
Abstract
Leao’s spreading depression of cortical activity is a propagating silencing of neuronal activity resulting from spreading depolarization (SD). We evaluated the contributions of action potential (AP) failure and adenosine A1 receptor (A1R) activation to the depression of evoked and spontaneous electrocorticographic (ECoG) activity after SD in vivo, in anesthetized mice. We compared depression with SD-induced effects on AP-dependent transmission, and synaptic potentials in the transcallosal and thalamocortical pathways. After SD, APs recovered rapidly, within 1–2 min, as demonstrated by evoked activity in distant projection targets. Evoked corticocortical postsynaptic potentials recovered next, within ∼5 min. Spontaneous ECoG and evoked thalamocortical postsynaptic potentials recovered together, after ∼10–15 min. The duration of ECoG depression was shortened 20% by systemic (10 mg/kg) or focal (30 µM) administration of A1R competitive antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX). ECoG depression was also shortened by focal application of exogenous adenosine deaminase (ADA; 100 U/mL), and conversely, was prolonged 50% by the non-competitive ADA inhibitor deoxycoformycin (DCF; 100 µM). We concluded that while initial depolarization block is brief, adenosine A1R activation, in part, contributes to the persistent secondary phase of Leao’s cortical spreading depression.
Introduction
Leao’s spreading depression of cortical activity 1 is the best-known consequence of spreading depolarization (SD), a wave of neuronal depolarization occurring in injured or excessively stimulated brain tissue.2,3 Leao 1 first described a profound, propagating depression of spontaneous and evoked cortical activity in rabbit cortex that persisted for >5 min after passage of the event wave-front. SD and resulting spreading depression have since been implicated in migraine aura 4 and reported in a wide range of experimental animal studies.3,5,6 More recently, SD has been observed in clinical recordings from strip electrodes placed in patients after stroke or traumatic brain injury, 7 where long-lasting depressions of spontaneous electrocorticographic activity (ECoG) are known to correlate with lesion expansion and adverse outcomes. 8
Compared with our detailed understanding of cellular depolarization during SD, 3 the mechanisms underlying the ensuing depression of activity remain relatively neglected. It is widely appreciated that near-complete depolarized membrane potential during SD initially depresses neurotransmission, by disrupting ion gradients and channels required for the generation and propagation of action potentials (APs).3,7,9 Evidence of depolarization block of APs has been demonstrated during SD in a variety of preparations, including transcallosal, 1 hippocampal, 10 brain stem, 11 cortical island, 12 and pyramidal tract preparations. 13 However, evoked synaptic transmission remains substantially impaired for several additional minutes, after the ability to conduct axonal APs has fully recovered. 14 In clinical studies, depression of spontaneous ECoG can continue for tens of minutes following SD, 8 seemingly much longer than the ∼5 min of synaptic depression reported in animals. Whether this apparent discrepancy is a consequence of species differences, anesthesia, disease process, or perturbations of systemic physiology in ill patients is unknown. It is possible that additional, unknown mechanisms contribute to the depression of spontaneous activity, as compared with evoked activity for the same event within the same subject.
When observed, delayed synaptic recovery has been speculatively attributed to activation of postsynaptic GABAA receptors, 12 dendritic AP failure, 10 and dendritic beading and/or spine retraction,15–17 likely downstream of postsynaptic Ca2+ influx.18,19 However, evidence for these mechanisms is limited to the initial minute or so after passage of the SD wave-front, as ionic gradients recover. Excitatory-inhibitory balance appears to be altered for a longer period, consistent with selective regulation of glutamatergic terminals, rather than a pan-neuronal process. 20 It is clear that secondary synaptic depression is exacerbated if metabolic status is compromised. 6 Thus, mediators related to unmet energetic demand are among possible candidates.
Adenosine is a low-energy metabolite which accumulates following SD in vitro and in vivo, 21 as a consequence of the unusually high metabolic demands of tissue repolarization. Activation of presynaptic G-protein coupled A1 receptors selectively inhibits excitatory transmitter release and suppresses evoked synaptic potentials, by a pertussis- and Ba2+-insensitive mechanism.22–24 We have previously found evidence implicating adenosine in long-lasting suppression of evoked synaptic activity after SD in brain slices 25 and associated with depression of spontaneous ECoG in vivo. 21 Despite correlative evidence, the degree to which adenosine contributes to the spreading depression of spontaneous ECoG in vivo has not yet been explicitly tested.
The present study was designed to directly compare the contributions of depolarization block, synaptic inhibition, and adenosine A1 receptor (A1R) activation to spreading depression of spontaneous cortical activity in vivo. Preliminary findings have appeared in abstract form.26,27
Materials and methods
Ethical approval
All animal procedures were reviewed and approved by the University of New Mexico Health Sciences Center Institutional Animal Care and Use Committee (IACUC; Animal Welfare Assurance #A3350-01, USDA Registration #85-R-0014), in compliance with US Animal Welfare Regulations and Public Health Service Policy on Humane Care and Use of Laboratory Animals. 28
Animals and surgical preparation for in vivo recordings
Experiments were designed, conducted, and reported in accordance with the ARRIVE guidelines. 29 Adult C57Bl/6 J mice (mean ± SEM 10.2 ± 0.7 weeks of age, 23.6 ± 0.7 g, n = 38 males and n = 20 females, Jackson Laboratories, Bar Harbor, ME) were maintained in standard group housing. Under continuous general anesthesia, individual mice were prepared for recording in a stereotaxic frame. Initial observations were made in isoflurane-anesthetized mice (0.9–1.2% delivered by nose cone at 2 L/min, n = 13 males, n = 7 females) as previously described.21,30 As isoflurane itself has CNS-depressant effects, 31 these initial findings were replicated and hypotheses further tested under urethane anesthesia (1.2–1.4 mg/g, i.p., n = 25 males, n = 13 females). Continuous systemic physiology was monitored non-invasively (Physiosuite, Kent Scientific Corporation, Torrington, CT) in urethane-anesthetized mice breathing room air, and values were stable over the course of experiments (pre-experiment heart rate 567.1 ± 2.1 bpm, spO2 86.3 ± 0.2%, post-experiment heart rate 571.1 ± 2.4bpm, spO2 87.0 ± 0.3%, n = 36). Non-invasive monitoring was verified by comparison to blood gas values and intraarterial manometry. Mean arterial blood pressure under these conditions was within the autoregulatory range (76.7 ± 4.4 mmHg, n = 4). After surgical preparation, a stabilization period of ≥60 min was allowed prior to experiments. Each mouse was euthanized by pentobarbital overdose at the conclusion of experiments, without awakening from anesthesia.
Stimulation/recording protocols
For initial experiments in isoflurane-anesthetized mice, burr holes (1–2 mm diameter, dura intact) were drilled bilaterally over the motor cortex (in mm, from Bregma: AP 2.0, ML ± 1.75) for continuous recording of direct current (DC) potential, ECoG, and transcallosally evoked excitatory postsynaptic potentials (EPSPs), and posteriorly (−2.0, ±2.0) for induction of SD by pin-prick with 25 G needle. As in our previous work, the depth of isoflurane anesthesia was titrated for breathing 2/s and absence of foot-pinch reflexes, and spontaneous ECoG recorded from these mice was characterized by a burst-suppression pattern of activity. 21 To evoke transcallosal postsynaptic potentials on the right hemisphere, we stimulated the left hemisphere with a parallel platinum bipolar stimulating electrode (FHC Inc, Bowdoin ME, 0.5 mm pole separation, 0.5 mm length, paired pulses, 50 ms inter-stimulus interval, 0.1 Hz, 70 µs, 3–5 mA). The stimulation intensity was selected to maximize transcallosal responses, and likely activated a large volume (rather than a single column) of the cortex. After a stable baseline was recorded, SD was induced in alternate hemispheres, at 20-min intervals (side of initial SD alternated by mouse) to characterize baseline rates of recovery, reported in the results section “Brief initial depolarization block of axonal APs, and longer lasting synaptic depression”. Then, in the same animals, 8-Cyclopentyl-1,3-dipropylxanthine (DPCPX, 10 mg/kg) or vehicle (DMSO 10% in normal saline, ≤0.3 mL total volume) was injected and after 20 min another pair of SDs was induced, 20 min apart. Post-treatment data are reported in the results section “Systemic A1 receptor antagonism”.
Subsequent experiments were conducted under urethane anesthesia, to eliminate the possible confound of burst suppression activity and to optimize the signal-to-noise ratio (SNR) for somatosensory stimulation. 32 Burr holes were drilled bilaterally over the barrel cortex (−0.5, ±3.0) for continuous recording of DC potential, ECoG, and transcallosal- and whisker pad-evoked potentials, and posteriorly (−2.0, ±2.0) for induction of SD by pin-prick. Again, a parallel bipolar stimulating electrode (FHC Inc, Bowdoin ME, 0.5 mm pole separation, 0.5 mm length) was placed on the left (−0.5, −3.0), to evoke transcallosal EPSPs on the right side (−0.5, +3.0). In addition, platinum subdermal needle electrodes (Type E2, Grass Instrument Company, Quincy, MA) were placed in the left whisker pad and stimulated (0.1 Hz, off-set 5 s from transcallosal stimulation, 70 µs, 3–10 mA) to evoke thalamocortical EPSPs in the right barrel cortex. Thalamocortical- and corticocortical-evoked potentials were readily identified time-locked to the relevant stimulus with appropriate synaptic delays (10–20 ms to peak following stimulation of left whisker pad, 5–15 ms following stimulation the left barrel cortex). SD was induced in alternate hemispheres, and the recovery interval between SDs was increased to 60 min to allow for complete recovery of whisker pad-evoked potentials (≥2 h between serial SDs on same hemisphere). Data from this group are presented in the results section “Spontaneous ECoG and thalamocortical synaptic activity recover later than corticocortical synaptic activity”.
Pharmacological experiments were conducted in urethane-anesthetized mice, using contralateral hemispheres to compare vehicle and drug exposures (within-animal design). A power analysis based on initial findings in untreated animals yielded a sample size of 7, to detect an arbitrary 33% change in the duration of depression of spontaneous ECoG. Mice were prepared for bilateral whisker pad stimulation (alternate stimulation, 0.1 Hz, off-set 5 s) and bilateral recording of ECoG and thalamocortical-evoked potentials in the barrel cortex (−0.5, ±3 mm). Drug or vehicle was pre-loaded into blunt recording electrodes (pulled on P-89 (Sutter Instruments, Novato, CA), then broken on a MF-83 microforge (Narishige, Tokyo, Japan) to 10–20 µm tip diameters, matched to ±2 µm within experiments) and delivered focally by gentle positive pressure (0–35 mmHg) applied through the microelectrode holders (MEH2SW15, World Precision Instruments, Sarasota, FL). Pressure ramps were controlled by a pipette perfusion pressure/vacuum generator (2PK+, ALA Scientific Instruments, New York), and the duration and level of each pressure step was matched to the control hemisphere. The hemisphere assigned to drug or vehicle treatment was alternated by animal and was assigned prior to the start of recording. The initial SD was always induced on the left, maintaining ≥1 h interval between SDs and alternating sides such that ≥2 h were maintained between serial SDs in the same hemisphere. These results are reported in the results section “ECoG depression modified by adenosine pharmacology”.
Electrophysiology
Glass micropipette recording pipettes (filled with normal saline or phosphate buffered saline) were placed at controlled depths (300–500 µm or 150–200 µm, as noted in text) under micrometer control. Signals were amplified (Neuroprobe 1600, AM Systems, Sequim, WA or Axon Axopatch, Molecular Devices, Sunnyvale, CA), low-pass filtered, A/D-converted (Power Lab 8/35 or 16/30, AD Instruments, Dunedin, New Zealand), and sampled at 1 kHz (for DC recordings), or were further amplified (100× gain), and sampled at 20 kHz (for ECoG and EPSP recordings). Spontaneous ECoG was band-pass filtered (0.5–40 Hz), and power was calculated during inter-stimulus intervals (1 s bins). Electrical and physiological signals were recorded simultaneously in LabChart 7.3.7 with Kent Device Enabler (AD Instruments, Dunedin, New Zealand). Durations of DC potential shifts were measured as the width at 80% recovery, and depressions of ECoG and EPSPs were measured as the time to 50% recovery, as previously described.21,25
Drugs and solutions
Urethane (Sigma-Aldrich, St. Louis, MO) was prepared as a 10% (w/v) solution in normal saline (0.9% NaCl) and injected intraperitoneally. Isoflurane was obtained from Clipper Distributing Co (St. Joseph, MO). DPCPX (Sigma-Aldrich) was prepared as a stock solution (30 mM in DMSO) and diluted 1:1000 in normal saline. Deoxycoformycin (Tocris Bioscience, Ellisville, MO) was prepared as a stock solution (100 mM in normal saline) and diluted 1:1000 in normal saline. Lyophilized adenosine deaminase (ADA) from bovine spleen (lot X4C14821, Worthington Biochemical, Lakewood, NJ) was reconstituted in phosphate-buffered saline (in mM, NaCl 137, KCl 2.7, Na2HPO4 10, KH2PO4 1.8 (pH 7.4)) and used at 100 U/mL.
Statistics
Data are presented as mean ± SEM. The unit of analysis (n) is noted throughout, as the number of SDs and the number of animals. Blinding was not done, and to minimize potential sources of bias (order or time effects, equipment changes or other sources of experimental drift), all experiments were conducted with interleaved controls throughout.
Statistical tests are specified in the text and figure legends, and included analysis of variance (ANOVA), repeated measures (RM)-ANOVA, paired and unpaired Student’s t-tests, with Bonferroni correction for multiple comparisons. In pharmacological experiments, the specified outcome measure was relative change from vehicle control (Veh), thus ratio paired t-tests were calculated. The distribution of each dataset was checked for normality (D’Agostino and Pearson omnibus normality test), and parametric tests were used throughout. No outliers were excluded. Rarely, mice died during surgical preparation, prior to the start of experiments, and did not contribute data. We used the statistical package Prism 6.05 (GraphPad, La Jolla, CA).
Results
The aim of this study was to evaluate mechanisms underlying spreading depression of spontaneous ECoG activity, a complex signal composed of axonal APs, postsynaptic potentials, and reverberating activity within and between cortical and subcortical circuits. We began by characterizing the effects of SD on each of these components, and the sequence of their recovery within individual animals.
Brief initial depolarization block of axonal APs, and longer lasting synaptic depression
EPSP failure in distant projection targets, indicating depolarization block
We first evaluated depolarization block in the transcallosal pathway (a preparation similar to Leao),
1
comparing its duration to spontaneous ECoG depression accompanying SD (Figure 1). We reasoned that when SD invaded somata and proximal segments of projection fibers, AP-dependent activity in distant projection targets should be interrupted. Thus, we could use evoked transcallosal EPSP responses in the right hemisphere as a surrogate for AP generation and transmission along fibers originating in the left hemisphere. We electrically stimulated the left motor cortex (2.0–1.75), to elicit EPSPs in the right motor cortex, then induced left-sided SD by pin-prick (Figure 1(a)).
Brief depolarization block of action potentials compared with longer lasting synaptic depression following SD. (a) Schematic configuration of stimulation and recording electrodes to isolate depolarization block from other mechanisms of synaptic depression. EPSPs were elicited by bipolar stimulation (0.1 Hz) in the left motor cortex and recorded in the homotopic right hemisphere. SD (indicated by stippled shading) was induced on the left in order to disrupt action potential (AP) generation/propagation along fibers originating in the left hemisphere. (b) SD involving the left hemisphere was confirmed by DC shift (top panel). AP-dependent output, assayed by evoked transcallosal EPSPs on the right, was transiently abolished during the DC shift, and recovered with a monophasic time course over 1–1.5 min (middle panel). Spontaneous ECoG (bottom panel) on the left was strikingly depressed for an interval of 6–7 min after AP conduction had fully recovered. Average ECoG power is displayed, in 1 s bins. Discontinuities in the data are the result of removing the 0.1 Hz stimulus artifact. (c) SD was then induced in the right hemicortex of the same animal (indicated by stippled shading), in order to evaluate local effects on synaptic elements in addition to axonal APs. (d) The DC shift, confirming SD, lasted ∼1 min (top panel). EPSPs were interrupted during SD and recovered more slowly than in panel B (∼2–3 min). The gray vertical bars in B&D indicate the duration of EPSP depression <50% baseline. Thus, EPSPs were disrupted appreciably longer when SD involved all synaptic elements, than when SD involved only axons. (e) Summary data from studies illustrated above (n = 13). The duration of axonal conduction block was brief (<1 min), whereas synaptic inhibition was significantly longer lasting (>2 min). However, both were substantially shorter than the depression of spontaneous ECoG (∼12 min). Recovery time to 50% baseline is shown for each component. RM-ANOVA p < 0.0001, Bonferroni-corrected t-tests ***p < 0.001, ****p < 0.0001, n = 13 animals.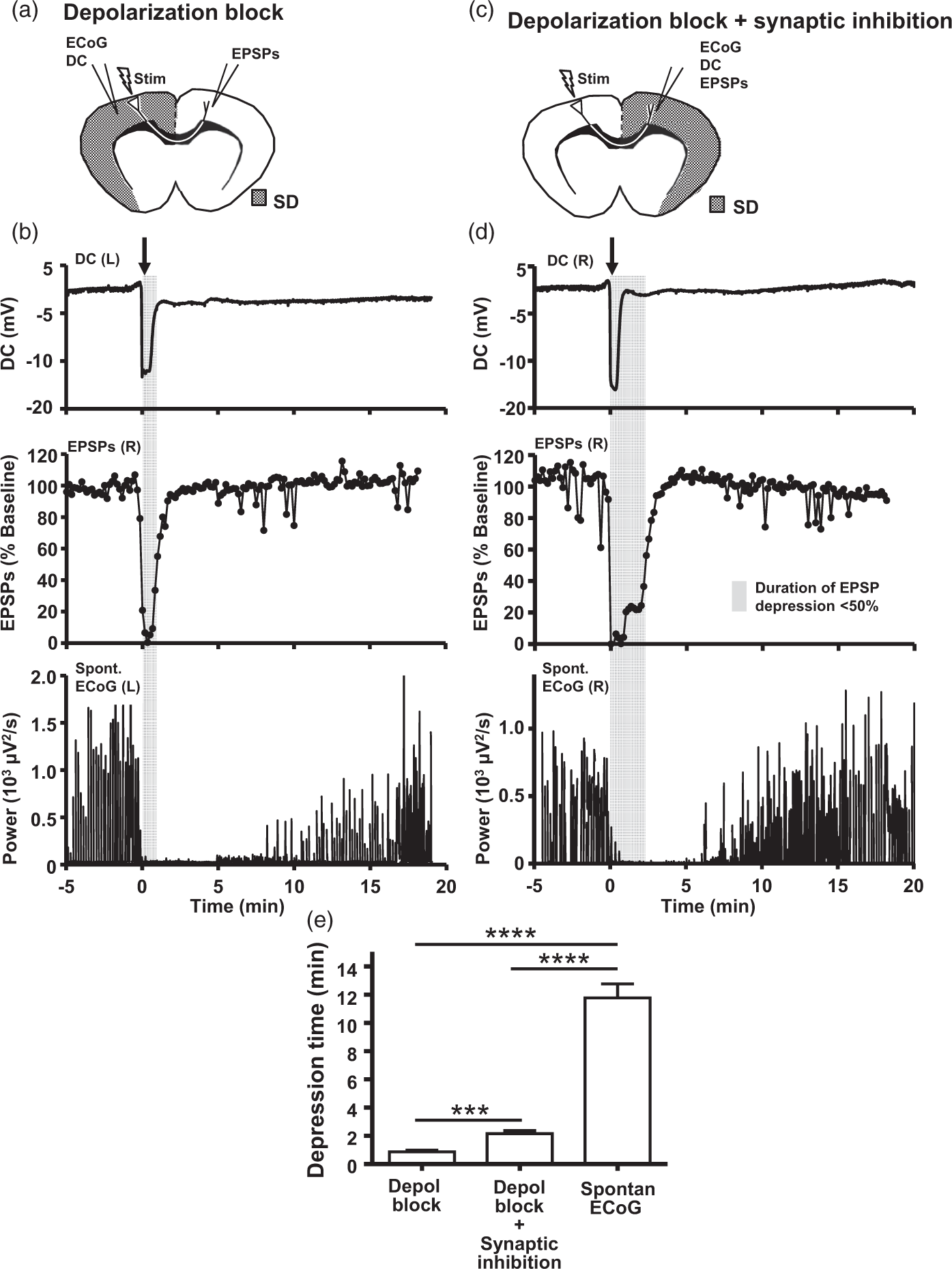
As expected, SD transiently interrupted AP-dependent output from the depolarized cortex. A typical time course of responses from a representative animal is shown in Figure 1(b). SD in the left hemisphere was confirmed by a negative shift in the extracellular DC potential, lasting 1–2 min (top panel). During the DC shift, AP-dependent output (to the remote, non-depolarized right hemisphere) was transiently interrupted (middle panel). This coincided with the cessation of spontaneous ECoG activity on the left, as expected (bottom panel). 33 Evoked activity in the distant target cortex then recovered promptly and monophasically, indicating the resumption of APs, also within 1–2 min. By contrast, spontaneous ECoG remained markedly depressed in the wake of SD for ∼8 min, similar to prior reports. 33 Thus, ECoG depression persisted for many minutes after neurons in the region had recovered the capacity to generate and conduct APs.
Prolonged synaptic depression
In the same preparations, we next quantified the local effects of SD on evoked synaptic transmission. Still stimulating the left motor cortex to evoke transcallosal EPSPs on the right, we induced SD in the right hemisphere (Figure 1(c)). In this configuration, pre- and postsynaptic elements (distal axons, presynaptic terminals, postsynaptic dendrites, and somata) participated directly in the SD event and were exposed to local diffusible factors accumulating in its aftermath. Thus, transcallosally evoked EPSPs served to report synaptic inhibition in addition to depolarization block. Figure 1(d) shows data from the same animal as Figure 1(b), this time in response to right-sided SD. Again as expected, SD abolished evoked and spontaneous activity (middle and bottom panels, respectively), simultaneous with the arrival of the DC shift (top panel). However, when SD occurred locally, synaptic responses recovered from SD significantly more slowly than axonal conduction, as can be appreciated from comparing the width of the shaded areas in Figure 1(b) vs. Figure 1(d). This implied that additional mechanism(s) in addition to AP failure contributed to synaptic depression following SD.
Spontaneous ECoG remained depressed despite relief of depolarization block and recovery of transcallosally evoked synaptic potentials
We had anticipated that APs and synaptic potentials were the essential elements required to permit recovery of spontaneous ECoG, and thus that ECoG would recover together with or shortly following evoked synaptic potentials. Instead, the duration of ECoG depression substantially outlasted both depolarization block (Figure 1(b)) and synaptic inhibition (Figure 1(d)). This finding was highly reproducible: in 13 animals depolarization block lasted 51.5 ± 7.2 s, whereas the additional effect of synaptic depression increased this to 128.5 ± 12.7 s, and spontaneous ECoG was depressed for 703.5 ± 59.1 s (Figure 1(e), RM-ANOVA p < 0.0001, Bonferroni-corrected t-tests p < 0.001, p < 0.0001, p < 0.0001, n =26 SDs (one each side) from 13 animals). Full restoration of both axonal APs and postsynaptic responses, at least in the transcallosal pathway, was not sufficient for recovery of spontaneous ECoG. This discrepancy was not attributable to either use of isoflurane anesthesia or recording position in the motor cortex, as we replicated our findings in urethane-anesthetized mice, and extended them to a sensory brain region (see below).
Presynaptic mechanisms contribute to synaptic depression
We next evaluated for evidence of presynaptic regulation of transmitter release after SD in vivo. We took advantage of the monosynaptic anatomy of the transcallosal EPSP, delivering paired-pulse stimulation to evaluate changes in the presynaptic release probability. Figure 2 illustrates changes in paired-pulse ratio (PPR) over the time course of depression of transcallosal EPSPs. Figure 2(a) plots EPSP depression and recovery for a representative passage of SD on the right, (with stimulation and recording electrodes configured as illustrated in Figure 1(c)). Figure 2(c) plots PPR for the same experiment, on the same time course. At baseline, we observed a slight paired-pulse facilitation of the S2 response (Figure 2(b), trace i), as is typical for this synapse.
34
During the DC shift indicating SD, no EPSPs could be detected, due to depolarization block of APs, and thus no PPR could be calculated (Figure 2(b), trace ii). However, immediately following SD, once APs had resumed, S1 and S2 responses were both depressed but facilitation of S2 was increased (Figure 2(b) trace iii), indicating a decreased probability of glutamate release from presynaptic terminals during this interval. The increase in PPR was highly reproducible during the period of synaptic depression (p = 0.018, paired t-test, n = 17 SDs from 17 animals, Figure 3(d)). These data suggest that, in vivo as in brain slices,
25
depression of evoked responses involves a presynaptic locus of regulation.
Paired-pulse ratio was elevated during depression of evoked synaptic activity. Presynaptic release probability was assessed by paired-pulse stimulation (50 ms inter-stimulus interval). (a–c) Results from a representative experiment, with recording configuration as in Figure 1(c). (a) Plot of normalized EPSP amplitude. Following SD, synaptic responses underwent depression and recovery (as in Figure 1(d), middle panel). (b) Responses to pairs of stimuli, at the time points indicated in (a), demonstrating modest paired pulse facilitation at baseline (i), absence of responses consistent with axonal conduction block during SD (ii), synaptic depression after SD (iii), and full recovery (iv). (c) Plot of paired-pulse ratios (S2/S1), from the same recording and on the same time scale as A. While both S1 and S2 responses were depressed in response to SD, the ratio of S2/S1 was increased, consistent with presynaptic inhibition of transmitter release. (d) Summary data from 17 such experiments. Paired t-test, p = 0.01. Spontaneous ECoG and thalamocortical-evoked potentials recovered together, later than corticocortical-evoked potentials. Representative experiment in a urethane-anesthetized mouse, demonstrating local effects of SD on spontaneous ECoG, whisker pad-evoked thalamocortical potentials and transcallosal-evoked corticocortical potentials. All recordings were from a single recording electrode in the barrel cortex. (a) Schematic of experiment (top) and representative responses (bottom) SD was verified by DC potential shift measured from the recording electrode (not shown). Corticocortical EPSPs (open circles) were depressed following SD and recovered within ∼5 min, as previously described for isoflurane-anesthetized animals (Figure 1(d) and Figure 2). Thalamocortical EPSPs were depressed following SD and recovered more slowly over tens of minutes (filled circles). Spontaneous ECoG recovered over tens of minutes (total power, expressed as % baseline, plotted by solid black line), similar to evoked thalamocortical activity (Pearson R2 = 0.56, p < 0.0001). (b) Summary data from 14 SDs from five urethane-anesthetized animals. RM-ANOVA p < 0.0001, Bonferroni-corrected comparisons vs. thalamocortical EPSPs *p < 0.05, ***p < 0.001. (c) Scatter plot indicating the relationship between depression of ECoG and whisker pad-evoked activity, measured as time to 50% recovery for each passage of SD in these subjects (R2 = 0.35, p = 0.0034, n = 22 SDs from five animals, including eight SDs during which transcallosal potentials were not elicited).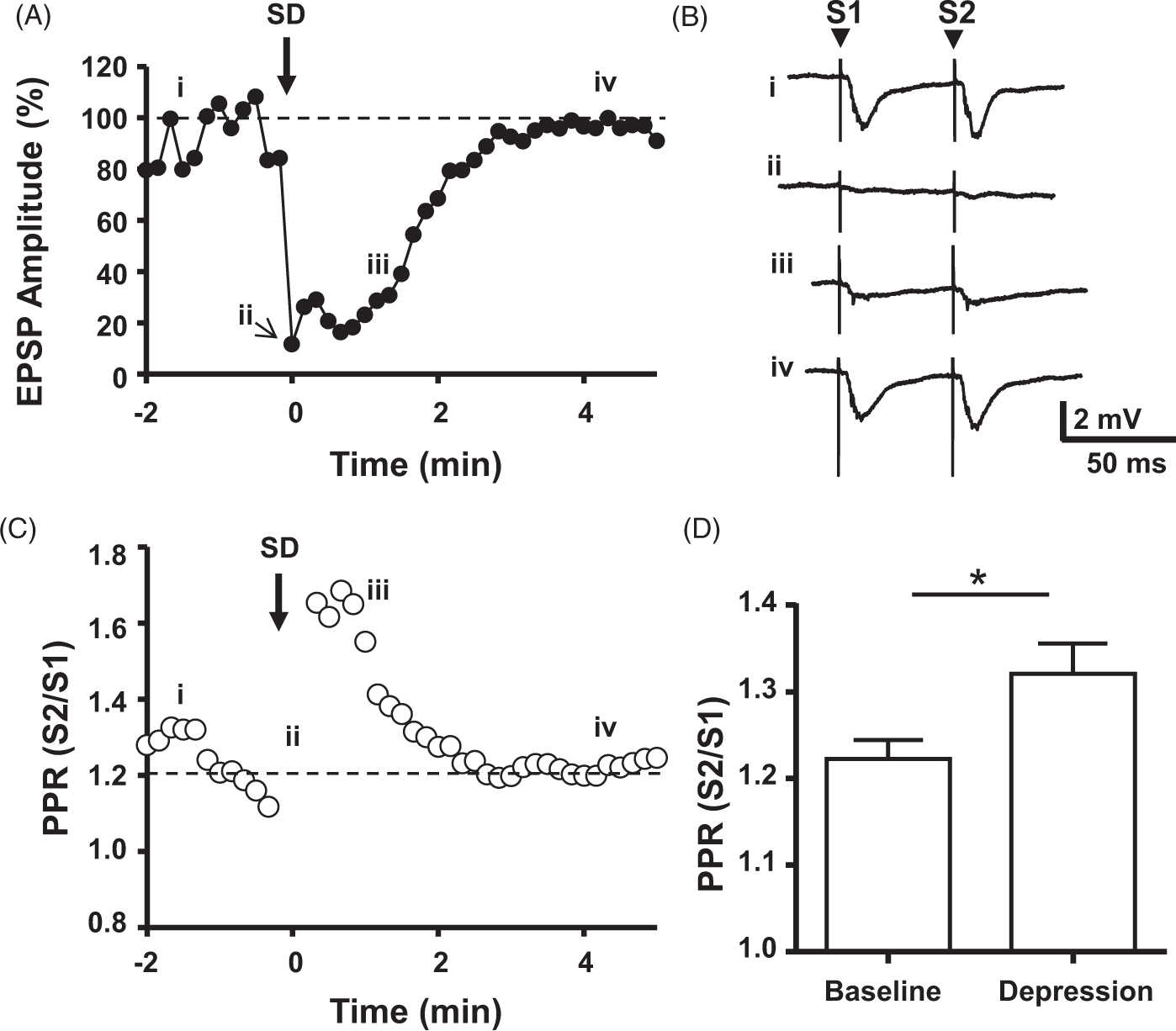
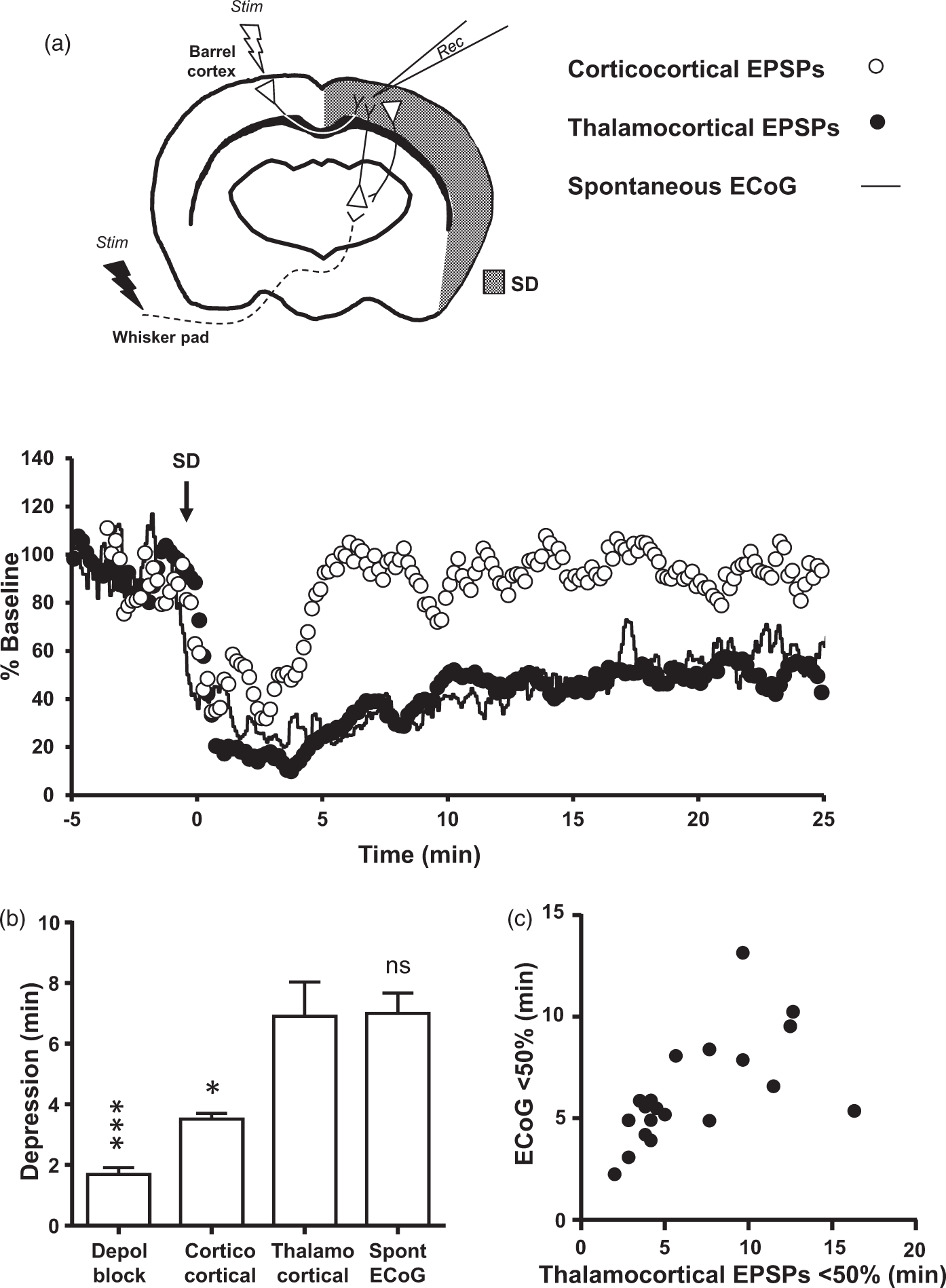
Spontaneous ECoG and thalamocortical synaptic activity recover later than corticocortical synaptic activity
Differential depression of somatosensory vs. corticocortical inputs
We considered two possible explanations for the early recovery of transcallosal evoked activity relative to spontaneous ECoG (Figure 1). One possibility is that by measuring evoked corticocortical synaptic activity, we underestimated depression of thalamocortical synapses, which make the predominant contribution to ECoG in anesthetized animals. 35 Alternatively, we considered that the discrepancy could reflect important differences between spontaneous and evoked activity. To distinguish between these two models, we conducted experiments comparing transcallosal-evoked EPSPs and spontaneous ECoG to whisker pad-evoked EPSPs (Figure 3). All responses were recorded simultaneously via a single extracellular recording pipette placed in the right barrel cortex (−0.5, ±3.0) (Figure 3(a), upper panel). If the cortex had recovered and was uniformly receptive, we expected to see both types of evoked activity recover together, earlier than spontaneous ECoG. If, however, the thalamocortical synapses within the cortex were selectively depressed, we expected to see spontaneous ECoG and thalamocortical-evoked potentials recover together, later than corticocortical potentials.
Figure 3(a) shows a representative experiment during a single passage of SD (lower panel). As expected, with the arrival of SD, spontaneous ECoG and both types of EPSPs were depressed. Corticocortical-evoked potentials (open circles) recovered relatively early, in this example within ∼5 min. By contrast, spontaneous ECoG in this animal (solid line) recovered slowly over tens of minutes, first attaining 50% of baseline at ∼13 min, and 95% of baseline at ∼30 min. Thalamocortical EPSPs (filled circles) recovered on a time course closely matching ECoG recovery. These findings were consistent across five animals, with corticocortical-evoked EPSPs recovering rapidly, as compared with whisker pad-evoked EPSPs and spontaneous ECoG (Figure 3(b)). In 22 SDs from 5 animals, the depression of thalamocortical-evoked potentials and ECoG was positively correlated (Figure 3(c), R2 = 0.35, p = 0.0034). In another animal, we stimulated the left forelimb and measured in the corresponding somatosensory cortex (+0.5, +2.25). Again, we observed brief depolarization block similar to DC shift duration (76.7 ± 10.8 s, 71.6 ± 10.8 s, n = 3 SDs from one animal), longer lasting synaptic block in corticocortical pathways (176.6 ± 14.7 s) and persistent thalamocortical depression (383.25 ± 176.8 s) similar to ECoG (496.67 ± 129.7 s).
We considered the possibility that the discrepancy could be due to proximity effects, i.e., that we were biased toward detecting depression of thalamocortical contributions to ECoG due to recording from a position near layer IV neurons receiving thalamocortical input (400 µm). Therefore, we conducted control experiments recording from a position closer to corticocortical inputs (150–200 µm deep (n = 2 mice). Again, we observed the same sequence of recovery. First, APs recovered, then corticocortical synaptic inputs, then thalamocortical synaptic inputs together with ECoG. Together, these results were consistent with a model in which ECoG depression is predominantly a manifestation of depression in the thalamocortical loop, at the level of the cortex.
ECoG depression modified by adenosine pharmacology
Finally, we evaluated the effects of adenosine pharmacology on the duration of depression of spontaneous ECoG, using both systemic and focal approaches to deliver pharmacologic agents (Figure 4).
Endogenous adenosine accumulation and A1R activation within the cortex contributes to ECoG depression. (a) Systemic administration of drug resulted in decreased duration of ECoG depression in mice treated with DPCPX (black bar) but not those treated with vehicle (white bar). Unpaired t-test, p < 0.05. (b) Schematic illustrating experimental setup to evaluate focal drug delivery. Bilateral continuous recordings of ECoG and evoked thalamocortical EPSPs were made in the barrel cortices of urethane-anesthetized mice undergoing alternating 0.1 Hz stimulation of each whisker pad (electric bolts). Vehicle and drug were focally delivered through the recording electrodes by pressure ejection (see Methods section). (c) Schematic illustrating the targets of drugs ejected into the recording region. Deoxycoformycin (DCF) is a non-competitive antagonist of adenosine deaminase, and was expected to increase the half-life and prolong the activity of endogenous adenosine accumulating in the extracellular space after SD. DPCPX is a competitive A1R-selective antagonist and was expected to decrease the action of adenosine at presynaptic receptors. (d, e) Representative ECoG responses to SD induced alternately in vehicle- and drug-treated hemispheres of the same animal. Depression was briefer in the DPCPX-treated hemisphere compared to within-animal vehicle control (Veh). Conversely, depression was prolonged in the DCF-treated hemisphere, relative to within-animal vehicle control. (f, g) Summary data of depression of spontaneous and whisker pad-evoked activity from hemispheres treated with DPCPX, DCF, or ADA, compared to control hemisphere within the same animals. *p < 0.05, **p < 0.01, Bonferroni-corrected ratio paired t-tests.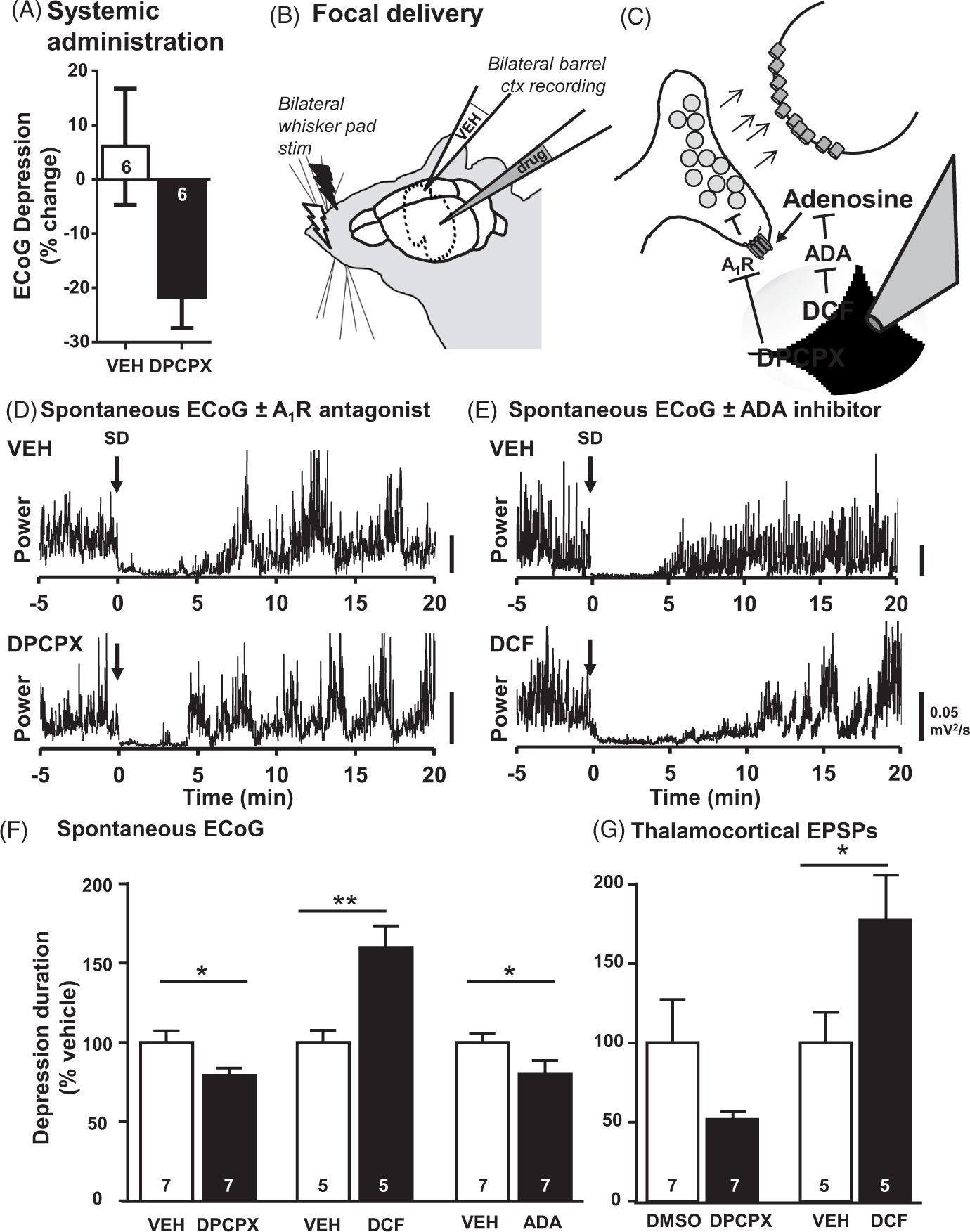
Systemic A 1 receptor antagonism
We first administered either the A1R antagonist DPCPX (10 mg/kg, n = 6) or vehicle (n = 6), by intraperitoneal injection in isoflurane-anesthetized mice (baseline characteristics described above, Section 1). DPCPX treatment significantly shortened ECoG depressions, from 11.84 ± 1.64 min to 8.95 ± 0.81 min (p = 0.0258, ratio paired t-test, n = 6 SDs in six animals). By contrast, depressions were stable in vehicle-treated animals (mean depression pre-injection 10.42 ± 1.67 min vs. post-injection 10.04 ±1.31 min, p = 0.8688, ratio paired t-test, n = 6 SDs in six animals). The relative change in depression time from baseline values in the same animals was significantly different in DPCPX- vs. vehicle-treated animals (Figure 4(a), −21.6 ± 6.37% vs. 6.0 ± 11.74%, p = 0.047, unpaired t-test), indicating that systemic administration of an A1R antagonist can reduce spreading depression in vivo. In addition, systemic A1R antagonist treatment tended to mitigate PPR increases after SD, measured at corticocortical synapses (PPR 1.45 ± 0.19 vs. 1.70 ± 0.35, DPCPX vs. vehicle, n = 6 per group), but this effect was not significant with this group size (p = 0.52).
Focal A 1 R antagonism
The simplest explanation for these results was that DPCPX accelerated recovery of ECoG by counteracting the effects of adenosine accumulation within the cortex. However, systemic DPCPX may have also acted on other elements of the thalamocortical circuit, or altered systemic physiology, for example, by elevating cardiac output and thus improving cerebral perfusion. Therefore, we further tested our hypothesis by focally applying DPCPX (30 µM) or vehicle to the cortex, through the recording electrodes (Figure 4(b) and (c)). For these experiments, the contralateral hemisphere served as a within-animal control (n = 7 animals, urethane anesthetized, with alternating whisker pad stimulation). As hypothesized, in the DPCPX-treated hemisphere of each mouse, depression of spontaneous ECoG activity was significantly shorter than the vehicle-control hemisphere (Figure 4(d) and (f), p = 0.013, paired t-test). DPCPX treatment had no significant effects on DC shift durations (47.5 ± 6.1 vs. 55.9 ± 9.7 s, DPCPX vs. Veh, p = 0.26 paired t-test) or baseline ECoG activity (1.0 ± 0.6 vs. 1.0 ± 0.6 ×10−6 µV2/s, p = 0.90, paired t-test). Consistent with the drug effect on ECoG depression, the depression of whisker pad-evoked thalamocortical activity also tended to be shorter in DPCPX-treated hemispheres (Figure 4(g)), although this did not reach significance (p = 0.08, paired t-test), largely due to poor SNR of EPSPs at baseline. Taking an average for each animal, DPCPX treatment was associated with a 20.6 ± 5.2% reduction in spontaneous ECoG depression time, relative to the vehicle-treated hemisphere (Figure 4(e), n = 7 animals).
Manipulation of adenosine deaminase (ADA)
Next, we focally applied the non-competitive ADA inhibitor deoxycoformycin (DCF, 100 µM) in order to increase the bioavailability of endogenous adenosine and prolong its effects. 36 Consistent with the hypothesis, DCF application prolonged ECoG depressions (Figure 4(e) and (f), p = 0.0006) and whisker pad-evoked potentials (Figure 4(g), p = 0.02), without altering DC shift durations (85.3 ± 25.6 vs. 59.4 ± 6.9 s, DCF vs. Veh, p = 0.22, n = 11 SDs from five animals). DCF slightly depressed baseline ECoG power (1.7 ± 0.4 vs. 2.6 ± 0.9 × 10−7 µV2/s, p = 0.04, n = 5 animals), and recovery times reported here were calculated from the baseline immediately prior to SD. Taking the average for each animal, DCF treatment was associated with a 53.4 ± 14.0% increase in ECoG depression time following SD (n = 5 animals).
Conversely, we focally applied exogenous ADA to accelerate degradation of adenosine and reduce the duration of its effects. 36 Consistent with the hypothesis, ADA (100 U/mL) decreased ECoG depressions (Figure 4(e), 5.74 ± 0.93 vs. 4.32 ± 0.45 min, PBS vs. ADA, p = 0.017, n = 14 SDs from seven animals), without altering the initial power (2.4 ± 0.8 vs. 2.5 ± 0.6 × 10−7 µV2/s, p = 0.79) or DC shift durations (65.6 ± 9.1 vs. 79.8 ± 22.0 s, Veh vs. ADA, p = 0.59); whisker pad responses were not collected in these animals. Normalized to within-animal vehicle controls, ADA treatment decreased the durations of ECoG depressions by 20.1 ± 7.1% (n = 7 animals).
Discussion
The results here identify a role for adenosine as a locally diffusible factor contributing to Leao’s spreading depression of cortical activity, in vivo. A quantitative comparison of the time courses of depolarization block, suppression of different types of evoked EPSPs, and ECoG depression following SD is also provided (Figure 5). These studies yielded the unexpected finding that somatosensory-evoked potentials were more sensitive to disruption than corticocortical-evoked potentials, which might predict subtle constellations of symptoms or functional deficits at different time points after SD. Understanding these mechanisms of ECoG depression will likely inform interpretation of clinical recordings of SD and its consequences.8,37
Schematic model illustrating mechanisms of ECoG depression. (a) Schematic summarizing putative alterations in synaptic physiology as a result of SD. SD impairs neurotransmission directly, through depolarization block of sodium channels conducting action potentials, as well as distortion of ion gradients required for pre- and postsynaptic transmembrane currents. Following SD, synaptic transmission is inhibited, at least in part due to reduced transmitter release (see PPR results in Figure 3), likely representing feedback regulation by adenosine resulting from metabolic depletion.
21
(b) Model of sequence and relative contributions of putative mechanisms. Early and absolute depression of activity by depolarization block is transient, and approximated by the DC shift duration. This period also likely encompasses phenomena such as prodromal increases in GABA
50
and dendritic beading and spine loss observed in conjunction with DC shifts.16,17 Longer lasting secondary depression of excitatory activity is mediated, at least in part, by adenosine and possibly other metabolic factors such as KATP channel activation and pH-dependent signaling. The degree and duration of this secondary depression likely varies by region and by circuit-specific sensitivity to adenosine. Under metabolic compromise such as in stroke, hypoglycemia, or hypoxia (indicated by dashed arrows), the duration of depolarization is prolonged, and the accumulation of adenosine and depression is increased.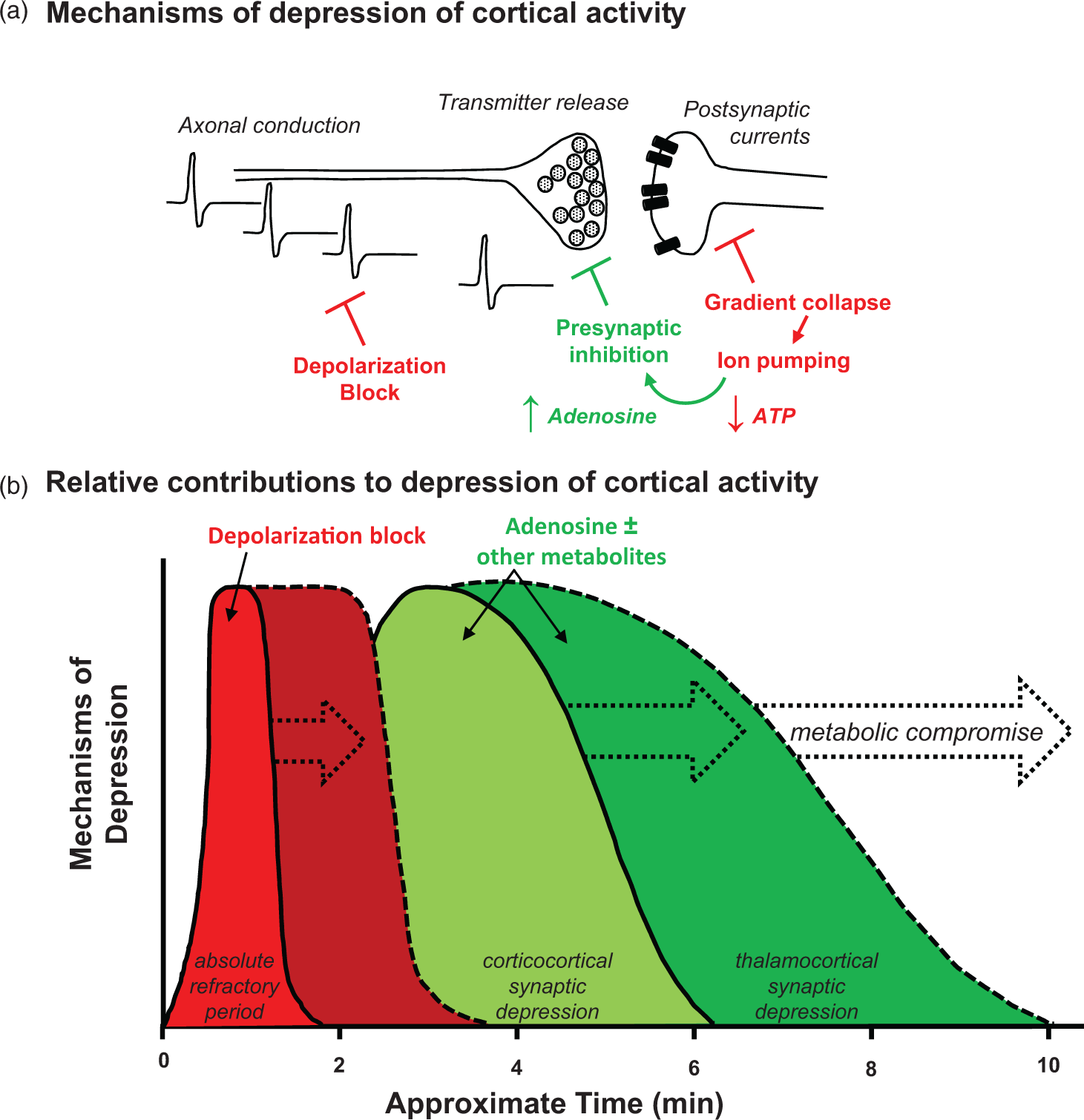
Depolarization block is brief compared with synaptic depression
Leao first recognized that tissue undergoing SD was refractory to electrical stimulation, 1 anticipating our modern molecular understanding of APs by nearly a decade. 38 Subsequent investigators identified depolarization block during SD, evaluating antidromic spikes in hippocampal 10 and pyramidal tract preparations.13,39 Evidence of depolarization block was also identified in a cortical island preparation12,14 and in brain slice studies11,25 and has been extensively modeled with Hodgkin–Huxley equations.40–42
It has long been appreciated that depolarization block is transient during SD, while synaptic failure is more prolonged.10,12,14 Similarly, we found that depolarization block could not fully explain the depression of evoked EPSPs in the transcallosal or thalamocortical pathways (Figures 1, 2, and 4). The current report is the first to our knowledge to directly compare depolarization block, synaptic inhibition and the duration of depression of spontaneous activity, all in the same preparations (Figures 1 and 3). Under these conditions, depolarization block was a minor contributor to the total depression of spontaneous ECoG activity, accounting for only 1–2 min out of ∼10–15 min of depression. In contrast, synaptic inhibition, (when evaluated in the relevant circuit, i.e., thalamocortical loop), could fully account for the entirety of ECoG depression (Figure 4). Thus, somatosensory-evoked potentials appear to be a reliable proxy for depression of spontaneous ECoG.
Adenosine receptor involvement
It has long been appreciated that the duration of ECoG depression relates to metabolic status, 6 and the present study suggests that adenosine is a key metabolite mediating this effect. Thus, administration of an A1R antagonist (DPCPX) shortened ECoG depression by ∼20% (Figure 4), as did focal supplementation with the adenosine degrading enzyme ADA. Either systemic or focal administration of DPCPX was effective, suggesting that adenosine acts locally in the cortex, rather than elsewhere in the thalamocortical circuit. Increasing adenosine bioavailability, without impairing other aspects of metabolism, was alone sufficient to prolong ECoG depression by ∼50% (Figure 4, endogenous ADA inhibition).
Earlier investigators had posed the hypothesis that adenosine contributes to spreading depression in vivo, but rejected it because neocortical adenosine applications failed to generate DC shifts pathognomonic of SD. 43 Adenosine accumulation prior to SD remained an important topic of investigation as a neuroprotectant molecule which could delay or prevent SD44–46 and thereby prevent damage during ischemia. The phenomenon of SD-induced adenosine release, and its physiologic role in spreading depression of cortical activity in vivo, remained untested. We can now appreciate that adenosine indeed accumulates to high levels in the cortex following SD in vivo, as a consequence rather than a cause of SD. 21
By comparison with hippocampal slice studies, where A1R blockade eliminates nearly all the secondary depression following relief of depolarization block, 25 the drug effects observed here in vivo neocortex were relatively modest. A1Rs are strongly expressed in the neocortex, 47 and localize to both presynaptic terminals and postsynaptic compartments. 48 The present results may significantly underestimate A1R contributions to SD-induced depression of activity, as the local drug concentrations achieved by focal pressure ejection in vivo are not known, and pipette concentrations of DPCPX could not be further increased, given solubility limits and our efforts to maintain vehicle concentrations ≤0.1%. 49 In conditions of compromised metabolic status where adenosine accumulates to higher levels after SD, 21 the A1R-mediated portion of spreading depression of cortical activity may well be much larger than described here. Additional metabolic candidates such as KATP channel activation and pH-sensitive signaling mechanisms remain to be evaluated and could also potentially contribute to synaptic depression, together with adenosine (Figure 5(b)). Together, these results were consistent with a role for activation of A1Rs by endogenous adenosine, accumulating and acting locally within the cortex, in the late phase of ECoG depression.
Possible sites of action
Dramatic decreases in neuronal excitability can be attributed to initial loss of membrane resistance during SD. 3 As intrinsic membrane properties recover after SD, other mechanisms must be invoked to explain the later phases of synaptic depression. Responses of layer V neurons to exogenous glutamate may be diminished following SD, 12 which has been interpreted as evidence of postsynaptic mechanisms in the depression of activity. Recent whole-cell recordings from Sawant-Pokam and colleagues have concluded that both pre- and postsynaptic mechanisms likely have roles, 20 although the relative contributions and mediators have yet to be fully determined.
A decreased probability of release (measured by miniature excitatory postsynaptic currents in layer II/III neurons) was recently reported after SD 20 and consistent with this, our PPR data from transcallosal projections similarly suggest that presynaptic mechanisms make a contribution to the period of sustained depression (Figure 2). We found that the A1R antagonist DPCPX tended to oppose PPR changes, however, this did not reach statistical significance and the number of animals needed to power the experiment appropriately was prohibitive. Adenosine remains a likely candidate for presynaptic effects, and future PPR studies should evaluate presynaptic contributions at the thalamocortical synapse that underlies ECoG depression (Figure 3), which is difficult to experimentally isolate in the intact animal. Future assessment could be conducted in high-throughput slice preparations, where deeper cortical elements can be accessed and monosynaptic inputs selectively stimulated.
Postsynaptic A1Rs may also contribute to adenosine effects, especially in deeper layers of the cortex. In response to bath-applied adenosine, layer V pyramidal neurons (but not layer II/III neurons) hyperpolarize and decrease in excitability. 51 Thus, while the relative contributions of pre- and postsynaptic A1Rs to total ECoG suppression are not identified in the present study, the relative importance each mechanism may depend on the cortical layer and cell type examined.
Differential sensitivity of corticocortical vs. thalamocortical circuits
We observed that synaptic transmission in corticocortical pathways recovered from SD much more quickly than in thalamocortical pathways (Figure 3), which is consistent with the finding that SD sharpens somatosensory receptive fields. 52 Our observations were not specific to the barrel cortex, as we observed the same pattern of recovery in the forelimb area of S1. Nor were they artifacts of electrode depth, as the same pattern of recovery was observed at both shallow and deep recording positions. The enhanced depression of evoked thalamocortical circuitry that we observed may be due to differential sensitivity to adenosine in deep versus superficial laminae of the cortex. 51 Supporting this possibility, a recent in vitro study of SD found that layer V pyramidal cells were more sensitive than layer II/III neurons to adenosine A1R effects. 53 In addition, there may be greater adenosine accumulation at greater depths within the cortex, with increasing distance from pial collaterals, particularly in conditions of metabolic compromise such as hypoxia, hypoglycemia, and stroke, where adenosine likely accumulates to higher levels after SD. The recent development of adenosine electrodes to distinguish local accumulation with better spatial and temporal resolution 54 may be helpful in addressing this question. Future studies could also be useful to identify current sources and sinks, and further characterize lamina-specific time courses of cortical recovery.
The experiments described here were conducted in anesthetized animals. While isoflurane and urethane anesthesia were not directly compared head-to-head, we could appreciate minor differences (i.e., longer corticocortical depression and shorter ECoG depression in urethane-anesthetized animals), which did not alter our main conclusions. Considering subjects’ sedation levels may be relevant when comparing studies or applying findings to analysis of spontaneous ECoG in the clinical setting. The current findings also raise the possibility that, in clinical settings where SD occurs with high incidence, adenosine receptor antagonists may be diagnostically useful to identify viable but depressed brain tissue. Adenosine receptor agonists, or agents that enhance endogenous adenosine accumulation, may contribute to therapeutic strategies to decrease SD frequency 46 in vulnerable tissue.
Conclusion
Findings presented here elaborate on the mechanisms linking SD to the phenomenon of depression in vivo. The direct within-animal comparison of time courses emphasizes the brief duration of depolarization block and provides a mechanism for the longer lasting sustained depression. Results presented here provide evidence that adenosine, acting on A1Rs, contributes to sustained synaptic impairment, and links the metabolic demand of repolarization to the sustained depression of spontaneous activity.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by NIH grants NS078805 (BEL) and NS051288 (CWS).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
BEL and CWS conceived and designed the experiments and interpreted the data. BEL conducted the experiments, analyzed the data, and drafted the manuscript. CWS critically revised the manuscript.