, we treated HUVEC with methyl-β-cyclo-dextrin, which extracts cholesterol at the cell surface, a reversible process to deplete caveolae at the membrane.
22
In comparison to VEGF-induced c-Src and Cav-1 phosphorylation in control-treated cells, the depletion of caveolae abolished both c-Src (Figure 4(d)) and Cav-1 (Figure 4(e)) phosphorylation. To further prove that the VEGF-induced Cav-1 phosphorylation is dependent on c-Src, we treated HUVEC with and without PP2, a specific inhibitor of the Src kinase family and studied its effect on Cav-1 phosphorylation. As expected, PP2 largely reduced the VEGF-induced c-Src autophosphorylation in HUVECs (Figure 4(f)). Moreover, it completely suppressed the VEGF-induced phosphorylation of Cav-1 as well (Figure 4(g)). Taken together, these data indicate that the VEGF-induced activation of c-Src is caveolae dependent and requires NDPK-B. Cav-1 is a downstream target of the VEGF-induced c-Src activation, which has already been linked to the VEGF-induced opening of AJ.
23
NDPK-B and caveloae are required for VEGF-induced phosphorylation of c-Src and Cav-1 in endothelial cells. HUVECs treated with control siRNA and NDPK-B siRNA were stimulated with or without VEGF (50 ng/ml) for 30 min at 96 h post transfection (a to c). HUVECs were treated with and without MβCD (5 mM, d and e) or PP2 (10 µM, f and g) and then stimulated with or without 50 ng/ml VEGF for 30 min. Phosphorylated c-Src and Cav-1 were detected in the cell lysate using phospho-Src- and phospho-Cav-1-specific antibodies. The phosphorylation was normalized to the respective protein content with levels in control siRNA treated cells set as 1. (a, d, f) Representative immunoblot and quantification of phosphorylated c-Src (P-c-Src) in HUVECs lysates (*p < 0.05 vs. Ctrl-siRNA, n = 3–6). (b, e, g) Representative immunoblot and quantification of P-Cav-1 in HUVECs lysates (*p < 0.05 vs. Ctrl-siRNA, n = 3–6). (c) Representative immunoblot and quantification of the NDPK-B depletion in HUVECs. γ-tubulin served as loading control (*p < 0.05 vs. Ctrl-siRNA, n = 3). Statistical analyses were performed by one way ANOVA with Bonferroni’s multiple comparisons.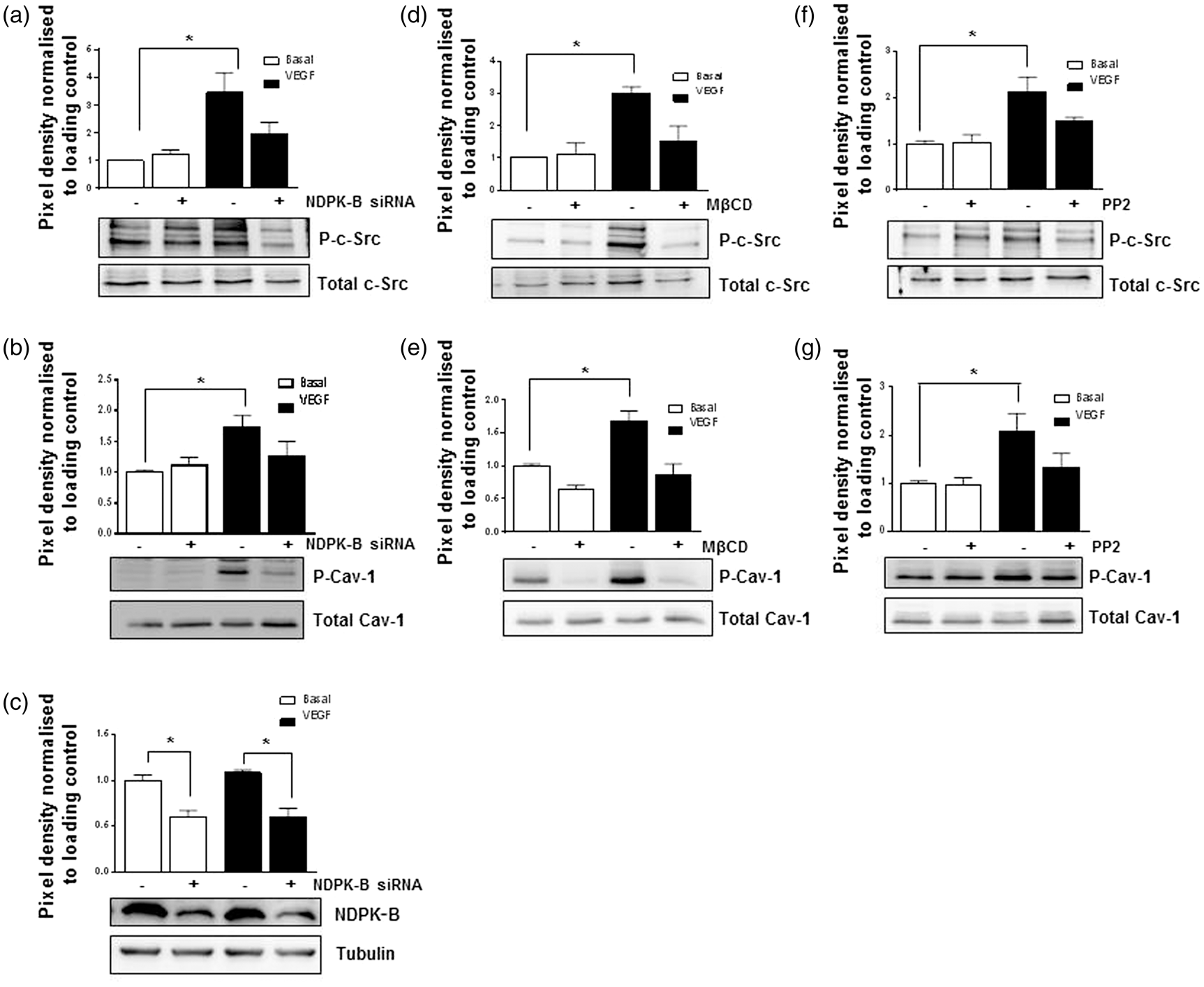
Cav-1-deficient mice show impaired retinal vascularization during physiological angiogenesis
We recently reported that NDPK-B-depleted zebrafish embryos show a severely impaired vessel angiogenesis.
3
As judged by the VEGF-driven physiological angiogenesis in the retina of newborn pups, the vascular development in NDPK-B deficient mice appeared to be unaffected. Nevertheless, when these NDPK-B−/− mice were subjected to oxygen-induced retinopathy (OIR) or hind limb ischemia, they showed a significantly decreased pathological angiogenesis.
3
Notably, Cav-1−/− mice suffer from severe defects in endothelial permeability and angiogenesis.
12
We therefore compared the development of the retinal vasculature of newborn Cav-1−/− mice to WTs and NDPK-B−/− mice. As shown in Figure 5, at postnatal day 5 (p5), the development of the retinal vasculature in NDPK-B−/− mice was similar to WT controls, whereas the vascular development in Cav-1−/− mice was significantly impaired reaching only 66% of the vascular network detected in WT controls. We therefore stained retinal whole mounts for vascular Cav-1 expression (Figure 5(e)). Interestingly, the amount of Cav-1 in NDPK-B-deficient capillaries was apparently higher than in the WT controls. Cav-1−/− mice did not show any staining in the retina confirming the specificity of the used approach.
Cav-1-, but not NDPK-B-deficient mice exhibit impaired postnatal retinal vascularization. Representative fluorescence images of WT (a, c, e), NDPK-B-deficient (a, e) and Cav-1-deficient (c, e) mouse retinas at postnatal day 5 (p5): (b, d) Quantification of the vascularized area. The total retinal surface area was set to 100%. Data were statistically analyzed by unpaired student’s t-test (*p < 0.05 vs. WT, n = 3–6, scale bar 250 µm). (e) Cav-1 expression was assessed by immunostaining with anti-Cav-1 and a TRITC-labelled secondary antibody (red). Retinal vessels were visualized with FITC-conjugated lectin (green).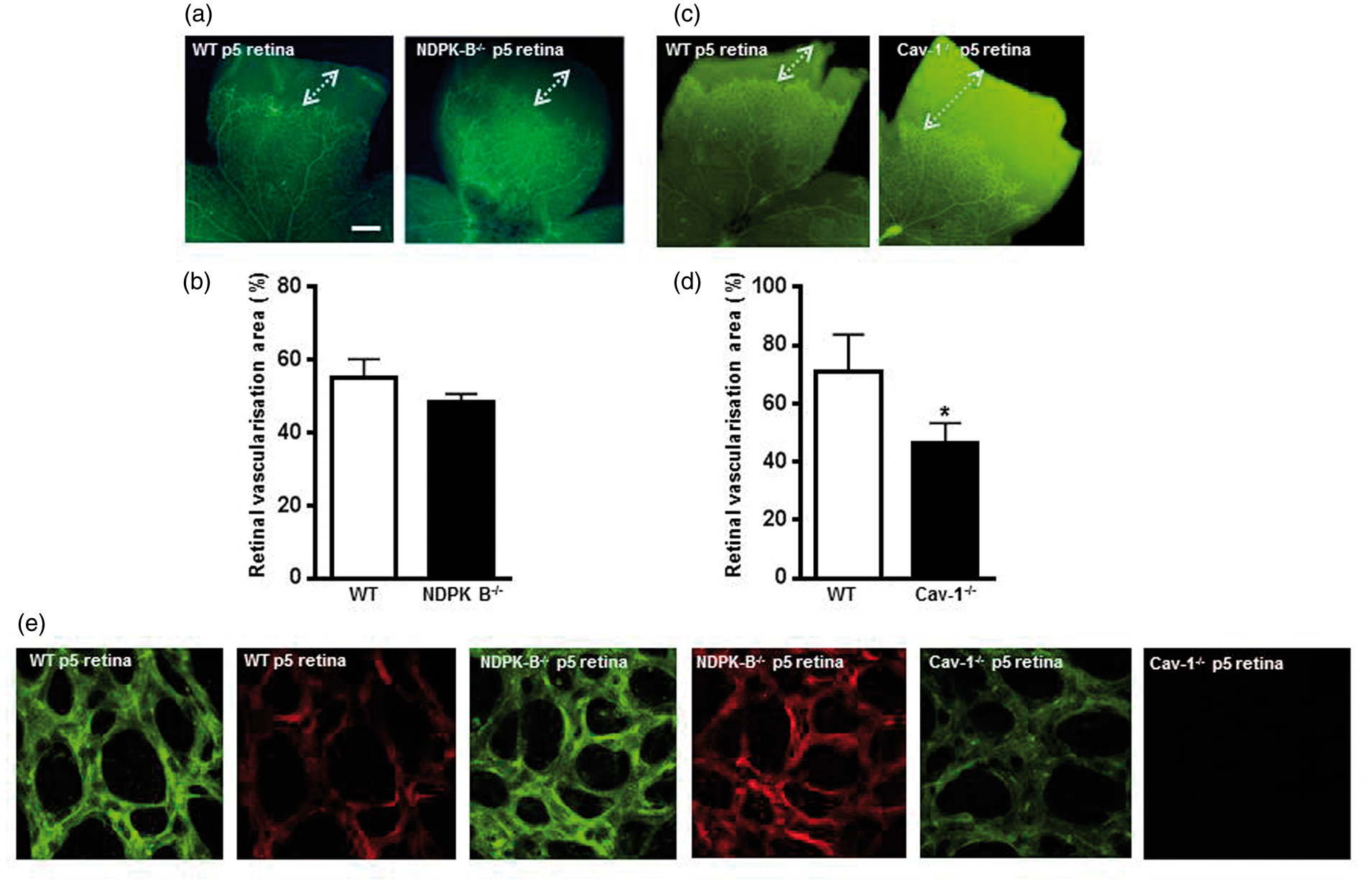
NDPK-B-deficient mice exhibit an increased intracellular vesicle and Cav-1 protein content in brain microvascular ECs
The differential Cav-1 expression between NDPK-B−/− and Cav-1−/− mice in the retina suggests that ECs of NDPK-B−/− mice might be able to compensate for the loss of NDPK-B and are able to partially restore their caveolae function. To test this hypothesis, we isolated and cultured mouse MBMECs from WT and NDPK-B−/− mice, which in vivo make up the blood–brain barrier (BBB) and are equipped with elaborate tight junctions (TJs). Moreover, brain ECs are characterized by a limited number of caveolae,
24
which are required for transcellular endothelial permeability via vesicle-mediated transcytosis.
25
In line with the reported data, MBMECs exhibited lower caveolae content when compared to other ECs, e.g. HUVECs (see Figure 2). Interestingly, we did not observe any significant differences in the caveolae content between MBMECs isolated from WT and NDPK-B−/− mice (Figure 6(a) and (b)). Nevertheless, upon a closer inspection of the MBMECs from NDPK-B−/− mice, we detected a significant 2.3-fold increase in intracellular vesicles, which might represent Cav-1 transporting vesicles (Figure 6(c) and (d)). In line with this interpretation, Western blot analysis revealed a significant 1.7-fold increase in total Cav-1 content in MBMECs from NDPK-B−/− mice when compared to WT MBMECs (Figure 6(e)). To verify that this upregulation of intracellular vesicles and the restoration of caveolae coverage is not an artefact of MBMEC isolation and culture, we analyzed the brain microvasculature of WT and NDPK-B−/− mice by electron microscopy. Indeed, a ∼2-fold increase in vesicular structures in the endothelium of NDPK-B−/− mice was detected (Figure 6 (f) and (g)).
Enhanced vascular permeability and endothelial caveolae content in the brain of NDPK-B deficient mice. Mouse brain microvascular endothelial cells. (a, c) Electron microscopy images of representative WT- and NDPK-B-deficient MBMECs isolated from 6–8 weeks old mice. (a) White arrows indicate caveolae at the membrane. (b) Quantitative analysis of the caveolae content at the cell membrane normalized to the length in µm. (n = 20–30, scale bar 50 nm). (c) White arrows indicate intracellular vesicles. (d) Quantitative analysis of the intracellular vesicle content normalized to the distance in µm. The vesicles were counted up to a depth of 250 nm from the cell surface along the cell length. Data were statistically analyzed by unpaired student’s t-test with Welch’s correction (*p < 0.05 against WT, n = 25–30, scale bar 50 nm). (e) Representative immunoblot and quantification of Cav-1 content of three independent MBMEC isolations from WT- or NDPK-B-deficient mice (four animals in each preparation). β-actin was used as loading control and for normalization. Data were statistically analyzed by unpaired student’s t-test (*p < 0.05 against WT). (f) Electron microscopy images of representative brain section of WT and NDPK-B−/− mice. White arrows indicate intracellular vesicles and caveolae in microvascular ECs. (g) Quantitative analysis of the intracellular vesicle content. Data were statistically analyzed by unpaired student’s t-test with Welch’s correction (**p = 0.0049 against WT, n = 3–4, scale bar 200 nm). (h) TEER (%) was measured with MBMECs isolated from WT and NDPK-B−/− mice. About 40 h post seeding, a confluent monolayer was formed (upper panels) and the TEER value reached a plateau in WT and NDPK-B-deficient MBMECs, and 72 h post seeding, the medium was changed and replaced by either control or VEGF-containing medium. Whereas the control monolayers remained tight, VEGF-induced junction disassembly decreased TEER similarly in WT and NDPK-B-deficient MBMECs. (i) The integrity of the brain vasculature in eight weeks old WT and NDPK-B-deficient mice was studied by an in vivo permeability assay. Mice were injected with TMR-3kDa dextran and its fluorescence in hemi-brain lysates per mg tissue of WT and NDPK-B−/− mouse brain was quantified after PBS perfusion, the mean value obtained from WT mice was set to 100%. Data were statistically analyzed by unpaired student’s t-test with Welch’s correction (*p < 0.05 against WT, n = 11).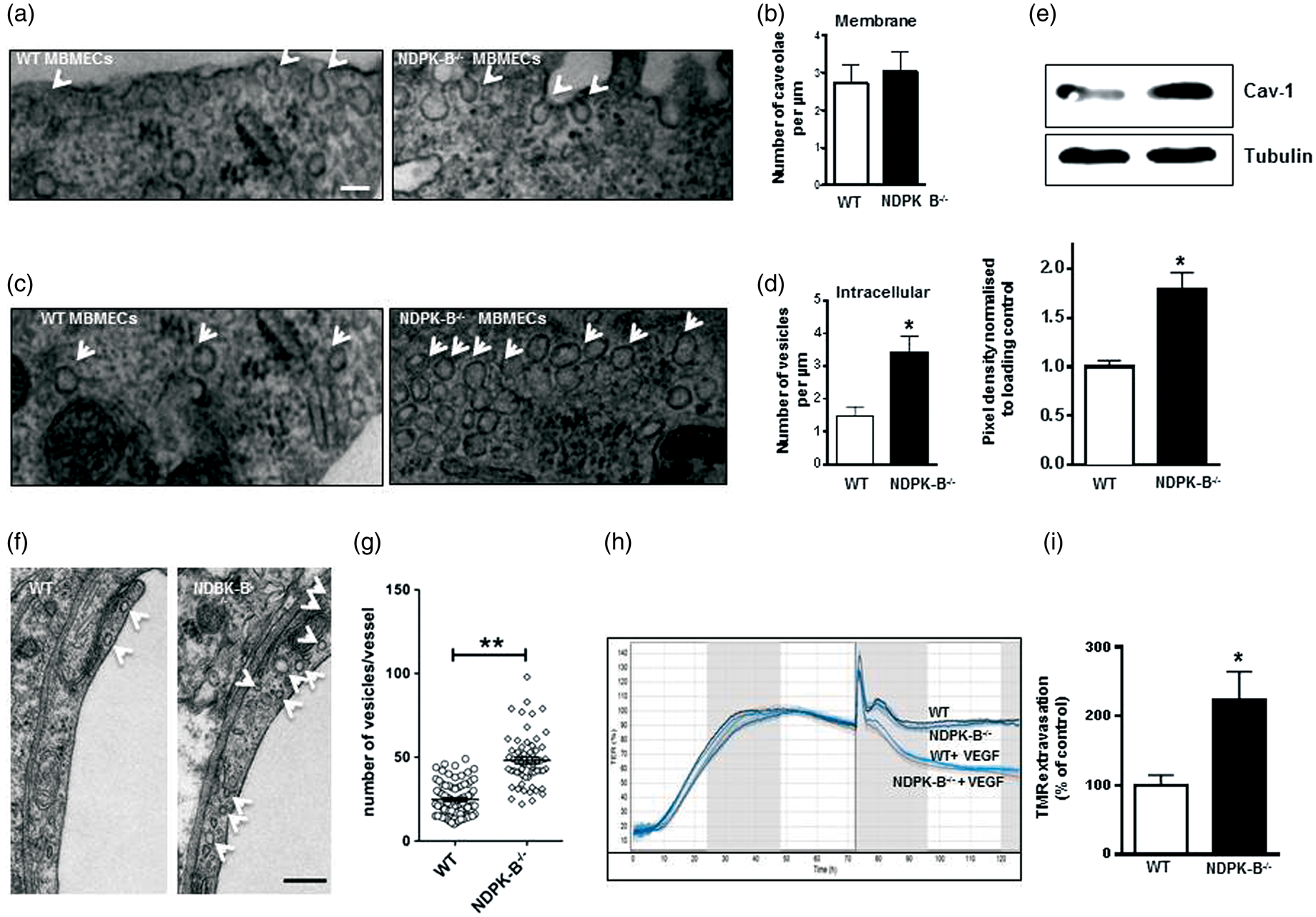
Using fluorescence microscopy, we further analyzed the localization of claudin-5, VE-cadherin and Cav-1 as markers for TJs, AJ and caveolae, respectively (Supplemental Figure 1). Whereas no alteration of claudin-5 and VE-cadherin localization was evident between in NDPK-B-deficient MBMECs, differences in the staining for Cav-1 were detected. Although similar amounts of Cav-1 seem to be present at the plasma membrane, an increase in the intracellular Cav-1 content in less defined structures was visible in NDPK-B−/− MBMECs. These findings are in line with the increase in the intracellular vesicle and Cav-1 content in NDPK-B-deficient MBMECs and suggest indeed an up-regulation of Cav-1-containing vesicles to compensate for the loss of NDPK-B in ECs.
NDPK-B-deficient mice suffer from an increased vascular permeability in the brain
To functionally assess the ability of MBMECs to form a tight monolayer, we performed TEER measurements. 15 As shown in Figure 6(h), a tight monolayer was formed by the MBMECs about 48 h after seeding. There was no difference between MBMECs obtained from WT or NDPK-B−/− mice. In contrast to monolayers of NDPK-B-depleted HUVECs, which exhibit a severely disturbed permeability and do not respond to stimulation with VEGF, 3 NDPK-B-deficient MBMEC monolayers responded normally to VEGF stimulation and showed the typical decrease in TEER.
As TEER only assesses paracellular permeabilty by ion fluxes but not transcellular permeabilty, we investigated the effect of NDPK-B-deficiency in an in vivo permeability assay. Mice were perfused with a fluorescent tracer (TMR-3kD dextran) and extravasation into the brain was quantitated in eight weeks old WT and NDPK-B−/− mice. Interestingly, NDPK-B−/− mice showed a significant 2.2-fold increase in relative fluorescence in the brain tissue when compared to WT controls (Figure 6(i)) whereas no difference was observed in kidneys used as a control for leaky endothelium (not shown). These results suggest an alteration of BBB function in NDPK-B−/− mice.
Discussion
The activation of ECs by angiogenic factors is pivotal for the formation of new vessels from pre-existing ones. Interestingly, 95% of the surface vesicles on the EC surface are made up of caveolae in which key signaling molecules like VEGFR-2 are localized. 22 Within the caveolae, Cav-1 acts as a scaffolding protein with both its N- and C-termini localized in the cytoplasm. By the caveolin scaffold domain, Cav-1 is able to interact with other proteins like eNOS and VEGFR-2. 26 Thus, caveolae have been implicated in a number of central processes in ECs like the regulation permeability and angiogenesis (see Sowa 27 for review). In line with previously published data obtained from in vitro and in vivo models,11,28 we observed a hampered postnatal retinal vascularization in Cav-1−/− mice, which depends on angiogenesis. Previous work from our laboratory indicated that NDPK-B is closely associated with Cav-1 and caveolae formation, as depletion of NDPK-B reduced Cav-1 content in cellular models as well as in the zebrafish.5,8 Herein, we obtained several lines of evidence that the previously reported impairment of VEGF-induced angiogenesis by NDPK-B depletion 3 is likely due to an impaired caveolae formation in ECs. Firstly, the amount of caveolae was drastically reduced in NDPK-B-depleted HUVECs. Secondly, Cav-1 did not reach the plasma membrane in sufficient amounts and thus, was not correctly localized in NDPK-B-depleted HUVECs. In line with this interpretation, NDPK-B and Cav-1 depletion additively or even synergistically inhibited VEGF-induced endothelial sprouting in the spheroid model. Therefore, the question arises how NDPK-B can actually contribute to caveolae formation. Noteworthy, NDPK-B was identified as a component required for the assembly of the COP II machinery in the ER. 29 Moreover, it was reported that positively charged residues on NDPK-B scaffold the ER membrane and thereby support ER functions. 30 Interestingly, after Cav-1 synthesis, Cav-1 monomers oligomerize to a complex within the ER.17,31 Eventually, Cav-2 is added to the complex, forming hetero-oligomers before exiting the ER. In order for Cav-1 oligomers to be transported to the membrane, they have to exit the ER in vesicles and enter the Golgi apparatus from which they are then transported in cholesterol enriched vesicles to the plasma membrane. Therefore, NDPK-B is possibly involved in this transport process during COP II assembly and ER exit. In line with this hypothesis, re-expression of NDPK-B in NDPK A/B-deficient cells rescued the transport of Cav-1 to the plasma membrane. 5 It is also noteworthy that the localization of Cav-1 in NDPK-B-depleted cells resembles the subcellular distribution of a defective caveolin mutant, which accumulates in ER-derived lipid bodies. 32
In our previous work, we further showed that the loss of NDPK-B in HUVECs increased the monolayer permeability and abolished the VEGF-induced opening of AJ.
3
Apart of being indispensable for caveolae formation, Cav-1 has been reported to be required for AJ assembly.
23
In addition, Kronstein et al.
10
showed that the phosphorylation of Cav-1 at Tyr14 is required for the thrombin-induced opening of AJ. Several reports show that Cav-1 has a Src-binding domain in its N-terminus, which enables phosphorylation on its Tyr14 residue by this non-receptor tyrosine kinase.33,34 As the VEGF-induced activation of c-Src has been shown to initiate a signal transduction cascade, leading to the phosphorylation of VE-cadherin and AJ disassembly, we hypothesized that VEGF, like thrombin, is able to initiate a c-Src-mediated Cav-1 phosphorylation, again contributing to AJ disassembly. We obtained several lines of evidence supporting this hypothesis. Firstly, stimulation of HUVECs with VEGF induced a concomitant autophosphorylation of c-Src and a phosphorylation of Cav-1 at Tyr14. Secondly, both phosphorylations are suppressed by the Src inhibitor PP2. Thirdly, disruption of caveolae by cholesterol or NDPK-B depletion abolished VEGF-induced c-Src and Cav-1 phosphorylation. Most interestingly, three other major signaling pathways initiated by VEGFR2 stimulation, the VEGF-induced activation of Erk1/2,
19
Akt
20
and eNOS
21
were not impaired in NDPK-B-depleted HUVECs. These data indicate that the localization of the activated VEGFR2 in caveolae is specifically required for the VEGF-induced opening of AJ and that the signaling cascade mediating this specific response
23
is therefore sensitive to NDPK-B depletion (see Figure 7). Most likely, all effects of NDPK-B depletion on VEGF-induced endothelial responses require the correct activation of this particular pathway.
Scheme of the pathway mediating the VEGF-induced disassembly of adherence junctions and its regulation by NDPK-B. As reported by Gavard and Gutkind,
23
the activation of the VEGFR-2 in ECs induces c-Src activation, which triggers a signaling cascade finally leading to the phosphorylation of VE-cadherin, its internalization and thus opening of AJs. Our data show that the phosphorylation of Cav-1 by c-Src in caveolae is essential for the functionality of this pathway. From our findings, NDPK-B ensures a correct Cav-1 transport to the plasma membrane, caveolae formation and VEGF-induced Cav-1 phosphorylation.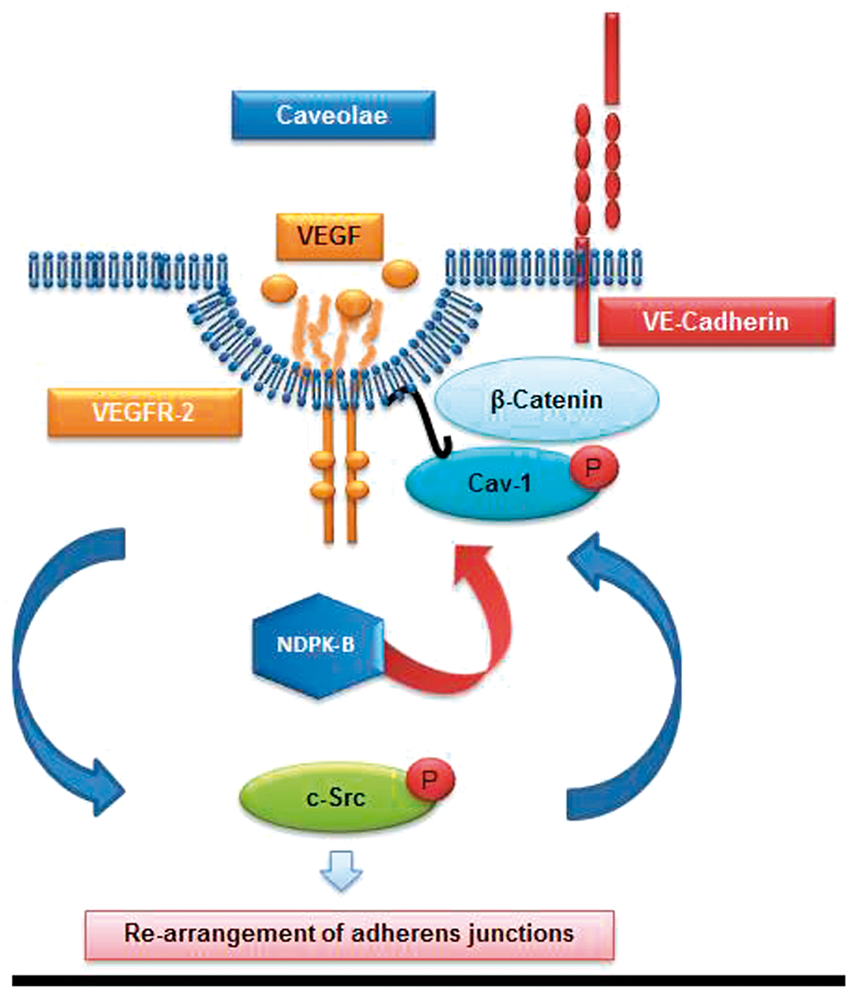
Interestingly, Cav-1−/− and NDPK-B−/− mice differ regarding the phenotype of the postnatal vascular development in the retina. Whereas the physiological angiogenesis in the retina of Cav-1−/− mice is attenuated, the vascular development of NDPK-B−/− mice appeared to be normal. At a first glance, these data argue against the hypothesis that NDPK-B is required for caveolae formation as described above. There is however, a similar discrepancy between the phenotype of NDPK-B−/− mice and the acute knockdown of NDPK-B in the zebrafish, using antisense morpholino oligonucleotides. 3 Like in the cellular models of acute NDPK-B depletion, the knockdown of NDPK-B severely impaired the angiogenesis-dependent physiological vessel formation in zebrafish embryos. As recently reported, there is however, a profound difference between the acute knockdown of a gene by morpholino-mediated knockdown and a genetic ablation of a gene by mutagenesis in the zebrafish. 35 In contrast to morpholino-induced gene knockdown, inducing permanent deleterious mutations as ablation of the respective gene goes along with considerable compensation. Similarly, compensation in mouse models with a permanent genetic ablation is a well-known phenomenon (see Kreiner 36 for a recent review), and hence we searched for a possible compensatory mechanism in NDPK-B−/− mice. Interestingly, the analysis of NDPK-B-deficient primary MBMECs as well as brain sections and retinal Cav-1 staining in NDPK-B−/− mice revealed a significant increase of the number of intracellular vesicles and of the Cav-1 protein content. This was accompanied by an unaltered amount of caveolae and unchanged paracellular permeability of an MBMEC monolayer. Of note, in contrast to ECs acutely depleted of NDPK-B in Feng et al., 3 NDPK-B-deficient MBMECs responded normally to VEGF stimulation. Although species differences between zebrafish and mouse as well as discrepant caveolae formation mechanisms between HUVECs, MBMECs and retinal ECs cannot be excluded, our data indicate that ECs obtained from NDPK-B-deficient mice appear to overcome the loss of NDPK-B via formation of an increased amount of Cav-1-containing vesicles. These vesicles sustain a sufficient transport of Cav-1 to the plasma membrane and thus restore the normal caveolae amount. Nevertheless, this compensatory mechanism is apparently not able to completely rescue EC function, as NDPK-B-deficient mice exhibit an impaired angiogenic response in the OIR model 3 and suffer from an increased leakiness of the brain vasculature (see Figure 6(h)). As recent evidence suggest that an increase in Cav-1 in ECs leads to enhanced transcellular permeability, 37 the observed increase in Cav-1 containing vesicles in MBMECs might contribute to the leaky vasculature rather by transcellular than paracellular permeability. We recently also demonstrated a decrease in pericyte coverage in the retina upon NDPK-B deficiency. 38 Such a loss of pericytes has been reported to contribute to BBB permeability mediated by the caveolae pathway, 39 and thus might represent another explanation for the increased permeability in brain ECs upon loss of NDPK-B. The importance of the paracellular pathway for the regulation of endothelial permeability is further underlined by increased vesicle formation in brain ECs and augmented permeability upon the genetic ablation of the omega-3 fatty acid transporter Mfsd2a of the major facilitator superfamily. 40
In summary, our data obtained from in vitro and in vivo models provide clear evidence for the importance of NDPK-B in Cav-1 transport and thus caveolae formation and function in ECs. We thereby identify NDPK-B as a novel molecular target in caveolae-dependent pathways in ECs. Our finding that some of the malfunctions associated with a lack of NDPK-B can be compensated offers an interesting opportunity for potential therapeutic interventions. NDPK-B−/− mice have a normal life span, and the total absence of NDPK-B activity seems to be well tolerated. Besides a moderate cardiac dysfunction 14 and vasoregression in the retina 38 with increasing age, no impairments have been reported. Thus, it might be speculated that interfering with NDPK-B function is less harmful than interference with Cav-1 function. Due to partial compensation, a basal functional state of caveolae might still be achievable despite of NDPK-B inhibition, whereas pathological functions like the formation of retinal neovascularization in diabetic retinopathy might be hampered. 3 Future work is therefore needed to clarify whether specifically interfering with the NDPK-B/Cav-1 pathway is indeed a therapeutic option.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the Deutsche Forschungsgemeinschaft SFB TR 23, (TP B6, B7).
Acknowledgments
We are grateful to Hiltraud Hosser, Anatomisches Institut, Heidelberg University for expert electron microscopy analysis. The authors thank Heinz Scheffel, Doris Baltus, Heike Rauscher, Kristina Stephan-Schnatz and Stefanie Gurnik for excellent technical assistance.
Disclosure of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
SG, KD, YF and JM performed experiments and analyzed data. YF, SL and TW designed research and analyzed data. SG, KD, SL and TW wrote the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.