
Abstract
Matrix metalloproteinases are versatile endopeptidases with many different functions in the body in health and disease. In the brain, matrix metalloproteinases are critical for tissue formation, neuronal network remodeling, and blood–brain barrier integrity. Many reviews have been published on matrix metalloproteinases before, most of which focus on the two best studied matrix metalloproteinases, the gelatinases MMP-2 and MMP-9, and their role in one or two diseases. In this review, we provide a broad overview of the role various matrix metalloproteinases play in brain disorders. We summarize and review current knowledge and understanding of matrix metalloproteinases in the brain and at the blood–brain barrier in neuroinflammation, multiple sclerosis, cerebral aneurysms, stroke, epilepsy, Alzheimer’s disease, Parkinson’s disease, and brain cancer. We discuss the detrimental effects matrix metalloproteinases can have in these conditions, contributing to blood–brain barrier leakage, neuroinflammation, neurotoxicity, demyelination, tumor angiogenesis, and cancer metastasis. We also discuss the beneficial role matrix metalloproteinases can play in neuroprotection and anti-inflammation. Finally, we address matrix metalloproteinases as potential therapeutic targets. Together, in this comprehensive review, we summarize current understanding and knowledge of matrix metalloproteinases in the brain and at the blood–brain barrier in brain disorders.
Keywords
Introduction
Matrix metalloproteinases
Matrix metalloproteinases (MMPs) are calcium-dependent zinc-endopeptidases of the metzincin superfamily.
1
Structurally, MMPs contain a conserved Zn2+-binding motif in the catalytic domain and several conserved protein domains (Figure 1).
2
MMPs are expressed as inactive zymogens with a pro-peptide domain (pro-MMPs) that must be removed for MMP activation. The pro-peptide is part of the “cysteine switch,” which is an intramolecular complex between a single cysteine in the pro-peptide domain and the zinc in the active site. Through cleavage of the pro-peptide, the cysteine dissociates from the complex, which activates the MMP enzyme and allows binding and cleavage of MMP substrates. MMPs also contain an amino-terminal signal sequence directing the peptide to the endoplasmic reticulum. In addition, all MMPs, except MMP-7 and MMP-26, have a hemopexin-like domain that is connected to the catalytic domain and is responsible for MMP interactions with substrates, endogenous inhibitors, and cell-surface molecules.
Matrix metalloproteinase structure. MMPs are divided into distinct structural groups: minimal-domain MMPs, hemopexin-domain-containing MMPs, gelatinases, and membrane-type MMPs. Minimal-domain MMPs contain an amino-terminal signal sequence (S) that directs them to the endoplasmic reticulum, a pro-peptide (Pro) that maintains them as inactive zymogens, and a catalytic domain with a zinc-binding site (Zn2+). In addition to the domains found in the minimal domain MMPs, hemopexin-domain-containing MMPs have a hemopexin-like domain that is connected to the catalytic domain via a hinge (h). This hinge region mediates the interactions with substrates, TIMPs, and cell-surface molecules. Gelatinase-type MMPs contain inserts that resemble collagen-binding type II repeats of fibronectin (FN). Membrane-type MMPs (MT-MMPs) have a domain that interacts with the membrane. Some MMPs also have a furin-cleavage site (F). MMPs found in the brain are highlighted in red.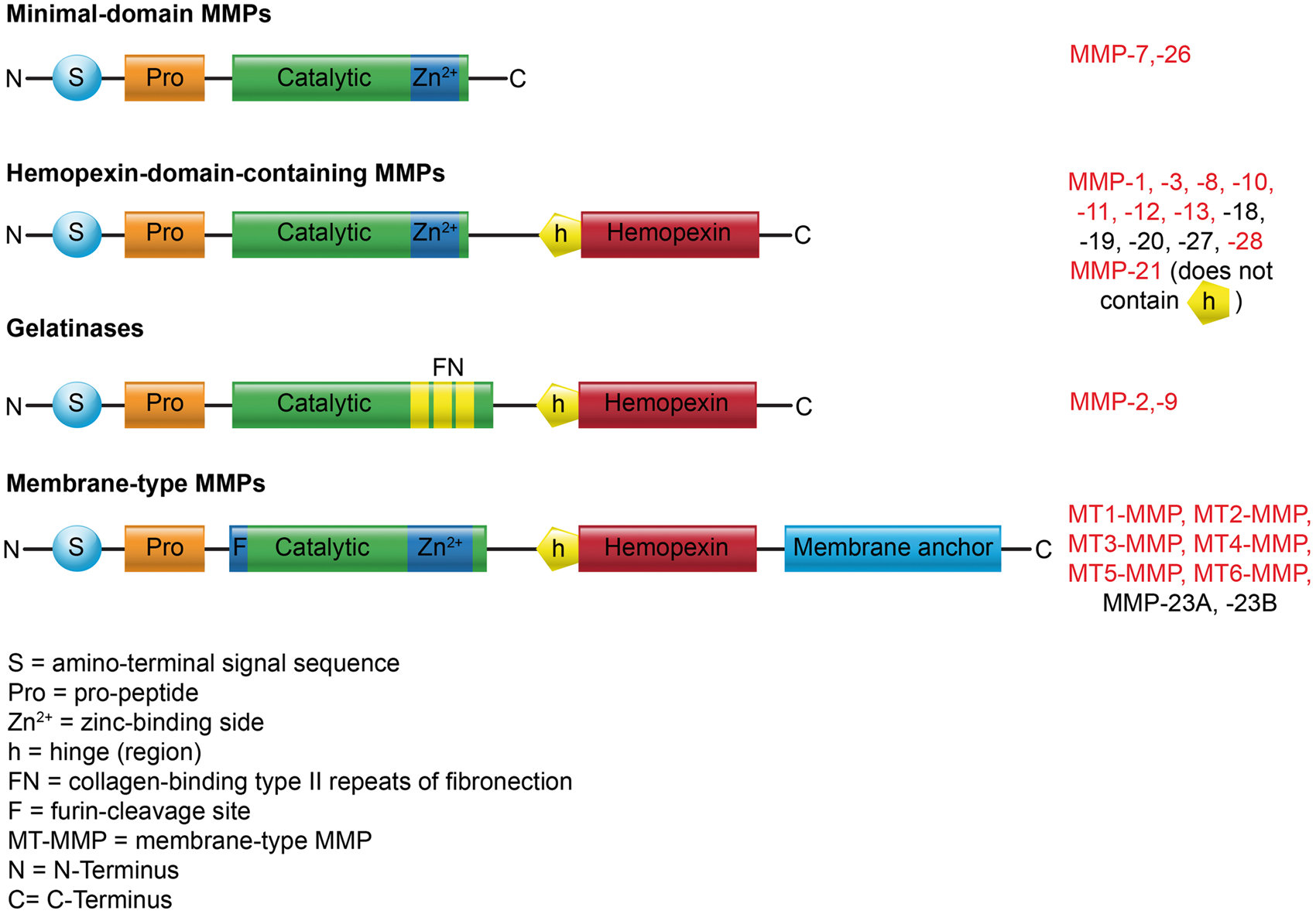
History
The first MMP (MMP-1) was identified by Jerome Gross and Charles Lapiere in 1962 in tadpole. 3 In 1968, the first human MMP was discovered in skin tissue. 4 Since then, a large family of MMPs has been described in various species. 1 In 1971, MMPs were shown to be biosynthesized as inactive precursors (zymogens) that require activation. 5 The first endogenous MMP inhibitor, tissue inhibitor of metalloproteinse-1 (TIMP-1), was identified in 1975 and as of today, four TIMPs (TIMP-1-4) have been described.6,7 In 1990, the “cysteine switch” MMP activation mechanism was discovered. 8 Since then our understanding of MMP biology has increased tremendously. Through the unraveling of the MMP catalytic cycle, we now know that MMPs also digest non-extracellular matrix proteins and contribute to the fine-tuning of cellular processes. In addition, new MMPs – MMP-20, MMP-26, and MMP-28 – have been identified over the last 25 years.9–11
Classification
MMPs in CNS disease.
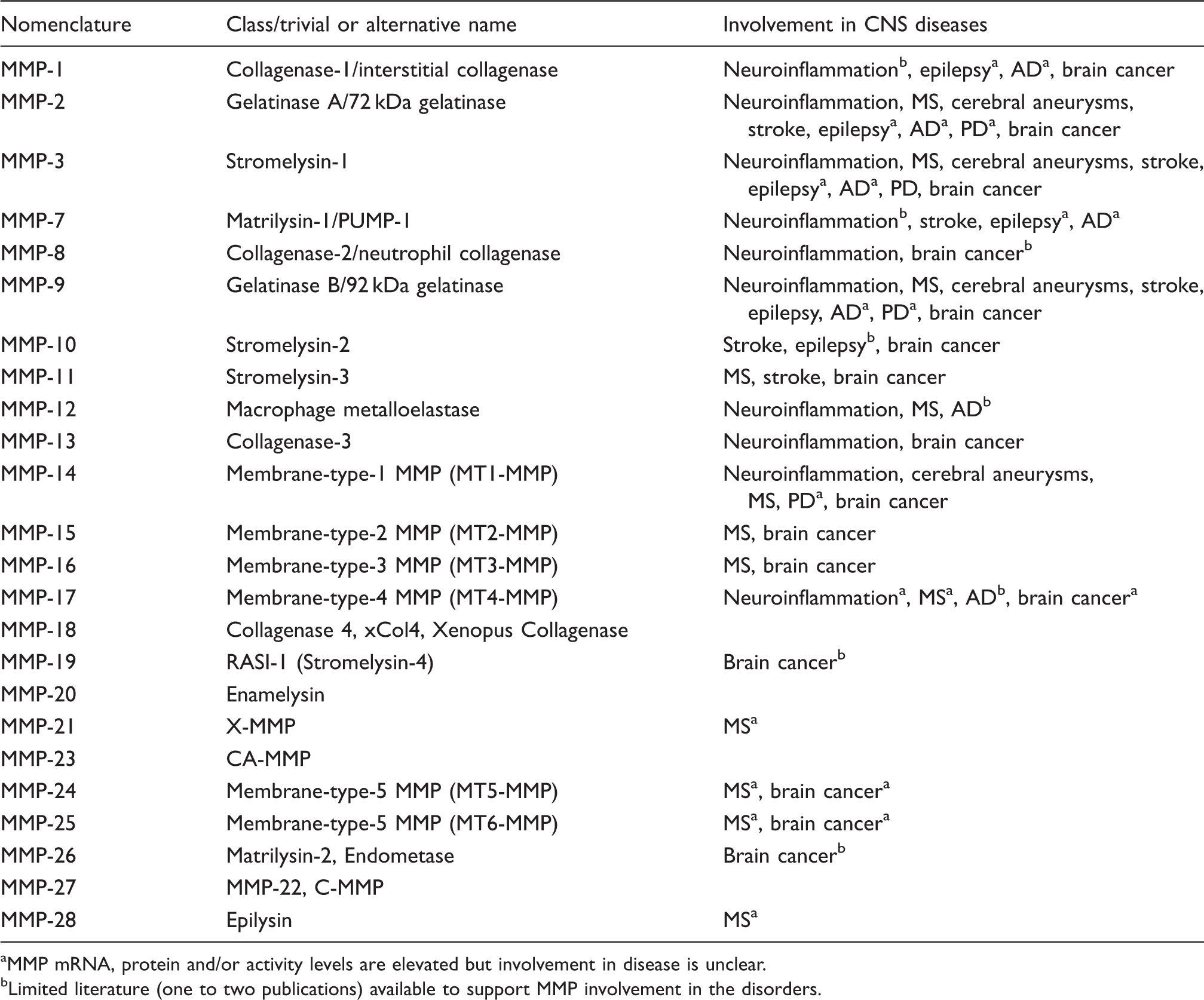
MMP mRNA, protein and/or activity levels are elevated but involvement in disease is unclear.
Limited literature (one to two publications) available to support MMP involvement in the disorders.
Function and role
MMPs play a role in normal physiological processes including tissue morphogenesis, cell migration, and angiogenesis. MMPs are also involved in pathophysiological processes including wound healing, inflammation, and cancer. Some speculate that MMPs cleave extracellular matrix (ECM) proteins to allow infiltrating cells, including leukocytes, metastatic, and transformed cells, to penetrate ECM barriers.14,15 However, this is controversial since the first in vitro studies that associated MMPs with the cleavage of ECM molecules were based on experiments using excessive amounts of MMPs. Today, evidence suggests that ECM cleavage is not a main function of MMPs in vivo. 16
Studies using mass spectrometry indicate that ECM molecules are MMP substrates and other studies show that blocking MMPs (e.g., MT1-MMP) prevents leukocytes from crossing artificial collagen and ECM layers.15,17,18 In these studies, it was demonstrated that fibroblasts and tumor cells tunnel through dense barriers of cross-linked type I collagen in vitro or in vivo via a virtually indistinguishable proteolytic process that requires MMPs. Furthermore, Ota et al. 19 showed that cancer cells utilize an MT1- and MT2-MMP-dependent basement membrane transmigration program to intravasate into the vasculature in vivo.
Other studies using MMP knockout (KO) mice and novel mass spectrometry techniques that allowed improved tissue analysis revealed a wide MMP substrate spectrum that includes cell surface molecules and soluble factors such as chemokines, cytokines, and cytokine receptors. 18 MMP-mediated cleavage of these substrates modulates their activity and represents an important mechanism of fine tuning cellular processes such as inflammation.18,20–22 In essence, MMPs are critical for remodeling processes in developing and regenerating tissues.15,18
Expression, activation, and regulation
All MMPs, with the exception of MMP-28, are ubiquitously expressed in mammalian organisms. MMP expression levels are generally low and only increase when needed, 23 except MMP-2 and MT1-MMP (and to a lesser extent MMP-9), which are constitutively expressed in the brain in both their pro- and activated forms.24,25 MMPs are generated and then secreted into the extracellular space in an inactive, latent pro-form (zymogen), which is activated through proteolysis of the N-terminal pro-domain (Figure 1). This process allows rapid regulation of MMP activity, and thus, controls the availability of cytokines and chemokines. Consequently, MMPs are pivotal in controlling fast cellular responses, such as cell migration during inflammation.
Under physiological conditions, most MMPs are activated by other MMPs or proteases in the extracellular space, but some MMPs are activated intracellularly by the enzyme furin, or by other mechanisms (e.g., phosphorylation). MMP inhibition, on the other hand, is mediated by tissue inhibitors of metalloproteinases, TIMPs, that coexist with MMPs.7,26 TIMPs inactivate MMP activity by binding to them, which under physiological conditions prevents excessive tissue degradation and injury. Under pathophysiological conditions, MMPs can be activated by reactive oxygen species and other factors (e.g., NO, hypoxia, pH) through a mechanism that likely involves auto-catalytic activation.27–29
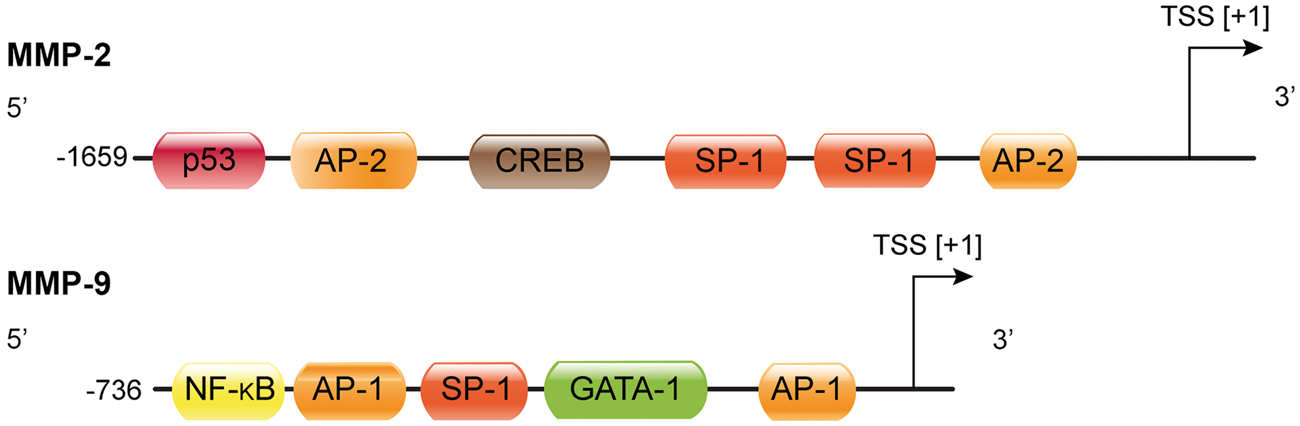
Little is known about transcriptional MMP regulation and what is known involves inflammatory signaling. Tumor necrosis factor-α (TNF-α) and interleukin-17 both stimulate MMP-9 transcription through the transcription factors activator protein-1 (AP-1) and nuclear factor-κB (NF-κB).30–32 This effect can be blocked with interferon-γ through NF-κB inhibition.
33
The endotoxin lipopolysaccharide (LPS) triggers reactive oxygen species production and p38 kinase phosphorylation, which activates AP-1 and induces MMP-9 transcription.
34
Figure 2 shows the MMP-9 promotor with one NF-κB and two AP-1 binding sites. MMP-2, on the other hand, is regulated by TNF-α and p38-MAPK acting through NF-κB, but not AP-1 (Figure 2), and a caspase 8-dependent pathway.
35
Together, MMP regulation is not fully understood and varies between cell types in a context-dependent manner.36,37
MMP-2 and MMP-9 promoter region with putative transcription factor binding sites. The boxes represent binding sites for the corresponding transcription factors. TSS: transcription start site; AP-1: activator protein 1; AP-2: activator protein 2; GATA-1: GATA-binding factor 1, erythroid transcription factor, globin transcription factor 1; SP-1: specificity protein 1; NF-κB: nuclear factor-κB, CREB: cyclic AMP response-element binding protein; p53: tumor protein p53 (modified after Peters et al.
38
and Rosenberg
39
).
MMPs in brain and blood–brain barrier
MMPs participate in many physiological and pathological processes in the brain and at the blood–brain barrier (Table 2). The blood–brain barrier is the capillary endothelium that separates blood from brain.
40
Physical barrier function is localized to three structures that are critical for barrier integrity: the brain capillary endothelium, the tight junctions between the endothelial cells, and the basement membrane (Figure 3).
40
First, the brain capillary endothelium is a barrier for small hydrophilic compounds. Second, tight junctions seal the clefts between adjacent endothelial cells, which prevents uncontrolled paracellular passage of solutes and makes the brain endothelium a low-permeability barrier.41–43 The major tight junction proteins in the brain endothelium are claudin-1, claudin-5, occludin, and zonula occludens-1. Third, the basement membrane, which is a specialized ECM, connects endothelial cells with pericytes and astrocytes to form the neurovascular unit and facilitates communication between the cells within this unit through receptors such as integrins and dystroglycans.44,45
Blood–brain barrier anatomy. The blood–brain barrier is formed by capillary endothelial cells that are linked by tight junctions, surrounded by a basement membrane, and astrocytic endfeet. Astrocytes provide the cellular link to neurons; pericytes are embedded in the basement membrane. In disease, MMP protein expression and activity levels are increased, which is thought to result in blood–brain barrier leakage, possibly through degradation of tight junction and basement membrane proteins. MMP effects in the brain and blood–brain barrier.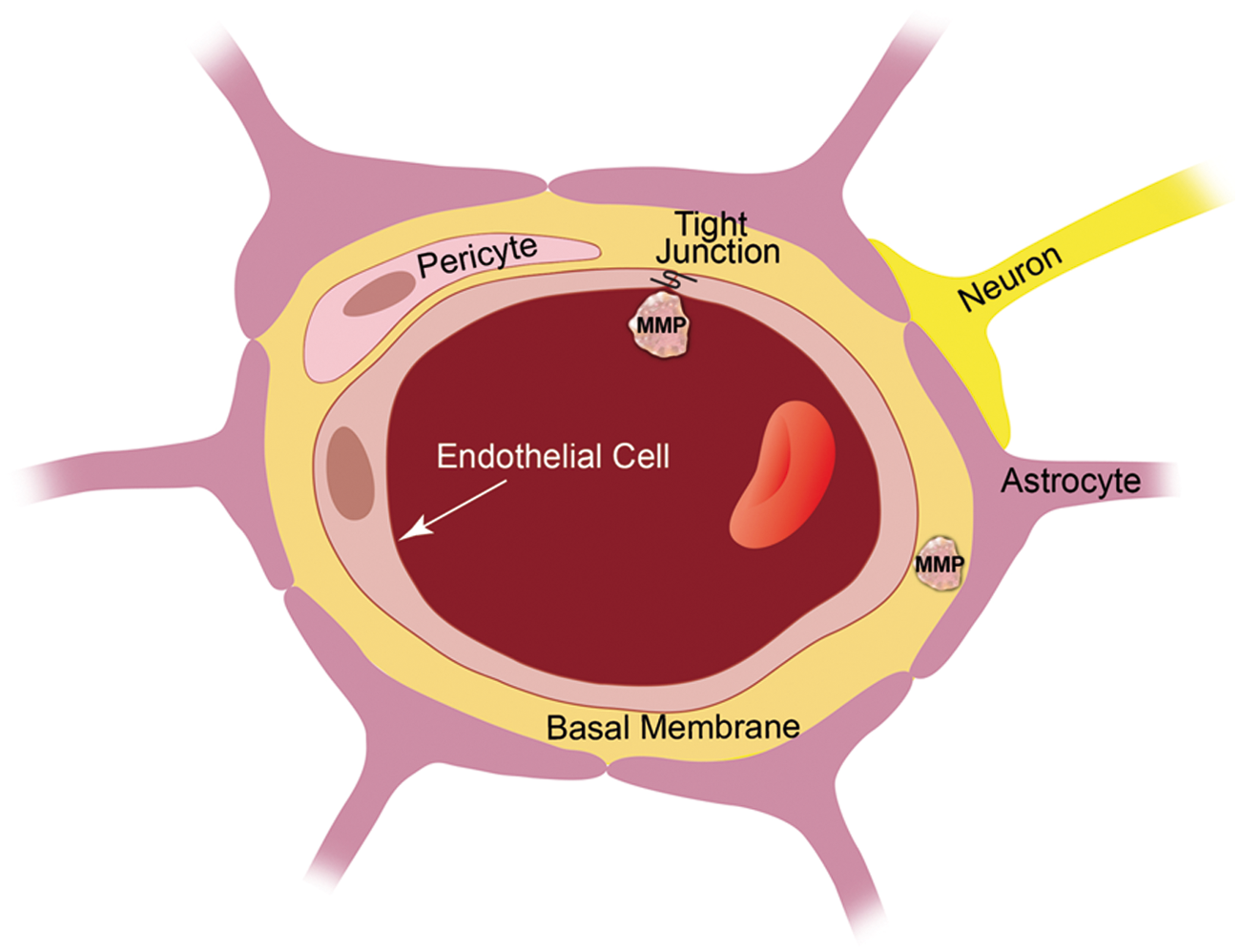
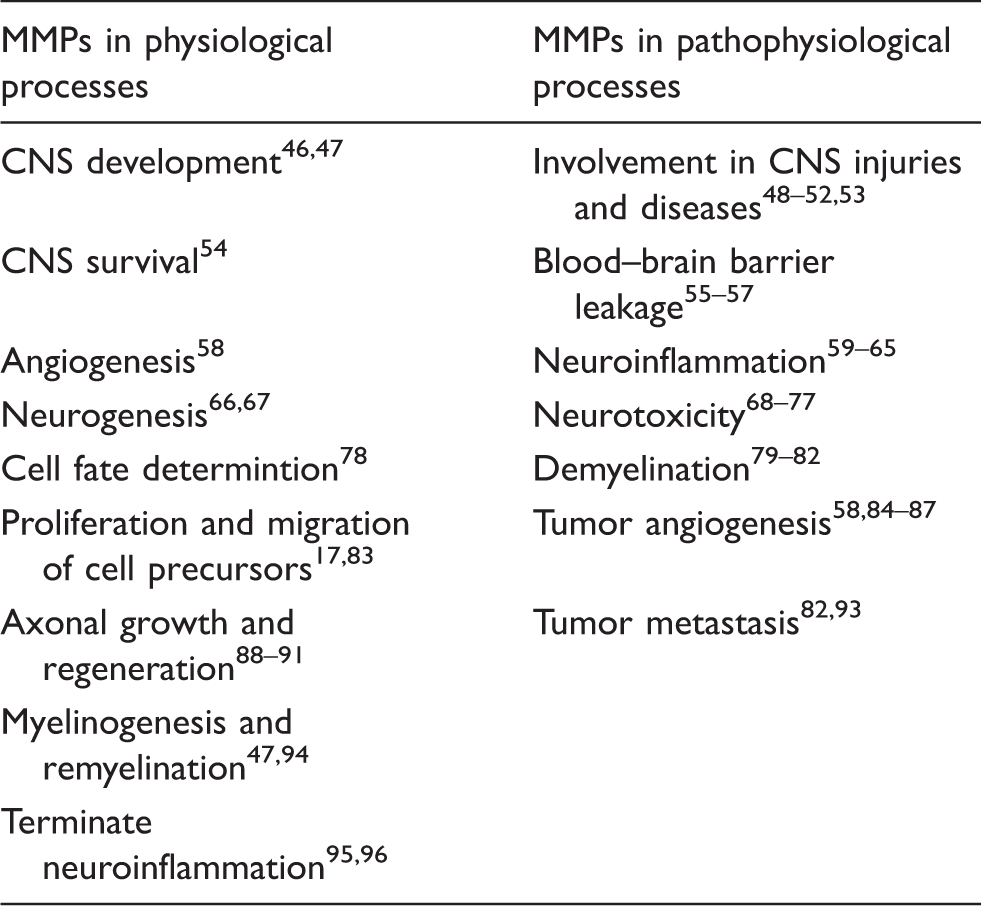
Endothelial cells, tight junctions, and basement membrane are critical for proper barrier function, and thus, for brain homeostasis and overall brain health. In turn, pathological disruption of the endothelium, tight junctions, or the basement membrane impairs barrier integrity, which can have severe consequences for the brain and contribute to disease progression.97,98 In this regard, it has been proposed that MMPs digest tight junctions and basement membrane proteins, and thus, are critical contributors to brain disease and directly affect brain health.99,100 However, little data exist to support this since it is technically challenging to demonstrate MMP activity in vivo. For example, Gu et al. 101 show increased MMP activity and higher permeability at the blood–brain barrier in stroke during reperfusion in vivo. Yang et al. 102 show increased MMP-2 and MMP-9 mRNA and activity levels after reperfusion in spontaneously hypertensive rats with middle cerebral artery occlusion (MCAO). The authors also observed blood–brain barrier leakage in the piriform cortex and disrupted tight junction proteins. Inhibiting MMPs prevented the loss of tight junction proteins, indicating that MMPs disturb barrier integrity by degrading tight junction proteins. 102 Thus, while technically challenging, first evidence showing that MMPs digest tight junctions and ECM proteins in vivo is emerging.
Methods and techniques to study MMPs
MMPs are mostly studied at the mRNA, protein, and activity levels. MMP mRNA expression has been demonstrated in several studies using real-time quantitative PCR and microarray analysis.103,104 MMP protein expression is usually determined by Western blotting, ELISA, or by immunohistochemistry. To detect MMP activity in vitro, a widely used technique – substrate zymography – is employed. Substrate zymography identifies MMPs by the degradation of their substrate and by their molecular weight.105,106 This method allows determining if the MMP is active or latent. All types of substrate zymography originate from gelatin zymography, which is used to detect the gelatinases MMP-2 and MMP-9. 107 To detect other MMPs, gelatin is replaced with another substrate such as collagen, carboxymethylated transferrin, or casein.104,108–111 Another method to detect MMP activity in vitro is by using fluorogenic MMP substrates. These artificial MMP substrates are composed of a fluorescent dye that is connected via a peptide to a quencher. Active MMP cleaves the peptide, and thus, removes the quencher resulting in fluorescence, which is a direct measure of MMP activity. 112
Currently, it is not possible to localize activity of most MMPs in tissues due to a lack of suitable reagents. An exception to this is gelatin in situ zymography, a method that allows detecting MMP-2 and MMP-9. Gelatin in situ zymography is a modification of substrate zymography in frozen tissue sections. In this method, an MMP substrate is transferred onto a frozen section of an unfixed tissue sample. The substrate is digested by active MMPs in a time- and dose-dependent manner, which is visualized with microscopy. 113 A further development of this method is in vivo zymography, 114 where substrates are used in a live animal to detect MMP activity in vivo. 115
MMPs in central nervous system disease
Neuroinflammation
Neuroinflammation is defined as an unspecific inflammatory event in the brain. All central nervous system (CNS) disorders discussed here – multiple sclerosis (MS), cerebral aneurysm, stroke, epilepsy, Alzheimer’s disease (AD), Parkinson’s disease (PD), and brain cancer – have a neuroinflammatory component that involves MMPs.
MMPs contribute to neuroinflammation through four mechanisms (Figure 4). First, MMPs activate neuroinflammatory pathways. This is done indirectly by activating enzymes that act on signaling molecules such as cytokines, cell surface receptors, cell–cell adhesion molecules, or clotting factors.59,60 Alternatively, MMPs directly activate neuroinflammatory pathways. For example, MT4-MMP has TNF-α-convertase activity through which transmembrane TNF-α is proteolytically converted into soluble, active TNF-α (Figure 4(1)).61,62 Second, MMPs themselves act as neuroinflammatory signaling molecules. Upon stimulation with LPS, apoptotic signals, or in PD, neurons secrete active MMP-3 into the interstitium, which triggers microglial activation and production and secretion of pro-inflammatory cytokines (Figure 4(2)).63–65 Third, MMPs contribute to neuroinflammation-mediated neurotoxicity by shedding death molecules like the Fas ligand, by affecting GABA and glycine levels, which modulate chloride channel activity, by stimulating glutamate receptor-mediated excitotoxicity, or by altering cell–ECM interaction.68–72 However, the exact mechanisms through which MMPs exert neurotoxicity are not fully understood (Figure 4(3)).73–77 And lastly, neuroinflammation-induced MMPs may proteolyze cerebrovascular basement membrane and tight junction proteins, which could compromise vascular integrity resulting in barrier leakage and extravasation (Figure 4(4)).55–57,79
MMPs in neuroinflammation. MMPs contribute to neuroinflammation through four mechanisms. (1) MMPs activate neuroinflammatory pathways and/or neurosignaling components. (2) MMPs act as signaling molecules themselves. (3) MMPs contribute to neuroinflammation-mediated neurotoxicity. (4) MMPs compromise vascular integrity resulting in blood–brain barrier leakage.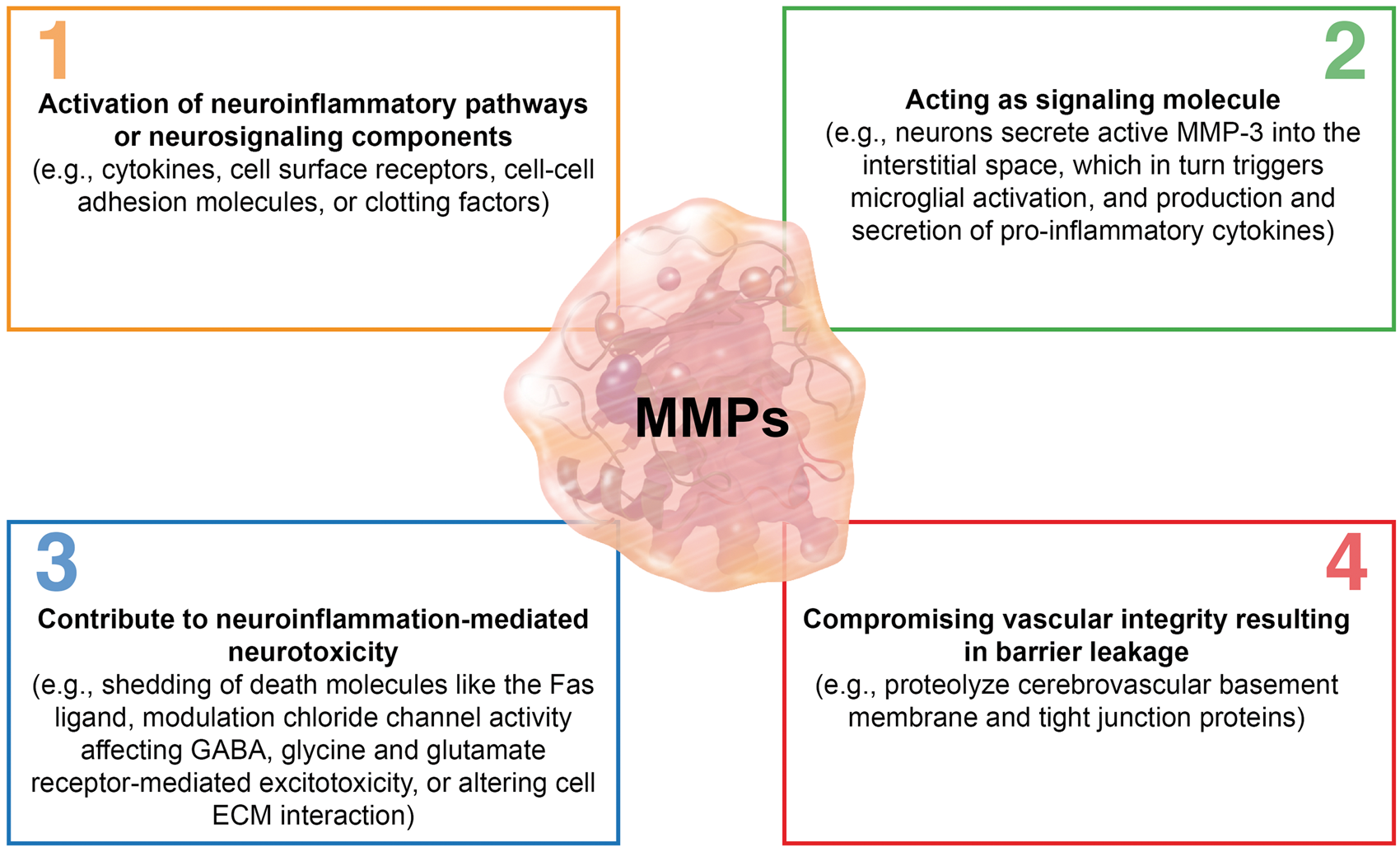
Together, MMPs are induced by and contribute to neuroinflammation through various mechanisms. MMPs also contribute to inflammation-induced barrier dysfunction and leakage. The combination of both – neuroinflammation and barrier dysfunction – promotes progression of several CNS disorders (MS, cerebral aneurysms, stroke, epilepsy, AD, PD, and brain cancer), which are discussed in the following sections.
Multiple sclerosis
MS is a neuroinflammatory auto-immune disease that affects about 1.3 million people worldwide. 116 In MS, the myelin sheaths that cover neuronal axons and nerve fibers in the brain and spinal cord are damaged, which disrupts communication and causes a wide range of disease symptoms. 117
The role of MMPs in MS has been studied extensively in animal models and human tissue.118–122 These studies revealed that MMPs digest myelin basic protein, which causes demyelination and drives MS progression (Figure 5(a)).79,81,82 Using the experimental autoimmune encephalomyelitis (EAE) animal model of MS, several groups analyzed MMPs in brain, brain capillary endothelial cells, spinal cord, lymph nodes, and spleen and showed that multiple MMPs were elevated during the EAE peak stage.119,123–128 Specifically, in EAE mouse and rat models, mRNA, and protein levels were increased for MMPs-2, -3, -7, -8, -9, -10, -11, -12, -13, -28, MT1-MMP, and MT6-MMP. In contrast, mRNA and protein levels for MT2-5-MMP and MMP-21 were decreased in lumbar and sacral spinal cord tissue of EAE mice.
129
While the consequence of decreased MMP levels, particularly those of MT-MMPs, is unclear, it is well established that increased MMP levels aggravate disease severity in EAE rodent models.118,119,125,127,128,130,131
MMPs in multiple sclerosis. (a) Brain endothelial cells and leukocytes secrete MMPs, which are thought to degrade tight junction and extracellular matrix proteins leading to extravasation of immune cells. (b) Leukocytes, microglia, neurons, and reactive astrocytes secrete MMPs, which demyelinate neuronal axons.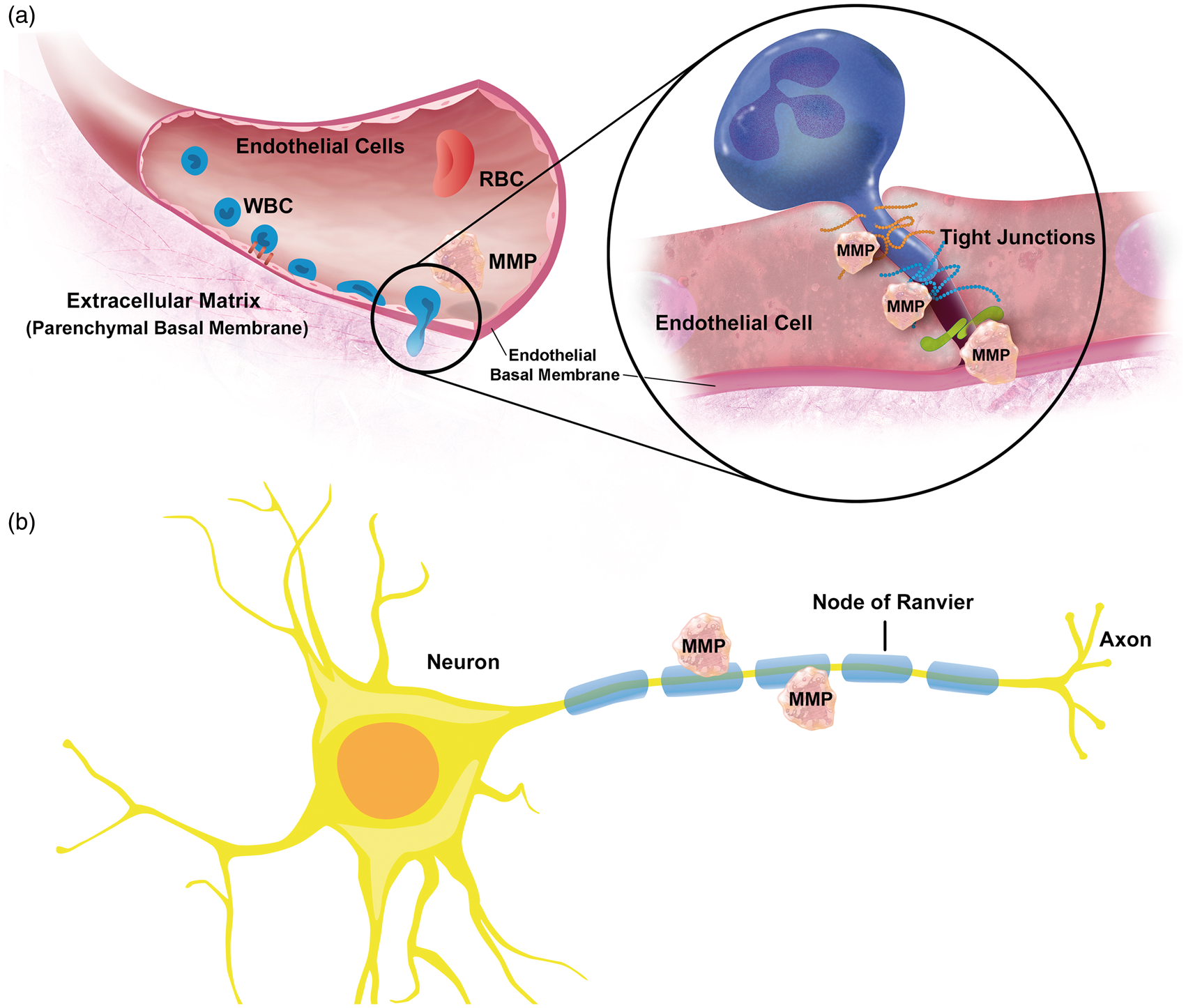
One characteristic of MS is leukocyte extravasation and transmigration across the brain endothelium into the CNS. MMPs may facilitate this process by activating adhesion molecules and degrading the basement membrane that surrounds blood vessels (Figure 4(1) and (4)). However, this is controversial since there is no conclusive evidence. Agrawal et al. 118 show selective MMP-2- and MMP-9-mediated cleavage of dystroglycan, which is a linker between astrocyte endfeet and parenchymal basement membrane molecules. This process is located at postcapillary venules, where extravasation occurs. 118 But this and other studies, for example, the study by Buhler et al. 119 suggest that MMPs are involved in immune cell extravasation into the brain during EAE.
Studies using brain tissue, serum, and cerebrospinal fluid (CSF) samples from MS patients consistently found increased protein levels for MMPs-2, -3, -7, -9, -12, -13, and MT1-MMP.80,120,132–138 In these studies, leukocytes were identified as a main source of MMPs, the best-studied of which in MS is MMP-9. MMP-9 mRNA, protein, and activity levels are increased in mononuclear blood cells, serum, and CSF, and correlate with barrier leakage and disease progression.121,139–143
Immune cells from the blood can cross the blood–brain barrier via a transcellular (likely no MMPs involved) or a paracellular (MMPs involved) pathway. For the latter, it has been reported that T cells, monocytes, and dendritic cells express and release active MMP-2 and MMP-9, which open the brain endothelial tight junctions to cross the barrier and migrate into the brain.118,144–149 After passing the tight junctions, MMP-2 and MMP-9 cleave the transmembrane receptor β-dystroglycan, which anchors astrocytic endfeet to the basement membrane. 118 In addition, in lesional MS tissue, MMPs-1, -2, -3, -9, and -19 have been detected in microglial nodules and microglial-like cells where they contribute to inflammation and further destabilize the blood–brain barrier.150,151
One MMP that currently gets attention in the MS field is MMP-12. MMP-12, also called macrophage metalloelastase, is assumed to be essential in the pathogenesis of MS, most likely due to its primary myelin- or oligodendrocyte-toxic potential and its role in macrophage extravasation. At the same time, MMP-12 does not seem to damage the blood–brain barrier or alter ECM remodeling and deposition.138,152 In contrast, data from other studies show that MMP-12 KO mice with EAE had a significantly worse maximum severity and disease burden compared with EAE wild-type control mice, suggesting that increased MMP-12 expression levels are protective in MS. 127 An additional study showed that wild-type and MMP-12 KO mice with EAE followed a relapsing-remitting course. 153 Although both mouse groups had a similar clinical onset, relapses in MMP-12 KO mice with EAE were more severe and their residual disability at remission was higher.
Thus, while it is clear that MMPs contribute to MS, it is less clear if this occurs by degrading the endothelial basement membrane, which would facilitate leukocyte extravasation and migration into the brain (Figure 5(a)). In the brain, leukocytes then release more MMPs that contribute to the overall MMP effect of axonal demyelination and neuronal cell death.
Cerebral aneurysms
An aneurysm is a blood-filled balloon-like bulge in the wall of an artery. The causes of brain aneurysms are manifold and include aging, atherosclerosis, hypertension, and severe head injury, all of which are accompanied by neuroinflammation. 154 Most cerebral aneurysms remain undetected until they rupture, which is life-threatening. 155 Therefore, it is critical to prevent rupture by securing intracranial aneurysms, which is accomplished by invasive brain surgery. 156 A less invasive approach would be to prevent aneurysms from forming, which requires understanding of aneurysm pathology. One theory states that MMPs degrade the vascular ECM, thereby contributing to localized ballooning of a blood vessel and leading to aneurysm formation and growth.157–159 For example, in human brain samples, protein expression levels of plasmin, MMP-2, MMP-9, and MT1-MMP were increased in the aneurysmal wall compared to normal cerebral arteries and overall MMP-2/MMP-9 proteolytic activity was higher in aneurysm tissue compared to control arteries. 157
Recent reports also indicate that MMPs are involved in vascular calcification,160,161 which could be another negative effect MMPs contribute to the pathological outcome of cerebral aneurysms. In a retrospective review, the authors showed that the presence of calcification in an aneurysm was the sole marker of adverse outcome. 162 Larger aneurysms tended to be more likely to be calcified, while size by itself did not have an adverse effect on outcome. Furthermore, in surgically securing intracranial aneurysms, calcified aneurysms are a significant source of morbidity. 162
One possibility to limit aneurysm progression and growth is through MMP inhibition, which could potentially reduce the need for invasive treatment.163,164 Pre-clinical studies show that MMP inhibitors block aneurysm formation and growth.164–166 Xiong et al. 166 demonstrated in a mouse model of Marfan syndrome that inhibiting MMP-2 and MMP-9 protein expression with doxycycline blocked ECM degradation, which significantly delayed aneurysm rupture. Nuki et al. 165 used a mouse model, where 70% of animals develop brain aneurysms and demonstrated that doxycycline reduced the incidence of aneurysms to 10%. The authors also showed a reduced incidence (40%) of intracranial aneurysms in MMP-9 KO mice, whereas over 60% of MMP-2 KO mice still developed cerebral aneurysms, suggesting that MMP-9 is critical for aneurysm formation. Aoki et al.167,168 used statins in rats, where cerebral aneurysms were induced by unilateral ligation of the common carotid artery and hypertension. Statin treatment decreased aneurysm size by 30–40% within one month likely through a mechanism that lowered MMP levels, which is thought to delay aneurysm formation and growth. 164 The therapeutic benefit of statins is attributed to their cholesterol-lowering, anti-inflammatory, and anti-NF-κB effects, which lower MMP activity. 164 Statins have also been studied in humans. Yoshimura et al. 169 conducted a retrospective study analyzing data from 117 patients with ruptured brain aneurysms and 304 patients with unruptured brain aneurysms to assess if statins prevent rupture. In this study, 9% of patients with ruptured cerebral aneurysms used statins, whereas 26% of patients with unruptured aneurysms used statins, which indicates that statins lower the risk of brain aneurysms to rupture.
Together, MMPs contribute to formation, growth, and rupture of cerebral aneurysms by digesting the ECM, which leads to ballooning of blood vessels. This suggests that inhibition of MMPs, especially MMP-9, could potentially prevent cerebral aneurysms.
Stroke
In 2012, stroke accounted for about seven million deaths worldwide, which is 12% of all deaths and makes stroke the number two killer. 170 With an additional 10 million people surviving a stroke each year, over 30 million people altogether survived a previous stroke. 171 Stroke is characterized by a loss in brain function due to limited cerebral blood flow (ischemic) caused by a blocked blood vessel, or due to bleeding into the brain parenchyma or subarachnoid space (hemorrhagic).
In stroke, MMPs have detrimental effects in the acute phase and beneficial effects in the post-stroke phase.
172
Detrimental effects are mediated by dysregulated MMPs and include neurovascular disruption and brain parenchymal damage (Figure 6(a)). Various studies in human and rat brains show that protein and activity levels of MMPs-2, -3, and -9 are increased after stroke and MCAO compared to control tissue.173,174 These changes in MMP protein and activity levels result in aberrant proteolysis that contributes to blood–brain barrier dysfunction and in part determines the extent of the infarct.132,173–177 In addition, studies using rat stroke models suggest that by degrading the basal lamina, MMPs predispose brain capillaries to rupture and hemorrhagic transformations after stroke.178–180 Other examples of detrimental MMP effects in stroke were shown in studies using rodent models of focal cerebral ischemia. In these studies, increased MMP-9 protein levels were detected in the acute phase (2–24 h) after stroke that coincided with an opening of the blood–brain barrier. In contrast, MMP-2 protein levels were increased several days after stroke,181,182 during which barrier leakage is presumably restored.
MMP-9 in stroke. (a) acute phase: endothelial cells and recruited leukocytes secrete MMP-9, which degrades the blood–brain barrier, the neurovascular basement membrane, and the ECM. (b) remodeling phase: astrocytes and neurons secrete MMP-9, which contributes to remodeling of the ECM in the neurovascular unit.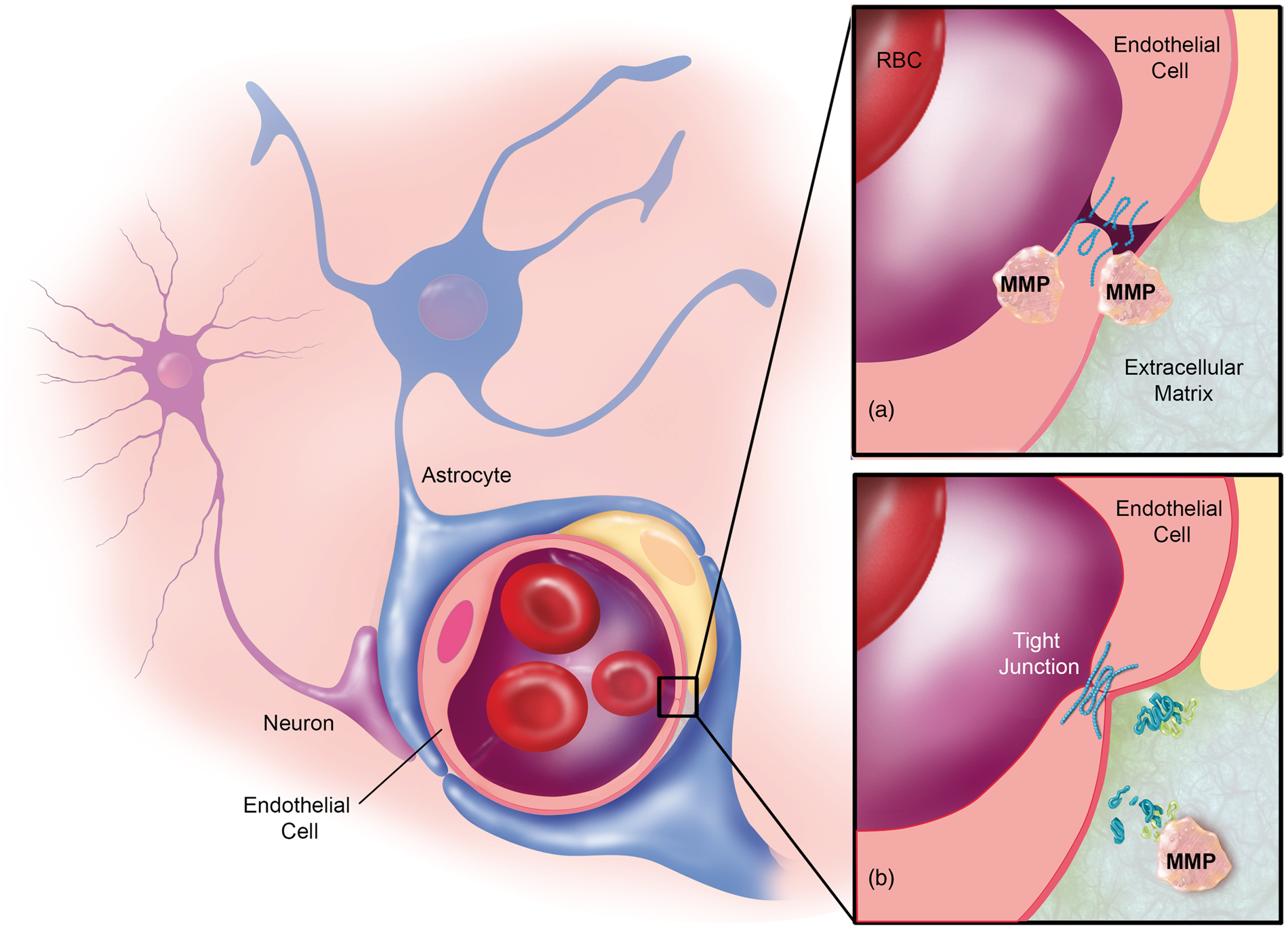
While most MMP research in stroke is focused on MMP-2 and MMP-9,183,184 other MMPs also have detrimental effects. Suzuki et al. 185 treated mice after thrombotic MCAO with tissue-type plasminogen activator (tPA) and observed an increased incidence of intra-cranial bleeding compared to mice not treated with tPA. The authors showed increased MMP-3 mRNA and protein levels in the capillary endothelium in the infarct region of tPA-treated mice compared to tPA-untreated control mice. Suzuki et al.185,186 concluded that in tPA-treated mice, MMP-3 digests the neurovascular basal lamina, thereby opening the endothelial barrier and contributing to intra-cranial bleeding. These findings suggest that MMP-3 has detrimental effects during tPA treatment or is induced by tPA, which is unfortunate since tPA is currently the only FDA-approved treatment for ischemic stroke.
However, MMPs also have beneficial effects during the recovery phase after stroke (Figure 6(b)). 187 Studies suggest that MMP-9, MMP-2, and MMP-7 remodel lesional ischemic and infarct tissue and participate in angiogenesis, vasculogenesis, and neurogenesis.17,83,94,188–190 Two mechanisms have been identified by which MMPs exert these effects. First, during tissue remodeling in the post-stroke recovery and healing phase, MMPs digest old ECM so that new ECM and tissue can be generated.2,187,191 Second, during ECM digestion, MMPs (mainly MMP-7 and MMP-9, but also MMPs-1, -2, -3, -10, and -11) increase the availability of growth factors (e.g., nerve growth factor, brain-derived neurotrophic factor, neurotrophin-3/-4, and vascular endothelial growth factor). This occurs through cleaving inactive growth factor precursors into their active form or through releasing active growth factors by ECM proteolysis.54,188 Increased levels of growth factors support tissue remodeling by stimulating angiogenesis, vasculogenesis, and neurogenesis, all of which are critical in stroke recovery. These findings suggest that during the remodeling and healing process, MMPs are involved in the migration of neuronal precursor cells to areas damaged by stroke.17,83
Together, MMPs are essential in stroke in both the acute phase and the post-stroke phase (Figure 6). In the acute phase, MMPs impair barrier integrity and damage the parenchymal tissue, whereas in the post-stroke phase MMPs contribute to the recovery process by remodeling lesional ischemic and infarct tissue and by participating in angiogenesis, vasculogenesis, and neurogenesis.
Epilepsy
The World Health Organization estimates that at least 65 million people worldwide suffer from epilepsy.192–194 Epilepsy describes various diseases and seizure syndromes, and patients are diagnosed with epilepsy after recurring, unprovoked seizures. 195
The role MMPs have in epilepsy is still unclear, but studies suggest MMPs contribute to epileptogenesis, epilepsy progression, and brain remodeling after seizures. For example, MMP-9 KO mice are less sensitive to chemically-induced seizures compared to wild-type mice, and conversely, human MMP-9-overexpressing rats are more sensitive to chemical seizure induction, suggesting MMP-9 affects epileptogenesis and/or seizure genesis.196,197
MMP levels are increased in the epileptic brain. In chemically induced animal seizure models and patients with temporal lobe epilepsy, MMP-9 protein and activity levels are increased in neurons of the parietal and frontal cortex, as well as the thalamus, the regions where the seizures originated.73,197–200 Li et al. 201 detected increased MMP-9 protein and activity levels in CSF from adult epilepsy patients with generalized tonic–clonic seizures compared to age-matched non-epileptic individuals. MMP-9 levels were also increased in serum from patients after seizures. Suenaga et al. 202 detected threefold higher MMP-9 protein levels in serum from children with encephalopathy after prolonged febrile seizures compared to serum from healthy children. The authors also found higher serum MMP-9 levels in children with prolonged febrile seizures but no encephalopathy, in children with simple febrile seizures, and in children with convulsive status epilepticus compared to healthy children. 202 Research suggests that increased MMP-9 protein and activity levels mainly serve two functions: first, MMP-9 contributes to seizure-induced neuronal cell death; and second, MMP-9 is critical in remodeling neuronal networks after seizures. Jourquin et al. 75 and Hoehna et al. 73 demonstrated neuronal cell death in areas with increased MMP-9 levels after seizures and showed that inhibiting MMP-9 reduced cell death. Other studies show that MMP-9 is involved in structural remodeling, mossy fiber sprouting, diminished seizure-induced pruning of dendritic spines, and decreased aberrant synaptogenesis.197,199,203
MMP-2 function in the epileptic brain is less understood than that of MMP-9. Jourquin et al. 75 demonstrated that MMP-2 does not contribute to neuronal cell death in epilepsy. However, it is conceivable that MMP-2 contributes to structural remodeling in epileptogenesis since MMP-2 mRNA, protein, and activity are increased after seizures.197,199,204
Several studies demonstrate blood–brain barrier dysfunction in epilepsy and seizure-induced barrier leakage.205–210 Additionally, barrier leakage itself triggers seizures, suggesting a pernicious feedback loop contributing to epilepsy progression.210–212 In this regard, MMPs most likely degrade tight junction and ECM proteins, which potentially contribute to barrier leakage after seizures.98–100 Li et al. 201 showed that increased MMP-9 protein and activity levels in serum and CSF of patients with generalized tonic–clonic seizures correlate with barrier leakage. Furthermore, barrier leakage is associated with leukocyte extravasation into the brain after seizures. Leukocyte extravasation is a complex, multi-step process that requires MMPs, in particular MMP-2 and MMP-9, both from the endothelium as well as from activated T cells and macrophages.188,147,149,213 Li et al. 201 demonstrated in patients with generalized tonic–clonic seizures that increased CSF leukocyte counts correlated with increased MMP-9 levels in CSF and the degree of barrier leakage. 201
Thus, MMP-2 and MMP-9 seem to contribute to seizure- and/or epileptogenesis, neuronal network remodeling, neuronal cell death, and barrier leakage after seizures. Little is known about other MMPs in epilepsy.
Alzheimer’s disease
AD is a neurodegenerative disorder that affects more than 20 million patients globally.214,215 Disease projections are grim and predict up to 100 million AD patients by 2050. 216 Despite all research efforts, AD etiology and progression are not well-understood and a cure or prevention is currently not available. AD pathology is characterized by brain accumulation of amyloid-β (Aβ), development of Aβ plaques, formation of neurofibrillary tangles, and neuroinflammation, all of which contribute to neurodegeneration.217,218 In this section, we first describe what is currently known about MMPs in AD and then describe the relationship between Aβ and MMPs.
Several groups have reported that MMP levels in rodent AD models are increased compared to control animals. Yan et al. 219 detected increased MMP-9 protein levels in brain slices from transgenic APPsw and amyloid precursor protein (APP)/PS1 mice compared to wild-type mice. Using 5xFAD mice, Py et al. 220 found increased MMP-2, MMP-9, and MT1-MMP protein levels in the hippocampus compared to control mice. MMP-2 and MMP-9 were primarily expressed in astrocytes; MT1-MMP was localized to neurons; MMP-9 and MT1-MMP were also detected in amyloid plaques. Consistent with these findings, several groups found overexpression of MMP-2, -3, and -9 mRNA and protein in postmortem brains from AD patients.221–224 Horstmann et al. 225 used zymography and detected MMP-2, -3, -9, and -10 activity levels in serum and CSF from AD patients and compared them to gender- and age-matched healthy control individuals. The authors found that MMP-3 activity was elevated by 40% in plasma and 60% in CSF from AD patients compared to control individuals. MMP-2 activity in the CSF of AD patients was decreased by 32% compared to CSF samples from healthy individuals, while activity levels in plasma remained unchanged. MMP-9 and MMP-10 activity were undetectable in CSF, MMP-10 activity was unchanged in plasma, and MMP-9 activity in plasma was decreased by 41% compared to healthy individuals. Lorenzl et al. 226 detected higher levels of proMMP-9 protein in plasma samples from AD patients compared to control individuals.
Some research aimed at clarifying the relationship between MMPs and Aβ. Deb and Gottschalk 227 observed in rat hippocampal and astrocyte cultures that Aβ40 induced protein expression and proteolytic activity of MMPs-2, -3, and -9. We demonstrated that exposing isolated rat brain capillaries to Aβ40 ex vivo increased MMP-2 and MMP-9 protein and activity levels. 228 We made similar observations in a transgenic mouse AD model (Tg2567 hAPP mice), where MMP-2 and MMP-9 levels in brain capillaries were elevated compared to capillaries from wild-type mice. Yin et al. 229 observed in APP/PS1 mice that astrocytes surrounding amyloid plaques secrete more MMP-2 and MMP-9. They also demonstrated that breeding APP/PS1 mice with MMP-2 or MMP-9 KO mice, or pharmacologically inhibiting MMP-2 or MMP-9 in APPsw mice increased Aβ brain levels by 1.5-fold compared to controls and increased Aβ half-life by about 50%. 229 Additionally, Yin et al. observed in the phosphate buffer-soluble fraction of cortex and hippocampus of MMP-2 KO mice increased murine Aβ40 and Aβ42 compared to age-matched wild-type mice, whereas Aβ40 and Aβ42 were unchanged in the phosphate buffer-insoluble fraction. In the cortex and hippocampus of MMP-9 KO mice, murine Aβ42 levels were increased in the phosphate buffer-soluble fraction of hippocampus and cerebral cortex compared to age-matched wild-type mice, while they remained unchanged in the phosphate buffer-insoluble fraction. These effects were due to decreased Aβ proteolysis and not to increased Aβ production. These findings suggest that MMPs potentially contribute to Aβ clearance. In this regard, MMPs-2, -3, and -9 proteolytically degrade Aβ.219,230,231,232 Ridnour et al. 233 showed that levels of Aβ1–16, a product of Aβ metabolism by MMP-9, and MMP-9 activity were decreased in brain lysates of hAPPSwDI mice lacking nitric oxide synthase compared to their littermates expressing nitric oxide synthase. Based on these data, the authors concluded that nitric oxide is potentially involved in clearing plaques through increasing MMP-9 activity. Yan et al. 219 demonstrated in brain slices of APP/PS1 mice in situ that MMP-9 digests both fibrillary Aβ42 as well as compact amyloid plaques. In another study, Wang et al. 234 examined in vivo the relationship between MMP-9 protein expression and Aβ plaques by deleting the MMP-9 gene in APP/PS1 mice. In the cortex and hippocampus of these APP/PS1 MMP-9 KO mice, Aβ plaques were larger in size and number compared to APP/PS1 mice with functional MMP-9. Finally, Liao et al. 235 demonstrated that MT1-MMP degrades both soluble and fibrillary Aβ peptide in a time-dependent manner in vitro and this effect is inhibited by the MMP inhibitors GM6001 and TIMP-2. The authors also showed that MT1-MMP degrades brain fibrillary amyloid plaques in another AD mouse model (hAPPSwDI mice) in situ.
These findings suggest an inverse relationship between MMP-2/MMP-9 and Aβ, where one would expect low MMP levels in the AD brain with high Aβ load. However, MMPs are upregulated in the AD brain, which is counterintuitive considering the above-mentioned findings. One explanation for this could be that MMP-mediated Aβ degradation is too low to prevent Aβ brain accumulation. While MMPs might be involved in processing Aβ and clearing plaques, they are likely not major players in this process.219,229
Thus, MMPs are increased in the AD brain, but the role of MMPs in AD is unknown. The current literature is unclear on whether changes in MMP levels contribute to AD progression or might have beneficial effects on the disease. While it is possible that MMPs play no major role in AD, studies show that MMPs could potentially be involved in processing Aβ and AD progression.
Parkinson’s disease
PD is a neurodegenerative movement disorder with more than six million patients worldwide. 236 PD was first described by James Parkinson 237 in 1817, but to this day, many aspects of the disease are unknown. At the molecular level, PD is characterized by accumulation of α-synuclein in dopaminergic neurons, resulting in the formation of Lewy bodies, cell damage, and neuronal death of dopaminergic neurons. PD is also accompanied by neuroinflammation aggravating the disease.238,239
In recent years, MMPs have received some attention in the PD field. Lorenzl et al. 240 determined protein expression and activity levels of MMPs-1, -2, and -9 in postmortem brain tissue from PD patients and age-matched control individuals. While the authors observed no change in MMP-1 and MMP-9, they detected a 50% reduction in MMP-2 activity levels in the substantia nigra of PD patients compared to control individuals. The authors suggested reduced MMP-2 activity could help neurite outgrowth of surviving dopaminergic neurons in the substantia nigra. 240
In addition to MMPs-1, -2, and -9, PD research has focused mainly on MMP-3. Three mechanisms have been suggested on how MMP-3 could be involved in PD. First, using in vitro cell lines and primary cultures of dopaminergic neurons from rat, Choi et al. 241 observed that active MMP-3 is released from apoptotic dopaminergic neurons and that MMP-3 protein levels are higher compared to healthy, non-apoptotic dopaminergic neurons. Using the MTPT mouse PD model, Chung et al. 238 found increased MMP-3 protein and activity levels compared to control mice resulting in apoptosis and cell death. MMP-3 was also involved in caspase-3 activation, specifically in apoptotic signaling upstream of caspase-3 and downstream of c-Jun N-terminal kinases.238,241–244
Second, MMP-3 might potentially be involved in α-synuclein cleavage. Sung et al. 245 showed that MMP-3 cleaves purified α-synuclein in vitro and that α-synuclein aggregation increased in the presence of MMP-3-cleaved α-synuclein fragments compared to a solution without these fragments. Furthermore, aggregates of α-synuclein fragments were more toxic in cell viability assays compared to aggregates of non-fragmented α-synuclein. Sung et al. 245 also demonstrated that MMPs-1, -2, -9, and MT1-MMP cleave purified α-synuclein as well, however, they were less effective than MMP-3. Moreover, Kim et al. 246 showed in microglia cultures and in a 6-OHDA mouse PD model that α-synuclein-induced MT1-MMP expression supports cell migration of reactive microglia into the pathological region, which accelerated PD pathogenesis.
Third, recent data indicate involvement of neuroinflammatory events such as microglial activation, T-leukocyte infiltration, and blood–brain barrier dysfunction in PD.238,247–249 This is consistent with data from Chung et al., 238 who showed infiltration of peripheral immune cells and brain uptake of FITC-albumin (70 kDa) in the MPTP mouse PD model, indicating neuroinflammation and barrier leakage. The authors showed in MMP-3 KO mice that barrier leakage was attenuated and the number of immune cells infiltrating the substantia nigra was decreased, demonstrating MMP-3 involvement in the MPTP mouse model.
In conclusion, MMP-3 seems to be involved in dopaminergic neurodegeneration, neuroinflammation, and barrier leakage in PD. More research will have to clarify the role MMPs play in PD and if MMP inhibition could be a valid therapeutic strategy.
Brain cancer
In 2012, more than 250,000 people were newly diagnosed with brain cancer and nearly 190,000 patients died worldwide.250,251 Survival rates for adult brain cancer patients are low. Even with aggressive therapy, median survival of patients with glioblastoma multiforme, the most common and deadliest malignant brain cancer, is only 12–17 months.252–254 Thus, to effectively treat brain cancer, new approaches and interventions are desperately needed.
MMPs are potential therapeutic targets in cancer because they play a role in cancer biology. In this regard, mRNA and protein overexpression of MMPs-1, -2, -3, -7, -8, -9, -13, and MT1-MMP has been observed in many malignant peripheral and CNS tumors, and a correlation between MMP expression, tumor aggressiveness, tumor stage, and prognosis has been demonstrated.255,256 Indeed, secreted MMPs (MMPs-1, -2, -3, 7-, -8, -9, -10, -11, -13) as well as membrane-bound MT-MMPs are critical for the formation of cancer metastases, their invasion into the brain, and the formation of secondary tumors, and MMPs participate in most of the steps of the metastasis process (Figure 7).84,92
Formation of metastatic cells at the primary tumor: Ii et al.
256
showed in primary tumors that MMP-7 converts the transmembrane cell–cell adhesion protein E-cadherin into a soluble protein resulting in ineffective binding between tumor cells, which allows cancer cells to split from the primary tumor and form metastases that can enter the bloodstream. Intravasation of metastatic cells into the blood circulation: Juncker-Jenson et al.
257
found that MMP-1 participates in the intravasation of metastatic cells from a human HEp3 epidermoid carcinoma graft in chick embryos. The authors showed that MMP-1 regulating endothelial permeability and transendothelial migration supported tumor invasion by activating the endothelial non-tumor/non-matrix receptor PAR1. The authors also used grafts with naturally acquired or experimentally induced MMP-1 deficiency and found that intravasation was decreased by more than 80%.
257
Metastatic cell adhesion to the brain capillary endothelium: it is unclear if MMPs participate in this step of the metastasis process but given the various functions MMPs have it is conceivable. Hummel et al.
258
showed that MMPs-2, -3, -9, and -12 are involved in the shedding of cell adhesion molecules (e.g., vascular cell adhesion molecule-1, intercellular cell adhesion molecule-1) from the plasma membrane of human brain endothelial cell cultures after TNF-α-induced MMP upregulation. MMPs might also contribute to shedding CD44 in metastatic cell adhesion to the capillary endothelium.
259
This is particularly interesting since CD44 is a cell-surface glycoprotein in endothelial cells, leukocytes, and many metastatic cancer cells, where it presents selectins thereby facilitating adhesion of the host cell to the brain capillary endothelium.260–262 Metastatic cell extravasation: MMPs contribute to metastatic cell extravasation and facilitate paracellular transmigration of tumor cells across brain capillary endothelial cells in vitro and the blood–brain barrier in vivo.183,263–265 Lee et al.
264
showed that MMP-2 contributes to the migration of breast cancer cells across the cell monolayer of an in vitro human blood–brain barrier model. Lorger and Felding-Habermann
265
demonstrated in vivo that breast cancer cells injected into the left internal carotid artery of BALB/c mice resulted in brain metastases. For paracellular extravasation, endothelial cell–cell contacts have to be loosened so that metastatic cells can migrate across the endothelium. This requires junction proteins to be degraded. Indeed, MMPs proteolyze tight junction and adherence junction proteins, thereby opening the paracellular route.99,100,255,263 Feng et al.
99
demonstrated in leukemic BALB/c nu/nu mice that leukemic cells secrete MMP-2 and MMP-9, which degraded the tight junction proteins zonula occludens-1, claudin-5, and occludin. Thus, MMPs are critical for leukemic and other cancer cells to cross the capillary endothelium and access the brain. Metastatic cell adhesion to the ECM: several membrane-type MMPs (MT1-, 2-, 3-, 5-MMP) shed the cell-surface glycoprotein CD44, which is critical in metastatic cell adhesion to the luminal endothelial membrane and the ECM on the basolateral side of the endothelium.259,266–268 CD44 is involved in presenting cytokines, chemokines, cell adhesion molecules, growth factors, and other proteins such as MMP-2 and MMP-9 to receptors on metastatic and endothelial cells and mediates signaling that regulates metastatic cell migration and invasion.267,269 CD44 also interacts with ECM proteins like fibronectin, thereby supporting metastatic cell adhesion to the ECM.
270
MMPs in ECM proteolysis: localized opening of the ECM (Figure 7) is necessary for metastatic cells to bypass it and MMP-2 and MMP-9 seem to support this process through ECM proteolysis. Wang et al.
271
compared MMP-2 and MMP-9 levels in human glioma samples with those from control individuals and showed that MMP-2 and MMP-9 levels in gliomas were increased and correlated with the degree of glioma malignancy. The authors also showed that MMP-2 and MMP-9 staining in gliomas was localized to the cytoplasm of tumor cells, endothelial cells, and their ECM. They concluded that by degrading the ECM, MMP-2 and MMP-9 are determining factors of glioma invasiveness and angiogenesis.
271
Other studies in cancer cells showed that CD44 captures MMP-2 and MMP-9 at the tumor cell surface, where MMPs then locally digest the ECM surrounding the tumor cells during extravasation.272,273 Other MMPs might also contribute to ECM proteolysis. While conclusive in vivo proof in support of this is still missing, there are many findings that point in this direction. Shiomi et al.
276
reported increased MT1-MMP mRNA and protein levels in resected glioblastoma tissue compared to non-tumor control tissue. MT1-MMP levels correlated with pro-MMP-2 activation and tumor malignancy and the authors speculated that MMP-2 and MT1-MMP likely contribute to glioma invasion through degrading brain ECM proteoglycans and glia limitans. Nakada et al.
274
observed increased MT1-MMP and MT2-MMP mRNA and protein levels in astrocytomas compared to control brain tissue and concluded that both activate MMP-2, which then degrades the ECM. Other groups detected that MT3-MMP directly cleaves ECM components such as type III collagen, proteoglycan, and interstitial collagens.275,276 These findings are key for cancer metastasis and invasion since they result in ECM digestion.
MMPs in cancer metastasis. MMPs participate in most steps of the cancer metastasis process. (1) Formation of metastatic cells at the primary tumor. (2) Intravasation of metastatic cells into the blood circulation. (3–6) Extravasation of metastatic cells across the blood–brain barrier into the brain. (7) Tumor cell migration in the brain. (8) MMPs contribute to the tumor microenvironment and tumor angiogenesis. Metastatic cell migration: for a secondary tumor to form, cancer cells require space when settling in new tissues. This space is likely generated through MMP-mediated ECM degradation and remodeling. In this context, Belien et al.
277
showed MT1-MMP digesting axonal myelin proteins that inhibit cell migration and neurite outgrowth. Given that invasive glioma cells preferentially migrate along white matter fiber tracts and that MT1-MMP degrades cell migration-inhibiting proteins that are embedded within white matter fiber tracts, these observations suggest that MT1-MMP facilitates cancer cell migration, thereby increasing glioma malignancy. MMPs in the tumor microenvironment and tumor angiogenesis: increasing evidence suggests MMPs establish and maintain a microenvironment that supports tumor survival and growth. MMPs facilitate cancer cell proliferation by regulating cytokines, growth factors, and cell adhesion molecules that attract tumor cells and support metastatic cell spreading.
255
Thus, increased MMP levels are considered to correlate with increased tumor malignancy, and studies report that MMP mRNA, protein, and activity levels are increased in cancer.255,256 Xu et al.
278
observed increased MMP-1 protein levels in gliomas compared to levels in resected brain tissue from epilepsy patients. Levels of MMPs-1, -2, -3, -7, -8, -9, -13, MT1-, 2-, 3-, 5-, and MT6-MMP were also increased in brain tumors compared to non-cancerous brain tissue.253,274,279,280 The same MT-MMPs activate proMMP-2 and proMMP-13, and activity and protein expression levels of those MT-MMPs correlate with pro-MMP-2 activation in gliomas, and thus, with tumor malignancy.256,279,280 MMPs also contribute to angiogenesis, which is critical for the tumor microenvironment because blood vessels supply tumors with oxygen and nutrients, thereby supporting tumor survival, growth, and increasing tumor malignancy.58,110,281 Angiogenesis is based on endothelial cell migration into surrounding connective tissues and MMPs are critical in this process.282,283 MMPs degrade the ECM, release ECM-sequestered pro-angiogenic compounds such as vascular endothelial growth factor, process growth factors, integrins, and adhesion molecules, thereby balancing pro- and anti-angiogenesis.58,110,281 Tumor-induced angiogenesis is important to sustain growth of solid tumors and the functional roles of MMPs in tumor angiogenesis are well established.85,93 For example, MMPs help recruit pericytes, which are localized in tumor blood vessels and are critical for the development of a functional vascular network. MMPs participate in several steps of the pericyte recruitment process. First, MMPs degrade the ECM to allow pericyte invasion. Second, MMPs stimulate pericyte proliferation and protect pericytes from apoptosis. Third, MMPs help recruit bone marrow-derived stem cells, which differentiate into pericytes.
86
Angiogenesis is essential for tumor growth and blocking angiogenesis is considered a valid strategy to control malignant tumors. Thus, MMPs can be beneficial in cancer due to their anti-angiogenic effect that is based on processing growth factors, integrins, and adhesion molecules. For example, tumor angiogenesis is reduced in integrin-α1-null mice compared to wild-type mice.
87
Integrin-α1-null mice overexpress MMP-9, which cleaves angiostatin from plasminogen, and angiostatin inhibits endothelial cell growth, resulting in tumor growth inhibition.
87
MMPs also influence the tumor microenvironment by increasing the permeability of the vascular endothelium in brain tumors, which is then referred to as “blood-tumor barrier”. Thus, the blood-tumor barrier is leaky compared to the healthy, intact blood–brain barrier, which helps supply the tumor with an increasing demand of nutrients.284,285 Noell et al.
286
showed increased MMP-2, -3, and -9 immunoreactivity in brain slices of human primary glioblastomas compared to non-tumor brain tissue and assumed blood–brain barrier leakage in this area. Studies from other groups detected increased proMMP-2 and proMMP-9 levels in the CSF of dogs with intracranial tumors compared to healthy dogs.287,288 Turba et al.
288
attributed increased CSF proMMP-9 levels to the recruitment of leukocytes by the tumors and speculated that MMP-9 likely facilitated leukocytes bypassing the blood–brain barrier in order to reach the tumor, and that those leukocytes secreted MMP-9 into the CSF.
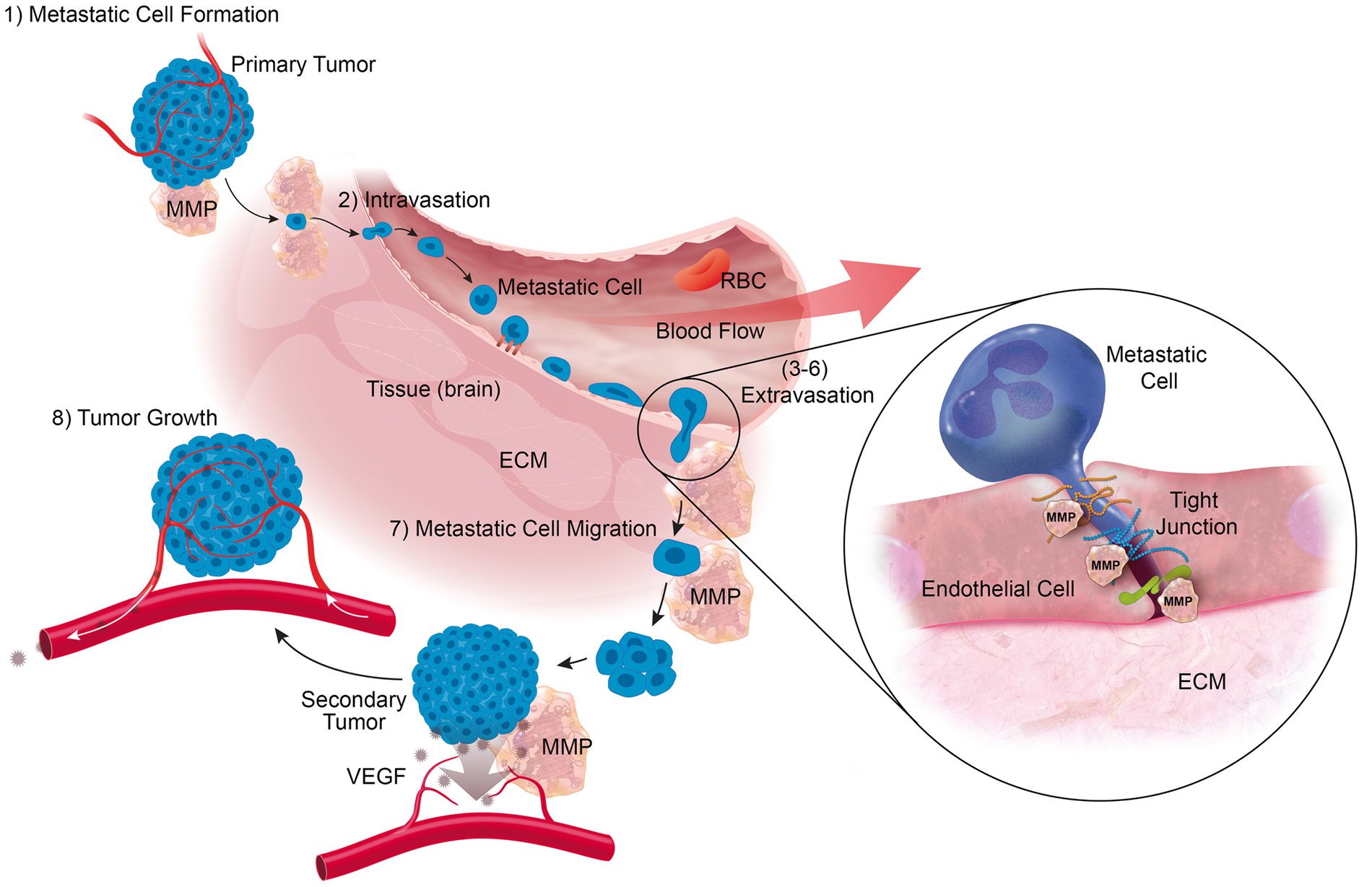
In conclusion, MMPs are critical in many aspects of brain cancer. Their main role lies in facilitating metastasis and angiogenesis, which makes them interesting targets for brain cancer treatment and prevention of secondary brain tumors.
MMP inhibition as a therapeutic strategy
Pharmacological inhibition of MMPs is a potential therapeutic strategy for the treatment of CNS disorders. MMP inhibitors such as marimastat, batimastat, and doxycycline could potentially be used in patients. Currently, however, the only FDA-approved MMP inhibitor is the tetracycline analogue doxycycline (Periostat®) for the treatment of periodontal disease.289–291 The major obstacle hindering the development of MMP inhibitors for clinical use in patients is our lack of knowledge and understanding of the complex MMP biology and the role MMPs play in CNS disorders such as cerebral aneurysm, epilepsy, AD, and PD. Nonetheless, there is a wealth of preclinical data that support MMP inhibition as a treatment strategy in MS, stroke, and brain cancer.
First, a number of MMP inhibitors decrease the incidence and severity of EAE in animal MS models.145,292–295 The MMP inhibitor Ro31-9730 suppressed EAE in rats, 295 and minocycline reduced MMP-9 protein and activity in T cells and suppressed EAE in mice. 292 Second, preclinical data from various mouse and rat cancer models including colon and breast cancer showed that batimastat reduced tumor growth, number, and occurrence of secondary lung and lymphatic metastases.296–299 Third, MMP inhibition with GM6001 or BB-94 in rodent stroke models shortly (hours) after stroke reduced edema, infarct size, and the number of hemorrhagic events.180,185,300,301 Long-term (days) MMP inhibition with the MMP-inhibitor BB-1101 for up to 48 h after stroke in rats reduced barrier leakage, but function in neurologic and behavioral tests did not improve. 302 Inhibiting MMPs with FN-439 or BB-94 in a rat stroke model for one week even aggravated ischemic brain injury and seemed to halt functional recovery. 191
Even though MMP inhibition seems promising in animal models, this has not or only partially been demonstrated in clinical studies. In an MS trial, 16 patients with relapsing-remitting MS were treated with a doxycycline/interferon combination for 4 months (NCT00246324 303 ). Doxycycline/interferon treatment reduced brain lesions, which correlated with reduced MMP-9 serum levels and improved posttreatment EDDS values with only one patient relapsing. Overall, doxycycline/interferon treatment was considered effective, safe, and well-tolerated; it was concluded that a trial with a larger patient cohort should be conducted. However, a report on a follow-up trial has thus far not been published.
In a stroke trial, minocycline was given with/without tPA to 60 patients within 6 h after acute ischemic stroke (NCT00630396304). The mean baseline NIH Stroke Scale score was 8.5 ± 5.8 (moderate stroke). Minocycline did not cause severe hemorrhages in tPA-treated patients, was concluded to be safe and well-tolerated up to 10 mg/kg, i.v. alone and in combination with tPA, and was considered ideal for a tPA combination therapy. In another clinical study, Lampl et al. 305 showed that minocyclin significantly improved patient outcomes. Specifically, NIH Stroke Scale and Rankin Scale scores were significantly lowered, and the Barthel Index was significantly increased. Moreover, participants are currently being recruited for a study testing the safety and efficacy of minocycline in acute cerebral hemorrhage (MACH trial; NCT01805895).
Lastly, several clinical studies testing MMP inhibition in brain cancer have been conducted. In two phase-II trials, a combination of marimastat and temozolomide was tested in recurrent GBM and gliomas.306,307 These trials showed that maristamat/temozolomide appears to increase progression-free survival (PFS): at six months, PFS was 39% for GBM (target PFS: 10%) and 54% for glioma (target PFS: 40%) compared to temozolomide treatment alone. Other brain cancer trials with MMP inhibitors showed no improvement. Prinomastat/temozolomide compared to temozolomide alone did neither improve the one-year survival rate nor PFS (NCT00004200, Pfizer). Likewise, phase I and II trials with the MMP inhibitor COL-3 in recurrent high-grade gliomas do not warrant further studies (NCT00004147308,309). In addition, clinical trials testing more than 50 MMP inhibitors for the treatment of cancer have all failed.310–322 Vandenbrouke and Libert 319 summarized the reasons for failure, which included the complexity of MMP biology and lack of MMP knowledge. The authors also listed suboptimal trial design, inadequate clinical endpoints, use of metabolically unstable MMP inhibitors, poor oral bioavailability, no effect, toxic adverse effects, and discrepancies between pre-clinical animal models and human patients. 319
In summary, while much effort has gone into MMP research over the decades, the field is still far away from a therapeutic breakthrough. Thus, more work remains to be done to test if MMP inhibition is a viable therapeutic strategy.
Conclusion and future perspectives
Here, we review and summarize current knowledge and understanding of the role MMPs play in health and disease in the brain, particularly the blood–brain barrier. While we know that MMPs participate in important neurophysiological functions and have basic understanding of their role in pathological conditions such as neuroinflammation, MS, stroke, and brain cancer, we know little about MMPs in other CNS disorders such as cerebral aneurysms, epilepsy, AD, and PD.
The picture that emerges is complex: a large number of diverse MMPs covers a broad spectrum of different functions in various physiological and pathophysiological processes resulting in a wide range of effects with varying impact. These MMP effects can be both beneficial and detrimental in the same disease depending on location, time point, and other factors. Thus, MMP expression and functional activity vary significantly and are context-dependent. However, one pattern all the diseases mentioned here have in common is neuroinflammation that involves MMPs.
From what we know today, MMP inhibition as an (additional) therapeutic option is problematic and has a history of failure, but is still discussed for the treatment of CNS disorders. Considering the knowledge gap in MMPs, more research needs to be done while avoiding the mistakes of the past. Specifically, research mainly focused on MMP-2 and MMP-9 needs to be expanded to other MMPs to understand the role each MMP plays in health and disease and to gain better insight into MMP biology in general. For example, elucidating the mechanisms responsible for MMP regulation could provide new opportunities for therapeutic intervention. In addition, specific and selective MMP inhibitors that can be used safely with no or only minor adverse effects need to be identified.
Clearly, we are only beginning to understand MMP biology in the larger context of health and disease. In the future, it will be critical to assess if MMPs can be attractive therapeutic targets to advance the treatment of neurodegenerative diseases.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institute of Neurological Disorders and Stroke: NINDS/NIH R01NS079507 (PI: BB, Co-I: AMSH) and the National Institute on Aging: NIA/NIH R01AG039621 (PI: AMSH, Co-I: BB) of the National Institutes of Health.
Acknowledgements
We thank Ms Paula Thomason for editorial assistance and Mr Tom Dolan for graphical assistance. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author’s contributions
RGR and BB wrote the manuscript; AMSH contributed to the content of the manuscript and critically revised the text, tables and figures for scientific content.