
Abstract
Neuroinflammation subsequent to developmental brain injury contributes to a wave of secondary neurodegeneration and to reactive astrogliosis that can inhibit oligodendrocyte progenitor differentiation and subsequent myelination. Here we evaluated the therapeutic efficacy of a small molecule antagonist for a TGFß receptor in a model of moderate perinatal hypoxia-ischemia (H-I). Osmotic pumps containing SB505124, an antagonist of the type 1 TGFß1 receptor ALK5, or vehicle, were implanted three days after H-I induced at postnatal day 6. Perinatal H-I induced selective neuronal death, ventriculomegaly, elevated CNS levels of IL-6 and IL-1α, astrogliosis, and fewer proliferating oligodendrocyte progenitors. Myelination was reduced by ∼50%. Anterograde tracing revealed extensive axonal loss in the corticospinal tract. These alterations correlated with functional impairments across a battery of behavioral tests. All of these parameters were brought back towards normal levels with SB505124 treatment. Notably, SB505124 preserved neurons in the hippocampus and thalamus. Our results indicate that inhibiting ALK5 signaling, even as late as three days after injury, creates an environment that is more permissive for oligodendrocyte maturation and myelination producing significant improvements in neurological outcome. This new therapeutic would be especially appropriate for moderately preterm asphyxiated infants, for whom there is presently no FDA approved neuroprotective therapeutic.
Introduction
Neuroinflammation inevitably follows perinatal brain injuries caused by neonatal encephalopathy, perinatal arterial ischemic stroke, premature birth and systemic infection, even though these clinical conditions have very different etiologies. The resulting neuroinflammation is regarded as an important contributor to the pathogenetic cascade, contributing to both the initial injury as well as to secondary neurodegeneration. 1 The majority of studies to date have focused on the production of the classic pro-inflammatory cytokines that include IL-1ß, IL-8, IL-18, TNFα and IL-6 as acute increases in these cytokines correlate with penultimate neurological impairments in children surviving perinatal brain injuries. 2
Microglial cells are the principal sentinels of the central nervous system (CNS) and they undergo a rapid transformation to an activated phenotype when the microenvironment of the brain is perturbed. After an acute injury, microglia possess a cellular phenotype likened to the M1 state of activated macrophages and they release many of the above-mentioned cytokines that propagate and sustain inflammatory processes. As the injury resolves, the microglia transition to an M2-like state where they release cytokines, such as TGFß1 and trophic factors such as glial cell-derived neurotrophic factor (GDNF) that are viewed as growth-promoting to enhance tissue repair and to promote inflammation resolution. 3 Given their central role in the production of pro-inflammatory cytokines, reactive oxygen species, matrix metalloproteinases and other cytotoxic molecules, a number of interventions targeted at reducing microglial activation have been tested in pre-clinical models of developmental brain injuries. 1
Reactive astrogliosis also occurs subsequent to brain injuries. Like the microglial reaction, the astroglial reaction is multifaceted. Astrocytes sense changes in their extracellular environment and adapt their activities to restore ionic homeostasis, scavenge free radicals and excess glutamate, produce energy substrates and trophic factors, restore the blood–brain barrier, and promote neovascularization. 4 However, in response to cytokines such as TGFß1, reactive astrocytes will produce extracellular matrix molecules that contain chondroitin sulfates or hyaluronan that interfere with developmental myelination and remyelination.5,6 Moreover, these extracellular matrix molecules may impair cerebrospinal fluid (CSF) absorption contributing to the development of hydrocephalus. 7
Using the Vannucci animal model of perinatal brain injury, we identified TGFβ1 as a cytokine that is produced during the subacute period of recovery from neonatal hypoxia-ischemia (H-I). In vitro, we and others have shown that TGFß1 activates the type 1 activin-like kinase receptor-5 (ALK-5) to stimulate the production of astrocytes from SVZ glial progenitors. 8 Increased levels of TGFβ1 in CSF subsequent to post-hemorrhagic hydrocephalus correlate with increased pathology in premature newborns9,10 as well as with poor outcome from endoscopic third ventriculostomy. 11 TGFß, which is associated with the M2-like microglial reaction, is often regarded as an anti-inflammatory cytokine, and indeed, antagonizing TGFß signaling in models of adult stroke exacerbates lesion size.12,13 However, other studies in the literature reveal TGFß to have a complicated and sometimes paradoxical effect upon CNS injury resolution. For example, in experimental autoimmune encephalomyelitis, inhibiting TGFβ1 signaling reduced T-cell infiltration and reduced the severity of disease. 14 This finding argues against the classic concept of TGFβ1 as an anti-inflammatory cytokine12,15 and suggests that the final outcome of its actions is context dependent. In this study, we tested the hypothesis that antagonizing the type I TGFß1 receptor ALK5 would inhibit astrogliosis and improve neurological function in a rat model of perinatal H-I.
Materials and methods
Neonatal hypoxia-ischemia
All experiments were performed in accordance with research guidelines set forth by New Jersey Medical School IACUC and these studies were in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23) revised in 1996 and the ARRIVE guidelines. Time pregnant Wistar rats were purchased from Charles River laboratories (Wilmington, MA). After normal delivery, the litter size was adjusted to 10 pups per litter. Animals were group housed and kept on a 12-h light:dark cycle with ad libitum access to food and water. Cerebral H-I was induced in 6-day-old rats (day of birth = P0; mean body mass = 15 g) as previously described. 8 We used an exposure time of 75 min in 8% oxygen to provide a moderate level of damage to the ipsilateral hemisphere. This model produces relatively reproducible damage, although there is some variability between animals. In this study, 45 out of 47 rats survived the HI event. The animals were randomly assigned to each experimental group. Twenty-one rats were used as shams controls. Sham-operated animals were anesthetized, and the carotid artery was isolated from the vagus (but not cauterized). Sham-operated animals were subjected to hypoxia. In previous studies from our laboratory, we have not observed significant differences between sham-operated animals and naive controls. For each experiment, sample sizes were chosen to minimize the number of animals needed while obtaining sufficient statistical power.
Drug administration
Intraperitoneal administration of SB505124
Six days after H-I, two different doses (1 mg/kg and 5 mg/kg) of the ALK5 pharmacological inhibitor, 2-(5-Benzo [1,3] dioxol-5-yl-2-tert-butyl-3H-imidazol-4-yl)-6-methylpyridine hydrochloride hydrate (SB505124) (Sigma; St. Louis, MO) SB505124, or vehicle (sodium citrate buffer with 30% DMSO v/v), were administered intraperitoneally (i.p.). Animals were euthanized 6, 12, and 24 h later.
Osmotic pump implantation
Three days after H-I, the rats were anesthetized with isofluorane (4% induction, 2% maintenance) and osmotic pumps (Alzet 1007D; Alza, Palo Alto, CA, USA) containing either vehicle or 10 mg/ml SB505124 dissolved in vehicle were implanted at a subscapular location. The insertion wound was sutured with 5-0 surgical silk. Animals were euthanized 3 days later for Western blot or 23 days later for immunohistochemical analysis. The optimal treatment duration and dosage of SB505124 was established from the experiments summarized in Figure 1, which were predicated upon previously published studies.
8
SB505124 inhibits Smad 2/3 phosphorylation after H-I. (a) SB505124 (1 mg/kg and 5 mg/kg) or vehicle was administered i.p. six days after H-I and the animals euthanized 6, 12, and 24 h later. (b and d) Brain homogenates from the ipsilateral (IL) contralateral (CL) and sham-operated hemispheres were analyzed by Western blot for pSmad 2/3. Equivalent protein loading was confirmed by probing with an anti-actin antibody. (c and e) Quantitative analyses of band intensities for pSmad 2/3 were performed using Western Lightening Chemiluminescence. Data represent average ± SEM, n = 6 H-I vehicle; n = 6 H-I SB505124 n = 6 sham. F. Rat pups were implanted with osmotic pumps containing either vehicle or 10 mg/ml SB505124 three days after H-I (n = 3 per group). Western blots for pSmad 2/3 were performed three days later. *P < 0.05, ***P < 0.001 by ANOVA followed by Tukey's post hoc test.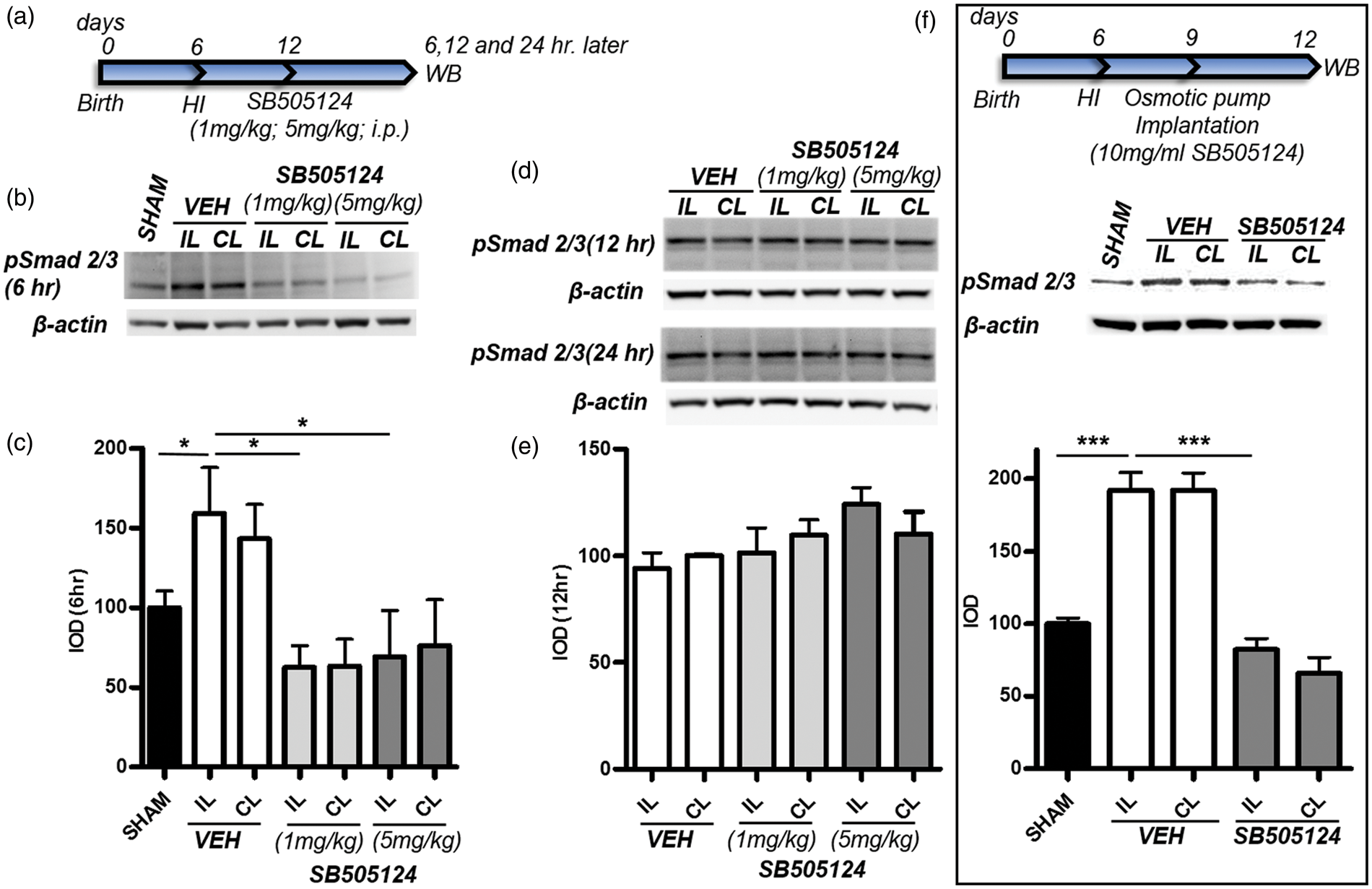
Intracortical injections of BDA for anterograde tract tracing
At 30 days after H-I, animals were anesthetized with a mixture of ketamine and xylazine and received four injections (0.5 µl each) of 10% biotinylated dextran amine (BDA) (10,000d; Invitrogen, Carlsbad, CA, USA) dissolved in PBS. BDA was introduced into the sensorimotor cortex of the ipsilateral hemisphere (coordinates were 1.25 and 2.25 mm lateral to the midline at 1 mm rostral and 1 mm caudal to Bregma at a depth of 1 mm). Seven days later, the animals were deeply anesthetized with a mixture of ketamine (75 mg/kg) and xylazine (5 mg/kg) before intracardiac perfusion with 3% paraformaldehyde in PBS. Coronal sections (20 µm thick) of the spinal cord from C2–C7 were collected (100 sections) and 10 sections were sampled from each animal in a random manner. Sections were stained with streptavidin-HRP (1:500; Thermo Scientific, Rockford, IL, USA) and developed using the ImPACT™ NovaRED™ kit (Vector Laboratories, Burlingame, CA, USA). From each section, a representative field from the contralateral dorsal funiculus of the spinal cord was acquired at 40 × magnification. The number of BDA labeled axons per mm2 was quantified using NIH image J software.
Behavioral tests
Modified neurological severity scoring (mNSS).

Note: For each item, the animals were awarded either a score of 1 or 0 according to the inability or capacity to perform the test or for the lack or presence of a tested reflex; thus, the higher scores indicate more severe behavioral deficits.
Histology and immunohistochemistry
5-Bromo-2-deoxyuridine (BrdU, Sigma) was administered at 50 mg/kg body weight intraperitoneally (i.p.) eight and nine days following H-I. Twenty-three days after H-I, animals were deeply anesthetized with a mixture of ketamine and xylazine before intracardiac perfusion with 3% paraformaldehyde in PBS. The brains were dehydrated in 70% ethanol and embedded into paraffin. Immunofluorescence staining was performed on 6 µm sections. The following antibodies were used: anti-BrdU (rat monoclonal, 1:30; Accurate, Westbury, NY, USA), anti-glial fibrillary acidic protein (anti-GFAP; rabbit polyclonal, 1:500; Dako, Carpinteria, CA, USA), anti-myelin basic protein (MBP) (1:1000; Covance, Princeton, NJ), anti-Iba-1 (1:200; Wako Chemicals, Richmond, VA, USA), and anti-Olig2 (1:250; Millipore, Temecula, CA, USA). Secondary antibodies against the appropriate species were incubated for 2 h at room temperature (Jackson, West Grove, PA, USA). 4,6-Diamidino-2-phenylindole (Sigma, 1 µg/ml) was used for 15 min to counterstain nuclei. For BDA staining, sections were incubated with streptavidin-HRP.
Damage evaluation
A total of nine sections per brain were used to evaluate the damage produced by the H-I insult. These sections were collected in a systematic manner (0.5, −1 and −3 mm from Bregma) and stained for cresyl violet. Ventricle size was determined from brain sections at 0.5 mm from Bregma captured by a Q-imaging Retiga-2000R CCD camera (Surrey, BC, Canada) connected to an Olympus BX51 microscope (Center Valley, PA). Using Image J, a ventricle size index was calculated as the ratio of the total area of the ipsilateral ventricle to the whole ipsilateral hemisphere. Stereological volume assessments of the cortex, striatum, hippocampus, and thalamus were produced using the Cavalieri principle associated with the counting point method as described elsewhere.21,22
Western Blot
Brain tissue from the affected striatum, corpus callosum, and cortex from the ipsilateral hemisphere as well as tissue from the contralateral hemisphere was dissected and placed into a lysis buffer. After solubilizing the tissue, 30 µg of protein sample was loaded onto a NuPage 4–12% Bis-Tris pre-cast gel (Invitrogen). Standard lanes were loaded with 5 µl MagicMark XP (Invitrogen) molecular weight standards. After gel electrophoresis, proteins were transferred to nitrocellulose membrane. The blots were rinsed and incubated overnight at 4 ℃ with gentle shaking with polyclonal antibodies to phosphorylated Smad 2/3 (Cell Signaling Technology (Beverly, MA, USA), 1:1000). The blots were incubated in HRP-conjugated secondary antibody and developed using Western Lightning chemiluminescence reagent (PerkinElmer, Wellesley, MA, USA). Equal protein loading and transfer were assessed by probing each blot with an anti-actin antibody (Sigma).
Determination of cytokines and chemokines in the brain tissue
Concentrations of TNF-α, IL-1β, IL-1α, IL-6, IL-10, IL-18, and G-CSF were simultaneously quantified in a single brain tissue sample using a MILLIPLEX™ rat cytokine/chemokine kit (LINCO Research, Inc., Saint Charles, MO, USA). Briefly, brain tissue was collected and flash frozen from sham-, SB505124- and vehicle-treated pups five days after H-I. The tissue was homogenized in a buffer containing 20 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 1 mmol/L PMSF, 0.05% Tween-20 and a cocktail of protease inhibitors (Roche Applied Science, IN, USA). Protein concentration was measured in each sample using a BCA assay. Cytokine and chemokine concentrations were determined according to the manufacturer's instructions.
Enriched microglial and astroglial cell cultures
Microglia and enriched type 1 astrocyte cultures were obtained using standard methods as described previously by Levison and McCarthy. 23 To further reduce microglial contamination from the astrocyte cultures, astrocytes were detached using 0.25% trypsin, collected by centrifugation (600 g, 10 min), resuspended in MEM-C and plated onto bacteriological plastic dishes for 20–30 min at 37 ℃. The un-attached astroglia were counted by trypan blue dye exclusion and replated into 60 mm dishes at a density of 1 × 105 viable cells/cm2 in MEM-C. The medium was replaced 24 h after plating with MEM supplemented with 1% FBS, 0.66 mg/ml BSA, 100 μg/ml d-biotin, 5 ng/ml insulin, 1 ng/ml selenium, 40 μg/ml iron poor transferrin, 2 mM glutamine, 15 mM HEPES buffer, and 100 U/100 μg/ml penicillin/streptomycin (MCDM). Cell cultures were treated with SB505124 (15 µM) or vehicle (2 h after media change) and stimulated with TGFβ1 (10 ng/ml) 30 min later. SB505124 and TGFβ1 were refreshed at 24 h. Culture supernatants were collected at 48 h after cytokine stimulation after removing cellular debris by centrifugation. The supernatants were concentrated (20 × for astroglia and 10 × for microglia) using Amicon Ultra centrifugal filters (10000 m.w. cutoff, Millipore, Billerica, MA, USA) following the manufacturer's instruction and stored at −20 ℃ until assayed.
Enzyme-linked immunosorbent assay
IL-6 content in the supernatants was assayed by sandwich enzyme-linked immunosorbent assay (ELISA) using matched antibody pairs from R&D systems (Minneapolis, MN, USA) for rat IL-6 (monoclonal anti-rat IL-6 antibody (MAB506) and biotinylated anti-rat IL-6 antibody (BAF506)).
Unbiased stereology
For stereological quantification of GFAP/BrdU and Olig2/BrdU, serial sections (6 µm) of the brain (0 ± 1000 µm Bregma) were collected (∼100 sections) and six pairs of consecutive sections were sampled from each animal in a systematic manner. For each pair of sections, at least 10 representative fields of the striatum were randomly acquired at 20 × magnification and cell counting was performed using the physical fractionator module of Stereo Investigator software (version 11.03, MBF Bioscience, Williston, VT, USA).
Statistical analyses
For non-parametric analyses, results were analyzed using the Mann–Whitney test. For tests that produced normally distributed data, the results were quantified and statistically analyzed using ANOVA followed by Tukey's post hoc analysis or using a Student's t-test. Error bars represent SEMs. Comparisons were interpreted as significant when associated with P < 0.05.
Results
Systemically administered SB505124 inhibits Smad 2/3 phosphorylation after H-I
H-I was performed on 6-day-old rat pups. Six days later, two different doses (1 mg/kg and 5 mg/kg) of the ALK5 pharmacological inhibitor, SB505124, or vehicle, were administered intraperitoneally (i.p.). Brain levels of phosphorylated Smad2/3 (pSMAD 2/3) were evaluated in homogenates from brain tissues extracted from the affected ipsilateral (IL) hemispheres as well as contralateral (CL) hemispheres 6, 12, and 24 h after i.p. injection (Figure 1(a)). At 6 days after H-I, there was a 1.6-fold increase in P-Smad2/3 in the IL hemisphere as compared to sham animals (Figure 1(b) and (c), P < 0.05). There was a trend toward increased levels of P-Smad2/3 in the CL hemisphere, which was not statistically significant (Figure 1(b) and (c), P > 0.05). Both doses of SB505124 significantly reduced P-Smad2/3 to normal levels (Figure 1(b) and (c), P < 0.05) 6 h after their administration, but the SB505124 was no longer effective by 12 h (Figure 1(d) and (e)). As it is impractical to inject SB505124 intraperitoneally to rat pups every 6 h in order to tonically inhibit ALK5, we transitioned to osmotic pumps loaded with SB505124, enabling the inhibitor to be delivered at a constant dose of 5 µg/h over seven days (Figure 1(f)). Osmotic pumps loaded with either vehicle or SB505124 were implanted three days after H-I and brain samples collected three days later. Western blot analyses confirmed that this administration route for SB505124 sustained the inhibition of Smad2/3 phosphorylation (Figure 1(f), P < 0.001). In addition to evaluating ALK5 signaling, we investigated signaling through the alternative TGFβ1 receptor, ALK1 that signals through Smad1/5 phosphorylation; however, SB treatment did not affect Smad1/5 phosphorylation levels (Figure 1(s)). Also, the activation of P38 that could potentially be affected by SB505124 at higher concentrations did not show significant changes (Figure 1(s))
Delayed SB505124 administration reduces the extent of brain damage and improves sensorimotor deficits after perinatal H-I
Between 14 and 20 days of age, H-I rats treated with vehicle had a lower mean body mass as compared to the shams (Figure 2(a), P < 0.05). By contrast, SB505124-treated H-I animals gained weight, reaching values not statistically different from the shams (Figure 2(a)). The brains from H-I vehicle and H-I SB505124-treated animals were evaluated to establish the extent of neuropathological changes in the striatum, neocortex, lateral ventricles, hippocampus, and thalamus from subjects at 23 days after H-I (postnatal day 29). H-I caused significant ventricle dilation. The ventricle size index increased by 17-fold as compared to the shams (P < 0.01 Figure 2(b) and (d)) in the vehicle group. SB505124 treatment alleviated ventricle dilation reaching values no different from the shams (P > 0.05). The total volume of the ipsilateral hippocampus and thalamus, two regions that are particularly vulnerable to degeneration after H-I, were significantly preserved in SB-treated rats (72% and 88% of contralateral hemisphere, respectively) as compared to the vehicle-treated rats (22% and 57% of contralateral hemisphere respectively, Figure 2(c) and (e), P < 0.05).
SB505124 administration reduces the extent of brain damage after H-I. Animals were implanted with osmotic pumps containing SB505124 or vehicle three days after H-I. (a) Body masses were measured during recovery from H-I. Data represent mean body masses from eight animals per group. (*P < 0.05, H-I vehicle vs sham). (b) Ipsilateral ventricle size index at P23. (c) Ipsilateral neocortical (CTX), hippocampal (HIP), thalamic (TH), and striatal (ST) volumes were measured and normalized to the volume of the contralateral structure. (d and e) Representative cresyl violet stained images illustrating attenuation of ventricle size dilation (arrows in (d)) and neuroprotection of the hippocampus and thalamus (asterisks in (e)) obtained with delayed SB505124 treatment. IL=ipsilateral; CL=contralateral. Significant differences were determined by ANOVA followed by Tukey's post hoc test. A minimum of six animals per group were analyzed, *P < 0.05, **P < 0.01.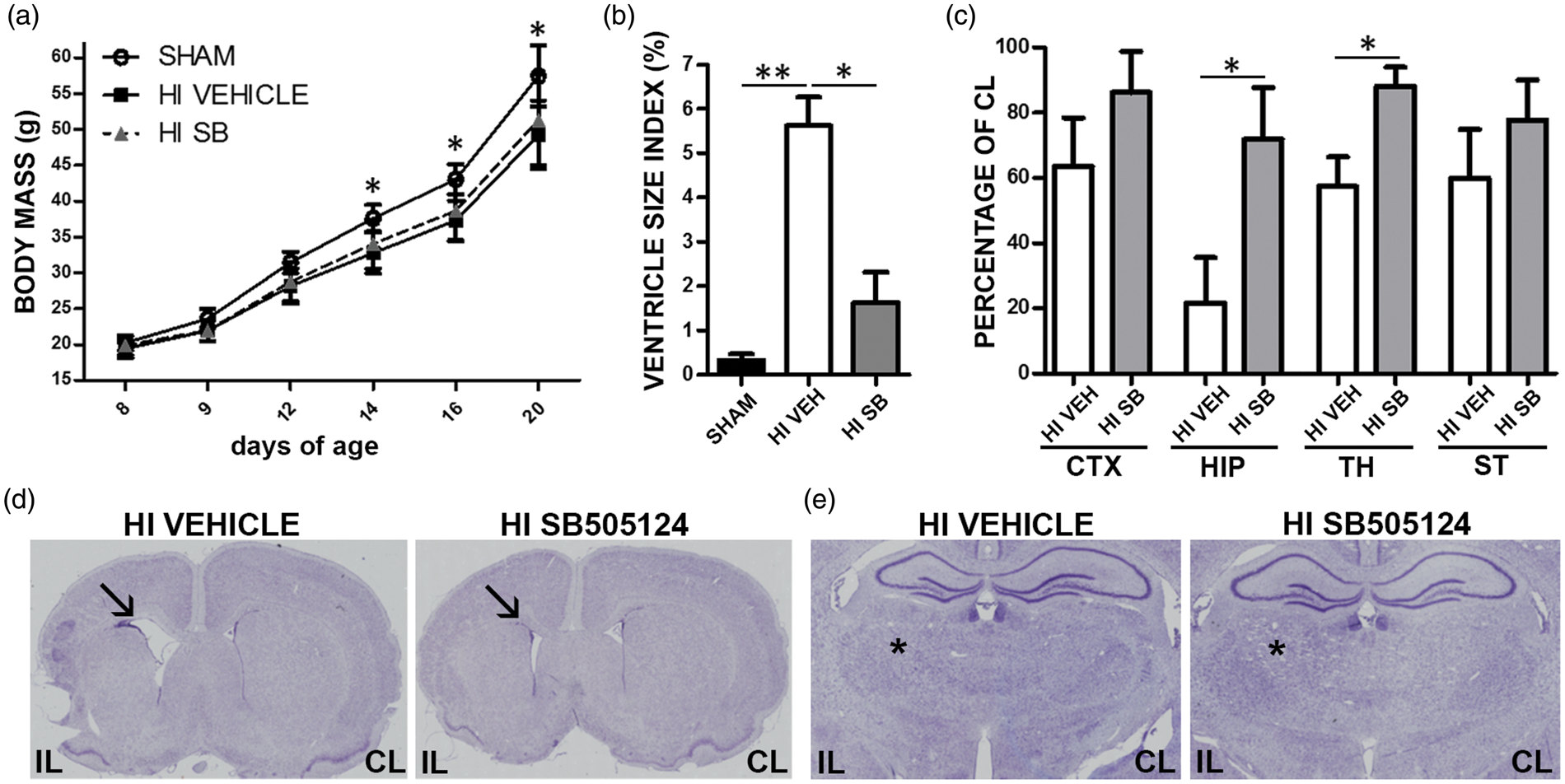
To determine whether the observed structural preservation obtained with SB505124 treatment translated to preserved sensorimotor function, we subjected the H-I rats to a battery of behavioral tests (Figure 3(a)). In the cylinder rearing test (CRT), which evaluates somatosensory asymmetry, the H-I injury produced motor deficits in the contralateral (left) forepaw, which were reflected in the significant preference to use the ipsilateral (right) forepaw (72% preference, Figure 3(b), P < 0.05). SB-treated rats showed an improved performance in the CRT with almost no paw preference (16% preference Figure 3(b), P < 0.05). Sensorimotor deficits were also evaluated with the Sticky Paper test. Here, H-I vehicle-treated rats took longer to remove the adhesive paper from the contralateral forepaw than the shams (37.6 s vs 10.2 s, respectively, Figure 3(b), P < 0.05). Deficits in performing this test were corrected by SB505124 treatment (12.5 s Figure 3(b), P < 0.05). There was no difference in the time to remove the label from the ipsilateral (non-affected) forepaw between any of the groups (data not shown). The Inclined Beam-Walking Test requires subcortical white matter integrity and H-I vehicle rats performed poorly exhibiting more foot slips than the shams (4 vs. 0.6 foot slips, respectively, Figure 3(b), P < 0.01; supplementary videos 1 and 2). SB505124 treatment after H-I significantly improved performance on this test, decreasing the mean number of foot slips (one foot slip, Figure 3(b), P < 0.01; supplementary video 3). The mNSS analysis which combines a number of tests to produce a single numerical score (Table1 and Figure 3(c)) similarly revealed improvement with delayed SB-treatment after H-I compared to vehicle treatment (1.5 vs 3.6, respectively Figure 3(c), P < 0.05).
Sensorimotor deficits after H-I are attenuated by SB505124 treatment. (a) Schematic representation of the experimental protocol. (b) Cylinder rearing, Sticky Tape Removal and Inclined Beam Walking tests were used to assess sensorimotor function (n = 6 per group). (c) Neurological deficits also were evaluated using the modified neurological severity scale, which evaluates nine tasks graded on a scale of 0–9 (normal score, 0; maximal deficit scored, 9). (d) The anterograde tracer BDA was injected at 30 days after H-I and animals euthanized seven days later. Representative photomicrographs of the dorsal funiculi of the spinal cords stained for BDA are provided. (e) Results of quantification of BDA labeled axons in the dorsal funiculus. Scale bar = 100 µm. IL=ipsilateral; CL=contralateral. Significant differences were determined by Mann–Whitney test in (c) and by ANOVA followed by Tukey's post hoc test in (b) and (e), and a minimum of six animals per group were analyzed, *P < 0.05, **P < 0.01.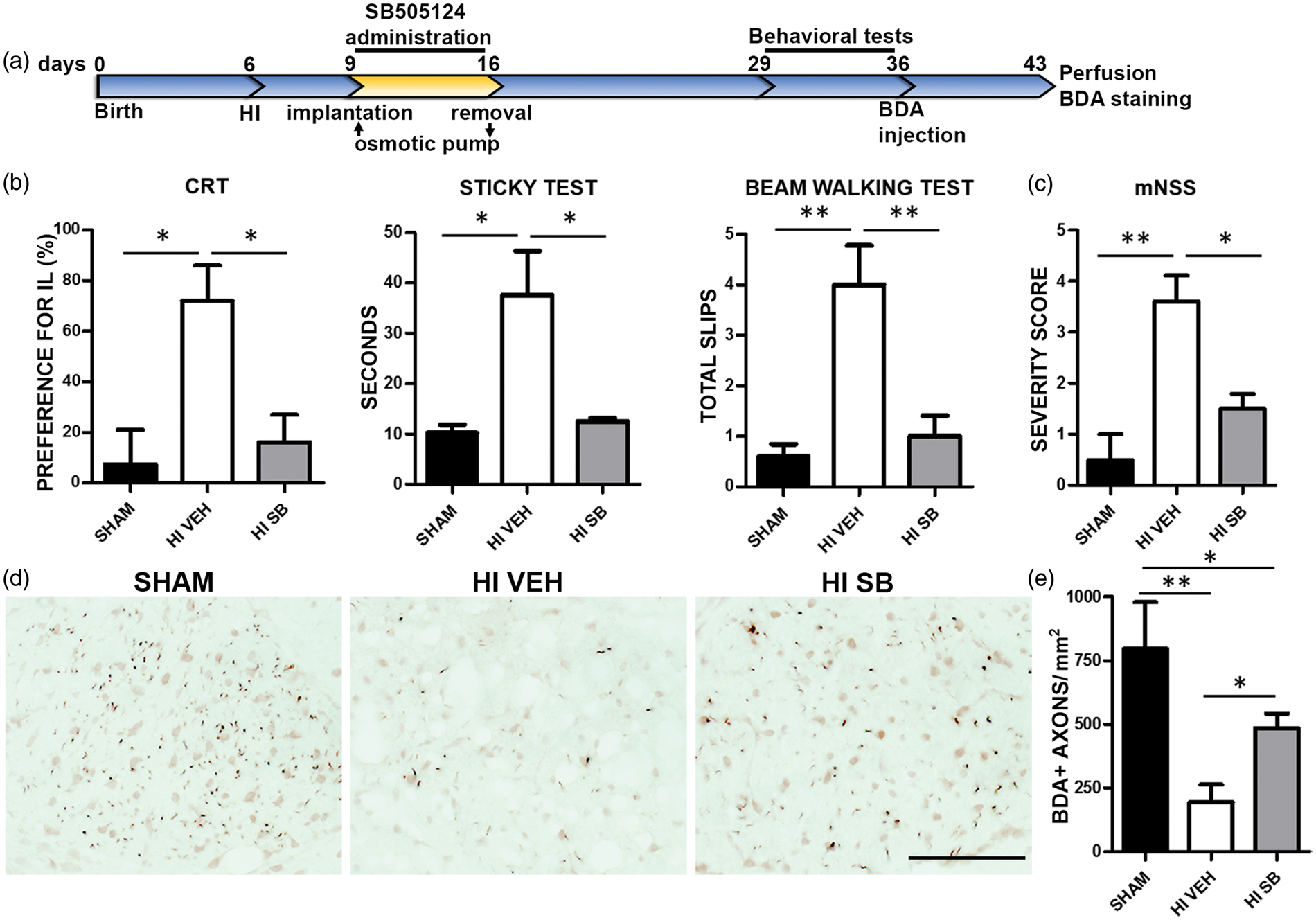
The most prominent behavioral improvements were seen in motor function. Therefore, we hypothesized that SB might prevent the degeneration of axons of the corticospinal tract (CST) which would contribute to the impaired motor function observed after H-I. To visualize the effect of SB505124 treatment on the connectivity of the CST, the ipsilateral motor cortex of vehicle- and SB-treated rats was injected with BDA 30 days after H-I (Figure 3(a)). In H-I vehicle-treated rats, the number of BDA-positive axons in the corticospinal tract of the contralateral spinal cord one week after BDA labeling was markedly reduced as compared to shams (194.3 ± 68.8 vs. 795.3 ± 182.7 axons/mm2, respectively, Figure 3(d) and (e), P < 0.01). Notably, significant more BDA labeled axons were present after SB505124 administration compared with vehicle-treated rats (484 ± 57.6 axons/mm2, Figure 3(d) and (e), P < 0.05).
Numerous studies have shown that there is an increase in the expression of pro-inflammatory cytokines after neonatal H-I.
24
To determine whether SB505124 treatment attenuated the production of pro-inflammatory molecules, we measured the concentration of multiple cytokines from brain tissue samples containing the injury core from animals under the different experimental conditions. At five days after H-I, levels of IL-1 alpha and IL-6 were elevated by 3.4 fold and 2.9 fold, respectively (Figure 4(a), P < 0.05). SB505124 treatment after H-I reduced both cytokines to levels present in sham-operated animals (Figure 4(a), P < 0.05). As both microglia and astrocytes produce proinflammatory cytokines after injury, we stimulated microglial- and astroglial-enriched cell cultures with TGFβ1 in the presence or absence of SB505124 and analyzed IL-6 content in the supernatant. Only the astrocytes responded to TGFβ1 by increasing IL-6 release, which was completely blocked by SB505124 (Figure 4(b)). Commensurate with injury and neuroinflammation, the number of Iba-1 + cells increased in the affected hemisphere of H-I vehicle group (6.9 fold increase as compared to shams, Figure 4(c) and (d), P < 0.01) and SB505124 treatment significantly attenuated the increase in Iba-1 + cells as compared to vehicle-treated animals, especially in the striatum (2.4 fold decrease, P < 0.05, Figure 4(c) and (d)).
SB505124 attenuates H-I induced inflammation. Three days after H-I, rat pups were implanted with osmotic pumps containing either vehicle or SB505124 and the animals were euthanized two days later. (a) Concentrations of 12 pro-inflammatory cytokines were determined using a Milliplex™ Multiplex assay. Only IL-6 and IL-1-alpha were significantly elevated in the ipsilateral hemisphere of H-I animals vs. Shams, and they were reduced by SB505124. (b) ELISA was performed for IL-6 on supernatants from astrocytes and microglia. Cell cultures were treated with SB505124 (15 µM) or vehicle and stimulated with TGFβ1 (10 ng/ml) or vehicle (CTROL) 30 min later. Media were collected 48 h after cytokine stimulation. LPS-stimulated microglia were used as positive control. (c) Iba-1 + cells in the ipsilatateral striata of H-I vehicle- and SB505124-treated animals as well as sham-operated animals after treatment. Values are expressed as the means ±SEM. n = 7 H-I veh; n = 7 H-I SB505124; and n = 6 sham; *P < 0.05, **P < 0.01 and ***P < 0.001 by ANOVA followed by Tukey's post hoc test. (d) There was a strong microglial response in the vehicle-treated H-I animals as revealed by Iba-1 immunostaining in the striatum. SB505124 significantly attenuated the microglial response. Scale bar represents 100 µm.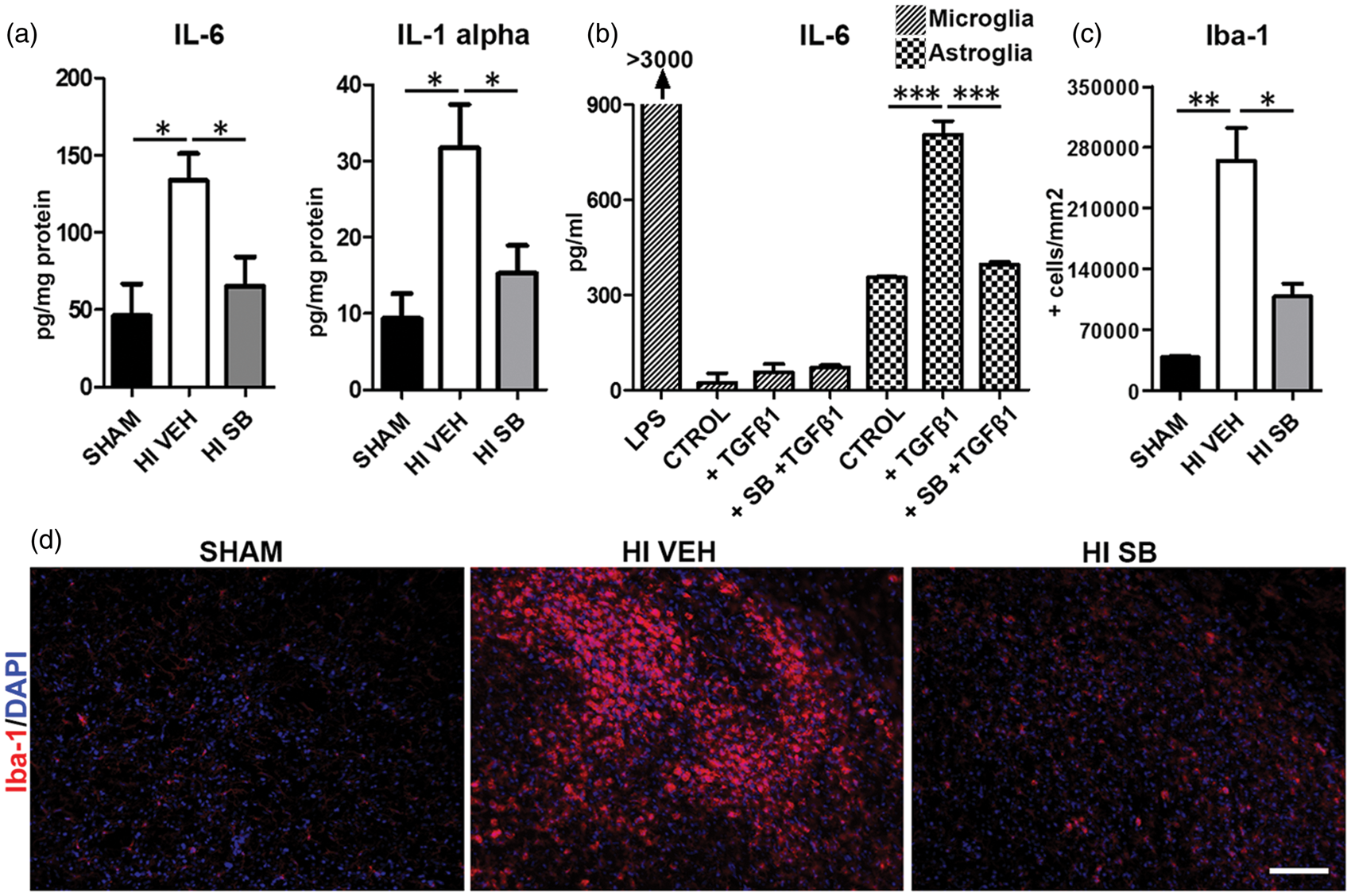
Aberrant glial development after H-I is prevented by inhibiting ALK5
We tested the hypothesis that inhibiting ALK5 after neonatal H-I would reduce astrogliosis and restore myelination. At three weeks after H-I GFAP immunostaining in the ipsilateral cortex, corpus callosum and striatum of vehicle-treated rats were significantly elevated compared to shams (two fold increase in cortex and corpus callosum and 2.6 fold increase in striatum, Figure 5(a) and (b), P < 0.01 and P < 0.001, respectively). MBP immunostaining was significantly reduced in these same areas (3.2 fold decrease in cortex and 2.3 fold decrease in corpus callosum and striatum compared to shams, Figure 5(a) and (b), P < 0.001 and P < 0.05 respectively). SB505124 treatment attenuated the H-I-induced increase in GFAP and restored MBP immunostaining to control values (Figure 5(a) and (b)). Based on these findings, we tested the hypothesis that inhibiting ALK5 after H-I would reduce the generation of astrocytes and restore the number of proliferating OPCs. BrdU was injected into H-I rats on P14 and P15 and animals were euthanized two weeks later (P29). Stereological counts revealed that the number of GFAP+/BrdU + cells was significantly higher in the ipsilateral striatum after H-I as compared to shams (5 fold increase, Figure 5(c) and (d), P < 0.05), whereas Olig2+/BrdU + cells were dramatically reduced (2.7 fold decrease, Figure 5(e) and (f), P < 0.05). Consistent with the results reported above, SB505124 treatment prevented the H-I-induced increase in BrdU-labeled GFAP + cells (4 fold decrease as compared to the vehicle-treated group, Figure 5(c) and (d), P < 0.05) and significantly restored the number of BrdU+/Olig2 + cells (1.62 fold increase as compared to the vehicle-treated group, Figure 5(e) and (f), P < 0.05).
SB505124 decreases H-I-induced astrogliosis and restores subcortical white matter myelination. (a). Coronal sections of the brain stained for GFAP and MBP from animals at 23 days of recovery that received vehicle or SB505124 by osmotic pump implanted three days after H-I. (b) The integrated optical density (IOD) was determined for MBP and GFAP immunoreactivity in the neocortex, striatum and the corpus callosum (CC) from ipsilateral (IL) and contralateral (CL) hemispheres as well as from sham-operated animals. Values are expressed as the mean ± SEM. n = 6 H-I vehicle; n = 5 H-I SB505124; n = 6 sham; *P < 0.05, **P < 0.01, and ***P < 0.001 vs. sham and ##P < 0.01 and ###P < 0.001 vs H-I SB by ANOVA followed by Tukey's post hoc test. (c and e) Animals were administered SB505124 or vehicle at three days after H-I. They received two BrdU injections at seven and eight days after H-I and were euthanized 15 days later (P30). Panels illustrate representative images of GFAP or Olig2 and BrdU staining in the striatum. Scale bar = 100 µm. Insets show higher magnification images. (d and f) Stereological counts were obtained for GFAP+/BrdU + and Olig2+/BrdU + cells in the striatum (n = 6 H-I veh; n = 5 H-I SB505124 and n = 6 sham; *P < 0.05 by ANOVA followed by Tukey's post hoc test.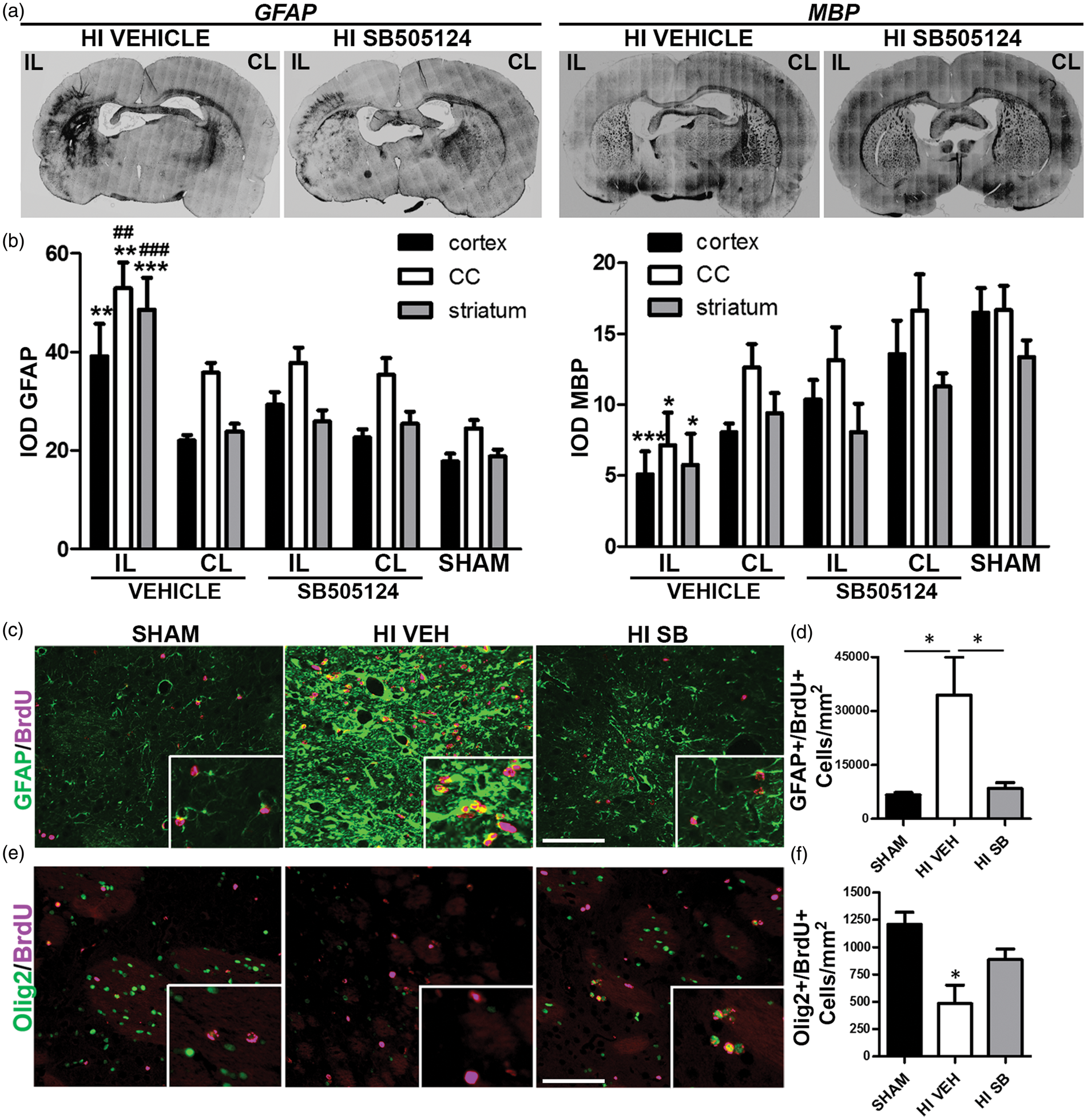
Discussion
Here we provide evidence that delayed administration of a TGFβ type 1 receptor inhibitor improves outcome after perinatal hypoxic-ischemic brain damage. Our data show that administering SB505124 beginning three days after H-I (1) reduces the levels of brain parenchymal pro-inflammatory cytokines; (2) reduces microgliosis and astrogliosis; (3) preserves hippocampal and thalamic volumes; and (4) preserves axons of the corticospinal tract which correlate with improved performance on a battery of sensorimotor tests.
TGFβ1 is a pleiotropic cytokine that affects a variety of biological processes in many organs. 25 Since we administered an antagonist to an important signaling receptor for TGFß, we were concerned that this might adversely affect the health of the animals or their growth. However, SB505124 animals gained weight and when we analyzed several peripheral organs, we found that rather than adversely affecting the lymphatic organs, H-I-treated animals that had received SB505124 exhibited a restoration in the thymus-body weight ratio vs. vehicle treated H-I animals normalizing the thymus weight to shams levels (data not shown). Indeed, in contrast to the cerebral pro-inflammatory response, cerebral ischemia depresses the peripheral immune system. 26 This phenomenon has been referred to as stroke or CNS injury-induced immunodepression syndrome27,28 and is attributed to cerebral production of pro-inflammatory cytokines and to local brain damage, which causes suppression of innate and adaptive immunity. In light of the reduced brain damage and reduced pro-inflammatory cytokines expression in SB505124 treated rats, it should not be surprising that peripheral lymphoid organs were not adversely affected.
Delayed secondary neurodegeneration has been previously demonstrated using the Vannucci rat model of H-I 29 and in models of adult stroke. 30 While most studies have focused on the inflammation that occurs within the first 24 h, there are reports of persistent neuroinflammation, which provide a plausible mechanism for progressive cell death. For example, elevated levels of pro-inflammatory cytokines and microglial activation have been demonstrated 14 days after neonatal H-I,31,32 and increased levels of IL-6 have been detected in the penumbra up to 14 days after middle cerebral artery occlusion in adult rats. 33 The signals that maintain this chronic neuroinflammation have not been fully established, but TGFβ1 is a strong candidate. TGFß1 expression is both delayed and prolonged after brain injury8,34,35 and several studies have shown that it can stimulate neuroinflammation within the CNS.14,36 Moreover, over-expression of TGFβ1 in the CNS of transgenic mice and direct intrathecal injection of TGFβ1 in animals produces hydrocephalus.37,38 In a model of germinal matrix hemorrhage (GMH), a small molecule antagonist for the type 1 TGFß1 receptor (SD-208) was shown to reduce GMH-induced ventriculomegaly and improve neurological deficits. 39 In our studies, SB505124 treatment similarly decreased H-I induced ventriculomegaly. Future studies are needed to establish how inhibiting ALK5 affects CSF production and reabsorption after H-I encephalopathy.
TGFβ1 can exert anti-inflammatory and pro-inflammatory actions both inside and outside the brain.40,41 The paradoxical functions of TGFβ1 have been described in the experimental models of arthritis, where injecting TGFβ1 locally into the joint exacerbates the inflammatory response and aggravates disease, 42 whereas systemically administered TGFß1 inhibits inflammation. 43 Contrary to the pro-inflammatory role for TGFβ1 demonstrated here, TGFβ1 signaling in astrocytes limits neuroinflammation in the subacute period after stroke and during toxoplasmic encephalitis.13,44 In these studies, transgenic mice that expressed a dominant negative type II TGFβ receptor in astrocytes were studied. Thus, both Smad and non-Smad downstream signaling pathways would have been inhibited. By contrast, in our studies, a small molecule kinase inhibitor that specifically targeted the type I TGFβ receptor ALK5 was used. Thus, not all TGFß signaling was antagonized. In addition, neurons and infiltrating T cells, not astrocytes, were found to be responsive to TGFβ1 signaling in the study reported by Luo et al. 14
Further studies are needed to more fully understand the contributions and signaling responses of the different cell types of the CNS to progressive neurodegeneration. As ALK5 is expressed by neurons, microglia, and astrocytes, 45 future studies will need to be employed using genetic approaches to delete ALK5 or Smad2/3 in neurons, astrocytes, and microglia to reveal their roles in the progressive neurodegeneration that occurs subsequent to perinatal H-I. Furthermore, an alternative TGFβ receptor, ALK1, is highly expressed in neurons after ischemic injury, and it was shown that this receptor mediates TGFβ1-induced neuroprotection through Smad 1/5 signaling. 46 It is possible that the preservation of the brain structures that we observed after ALK5 inhibition are the result of increased ALK1 activation in neurons. However, we did not observe an increase in Smad 1/5 signaling in the brains of animals treated with SB505124, suggesting that this is not the mechanism of neuroprotection afforded by SB505124 administration.
Several cytokines remained elevated in the affected hemisphere five days after injury as previously reported. 32 SB505124 reduced the levels of both IL-1 alpha and IL-6 in the brain parenchyma. The first peak of IL-6, at 6 h after H-I, has been attributed to microglial production. 32 Since SB505124 reduced IL-6 production from astrocytes stimulated with TGFβ1, and had no effect on microglial production, it is likely that in vivo astrocytes and not microglia are the main source of IL-6 during the subacute phase. In the clinical literature, it has been well established that increased levels of IL-6 correlate with the extent of brain lesion and severe motor deficits in newborns with hypoxic ischemic encephalopathy, 2 and studies have demonstrated a direct toxic effect of these cytokines in the brain. For example, IL1 receptor antagonist treatment reduces neuronal death in a model of middle cerebral artery occlusion, 47 and prolonged elevation of IL1ß systemically produces long-lasting myelination defects. 48 Furthermore, transgenic mice that overexpress IL-6 in the brain develop a neurologic syndrome characterized by seizures, tremor, ataxia and hind limb weakness, the severity of which correlates to the levels of cerebral IL-6. 49 In addition, these mice show neuronal degeneration in the hippocampus and cerebellum and severe astrogliosis and microgliosis. The expression of proinflammatory cytokines from reactive astrocytes could help to maintain a chronic state of inflammation that leads to progressive neurodegeneration. In this sense, a reduction in astrocyte proliferation by SB505124 may be responsible for the preservation of hippocampal neurons and corticospinal tracts. A related in vitro study demonstrated that IL-6 induced cell cycle withdrawal and maturation of OPCs. 50 Extrapolating from the results of this last study, elevated levels of IL-6 could deplete the pool of proliferating OPCs and stimulate the premature maturation of the existing OPCs into myelinating oligodendrocytes, thereby contributing to hypomyelination.
Since previous studies reported that TGFβ1 could induce mast cells degranulation to exacerbate excitotoxic brain lesions, 51 we asked whether mast cell stabilization was contributing to the neuroprotection observed after SB505124 treatment. However, we found no evidence of decreased mast cell degranulation in coronal sections stained with toluidine blue (Goyal and Clausi, unpublished data). Other studies have established an important role of TGFβ1 in endothelial cell plasticity, 52 a process that is essential for tissue regeneration. 53 In contrast to glial cells that only express ALK5 receptor, endothelial cells express both ALK5 and ALK1 TGFβ receptors. 45 ALK1 activity is not affected by SB505124, 54 and as expected we did not find any difference in brain levels of P-Smad 1/5 between the experimental conditions. Since angiogenesis is tightly regulated by the balance of P-Smad 1/5 and P-Smad 2/3, 52 we investigated the effect of SB505124 in this process. SB505124- and vehicle-treated H-I animals showed a robust increase in angiogenesis in the affected hemisphere, but no difference was found in blood vessel density between treatments (data not shown).
Studies have shown that the p38 mitogen-activated protein kinase (MAPK) is activated by oxidative stress leading to increases in superoxide generation and neuronal death. 55 SB505124 has been shown to inhibit p38 MAPK alpha with an IC50 value more than 200 times the IC50 value for ALK5. Since it was plausible that SB505124 could inhibit p38 MAPK, we evaluated the brain levels of the phosphorylated (active) form; however, there was no significant difference in active P38 between vehicle- and SB505124-treated HI animals; therefore, it is highly unlikely that the concentration of SB505124 used in our study inhibited downstream p38 MAPK signaling in the brain.
Therapeutic hypothermia has been established as the standard of care for infants with moderate hypoxic-ischemic encephalopathy. Mild hypothermia has been shown to reduce mortality and to improve early neurologic outcome when started within the first 6 h after birth; however, significant neurologic deficits persist into childhood. 56 Moreover, neonatal brain injury often eludes diagnosis, and therefore, a number of infants who would have benefited from hypothermia do not receive this intervention. 57 As is widely acknowledged, there remains a need to find new neuroprotective therapies to reduce adverse outcomes after hypoxic-ischemic encephalopathy. Our data suggest that SB505124 be considered as a potential therapeutic either alone, or after pre-clinical evaluation, as an adjunct to hypothermia to improve neurologic outcome. Since delayed inflammation and glial scar formation are also critical events in other forms of developmental brain injuries as well as for injuries sustained by adults such as stroke, spinal cord injury and traumatic brain injury,58,59 SB505124 should be evaluated in animal models of these pathologies as well.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Institutes of Health HD052064 and the Leducq Foundation awarded to SWL.
Acknowledgements
This work was presented in part at the 2013 Euroglia meeting. We thank Dr Isis Ornelas and Lauren Mursch (Department of Neurology and Neuroscience, New Jersey Medical School Cancer Center, Rutgers Biomedical and Health Sciences, Newark, New Jersey) for providing the rat glial cultures. Our thanks are also extended to the students in the Professional Skill course of the GSBS-Newark division for their critical reviews and constructive comments.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
SWL contributed to the study conception and design; MGC performed the experiments and quantifications. Both authors contributed to the interpretation, analysis of the data, and in writing the paper.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.