
Abstract
Advancements in molecular biology have led to a greater understanding of the individual proteins responsible for generating cerebral edema. In large part, the study of cerebral edema is the study of maladaptive ion transport. Following acute CNS injury, cells of the neurovascular unit, particularly brain endothelial cells and astrocytes, undergo a program of pre- and post-transcriptional changes in the activity of ion channels and transporters. These changes can result in maladaptive ion transport and the generation of abnormal osmotic forces that, ultimately, manifest as cerebral edema. This review discusses past models and current knowledge regarding the molecular and cellular pathophysiology of cerebral edema.
Introduction
Historically, the goal of brain protection following injury has been to reduce neuronal damage. Edema, the inevitable accompaniment, was considered a secondary event. A renewed interest in edema, its molecular antecedents and its importance in all but the smallest ischemic insults has shifted this paradigm. Strikingly, recent advancements in understanding molecular mechanisms of edema formation suggest that the translation of novel treatments for edema may be closer at hand than the translation of treatments for neuronal demise.
Cerebral edema is a pressing clinical problem. Cerebral edema and brain swelling inevitably accompany ischemic infarcts and intracerebral hemorrhages and, when severe, may increase mortality to nearly 80%. 1 Even in non-life-threatening stroke, the magnitude of brain swelling is strongly predictive of patients’ functional outcome. 2 Cerebral edema and brain swelling occur in 20–30% of patients with acute liver failure, and increase mortality to ∼55%. 3 Cerebral edema and brain swelling after traumatic brain injury are estimated to account for up to 50% of patient mortality. 4
Currently approved treatments for cerebral edema—decompressive craniectomy and osmotherapy—were developed prior to any knowledge of modern cerebral edema pathophysiology. These therapies attempt to manage downstream end-stage events without directly attenuating the underlying molecular mechanisms of cerebral edema. New advances have shed light on heretofore poorly understood cellular and molecular pathophysiology of cerebral edema, and have led to clinical trials of antagonists of key molecular events in cerebral edema formation. It is now understood that cerebral edema evolves in stages, where each stage is characterized by distinct morphological and molecular changes. Cytotoxic edema, or cellular swelling, manifests minutes after acute central nervous system (CNS) injuries. Ionic edema, an extracellular edema that occurs in the presence of an intact blood brain barrier (BBB), forms immediately following cytotoxic edema. Vasogenic edema, an extracellular edema that includes extravasation of plasma proteins, manifests hours after the initial insult.
This review is intended to serve as a foundation and a reference from which researchers and clinicians might extend their molecular understanding of cerebral edema formation and clearance. Here, the most current cellular and molecular models of cerebral edema formation, transport, and clearance are discussed.
Models of the cerebral vasculature
The blood–brain barrier
The term BBB refers to a complex of cells that separates the brain interstitium from the luminal contents of the cerebral vasculature (Figure 1). In addition, the term can be used to describe the functional consequence of these cells, namely, the relative independence of brain interstitial fluid (ISF) composition from that of blood plasma, an arrangement that was likely evolved to tightly regulate perisynaptic electrolyte homeostasis, thereby ensuring accurate synaptic integration.
5
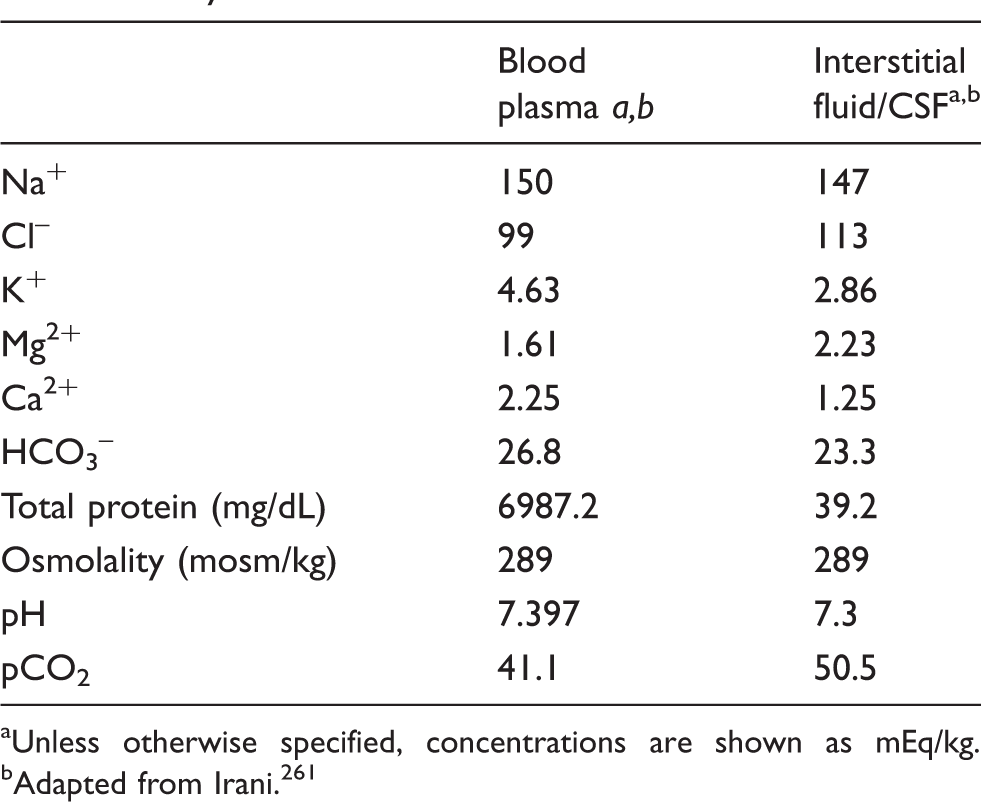
Brain ISF, which freely communicates with cerebrospinal fluid (CSF), is optimized for neuronal activity and differs from blood serum in that it contains higher concentrations of Cl– and Mg2+ and lower concentrations of K+, Ca2+, and HCO3– (Table 1).
6
The solute composition of plasma and brain ISF in adult humans is shown in Table 1.
Anatomy of cerebral arterioles (top) and capillary (bottom). The innermost layer of arterioles and capillaries is composed of a continuous layer of endothelial cells, linked by tight junctions, and bounded externally by a layer of basement membrane that contains pericytes; arterioles, but not capillaries, travel inside the perivascular Virchow Robin Space (VRS); the outermost layer of the blood brain barrier is composed of astrocyte endfeet, the terminal pads of large astrocyte processes. Mean concentrations of select solutes in plasma and CSF of healthy adult humans. aUnless otherwise specified, concentrations are shown as mEq/kg. bAdapted from Irani.
261
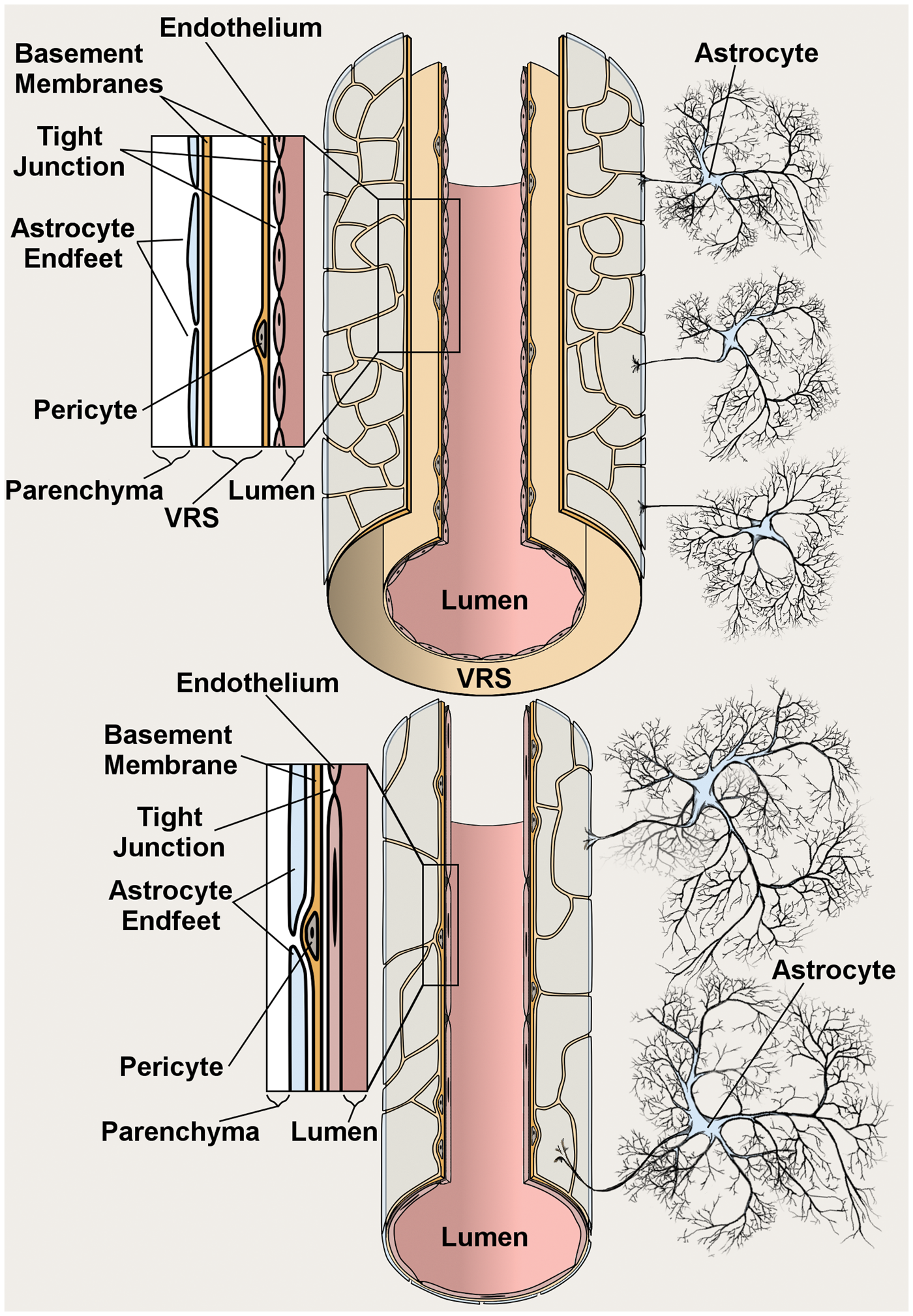
Various aspects of the BBB functionality are distributed among its constituent cell types. The innermost layer of the BBB is comprised of a monolayer of endothelial cells that directly contacts the circulating blood. Excepting select fenestrated capillary beds in the circumventricular organs, 7 vertebrate cerebral endothelial cells are interconnected by tight junctions that collectively form the “physical barrier” of the BBB. Notably, interendothelial tight junctions physically divide the brain endothelium plasmalemma into luminal and abluminal membrane faces, which allows for polarized localization of transporters and channels 8 akin to secretory epithelium. 9
External to the endothelium is a layer of basement membrane, a connective tissue composed of extracellular matrix proteins including collagens, laminins, heparin sulfate proteoglycans, fibronectin, vitronectin, nidogens, perlecan, and agrin. 10 Pericytes, a mesenchymal cell type that contributes to cerebral blood flow regulation, 11 and increases BBB “tightness”,12,13 are embedded within the endothelial basement membrane at varying intervals along the vessel.
In vessels larger than capillaries, the endothelial basement membrane is bounded externally by the perivascular Virchow Robin space. The Virchow Robin space is a CSF filled extension of the subarachnoid space that is internally bounded by the endothelial basement membrane and externally bounded by a second, glial, basement membrane. The Virchow Robin space follows penetrating arterioles into the brain parenchyma, becomes fenestrated close to the capillary bed, and eventually disappears at the level of brain capillaries, where astrocyte endfeet directly contact the vessel wall.14,15
Astrocyte endfeet, the terminal pads of large astrocyte processes, comprise the outermost layer of the BBB at all levels of the vasculature, including capillaries. Astrocytes are supportive cells that completely fill the brain parenchyma, and in grey matter, are arranged in a three-dimensional matrix with nonoverlapping spatial domains. 16 Nearly all astrocytes extend at least one process that contacts a vessel with an astrocyte endfoot. 16 The parenchymal surface of cerebral vessels are completely covered by a mosaic of astrocyte endfeet that are separated by gaps of approximately 20 µm. 17 The astrocyte endfoot is a specialized membrane domain that is enriched in transporters and channels involved with brain ISF homeostasis.
Recently, the Virchow Robin spaces and the astrocyte endfeet were identified as key anatomical components of the so-called “cerebral glymphatic system”. 18 This system was conceived to account for CSF movements observed in the healthy brain that may function to clear solutes such as amyloid β from the parenchyma and facilitate transport of small lipophilic molecules, particularly during sleep.18–20 While some controversy exists regarding details of this model, 21 these observations are clearly impactful, in that they give potential insight into the function of the astrocyte endfoot syncytium with regards to brain fluid movements.
The neurovascular unit
The BBB is not a static barrier, but rather it dynamically alters its properties in response to neuronal activity. The term “neurovascular unit” is used to reflect the communication between components of the BBB and cells in the greater brain parenchyma. 22 The neurovascular unit is a system composed of neurons, glia, endothelial cells, vascular smooth muscle, and immune cells that functions in part to trigger the hemodynamic responses to neuronal activity,23,24 regulate nutrient influx to support neuronal metabolism, 25 and modulate neuronal remodeling.
The term “neurovascular unit” highlights the dependence of neurons upon other central CNS cell types. One example of this dependency is the astrocyte-neuron lactate shuttle. 25 Neurons also rely on astrocytes for neurotransmitter recycling 26 and maintenance of neurons’ antioxidant capabilities through the production of ascorbic acid. 27
Historical perspectives
Historical models of cerebral edema
In the mid-to-late 1700s, cerebral edema was beginning to be recognized as an entity distinct from acute hydrocephalus, which theretofore was believed to etiologically underlie all cases of excess intracranial water. This recognition was driven by the observations of Robert Whytt (1714–1766) and George Cheyne (1671–1743) that excess intracranial fluid can occur without enlarged ventricles, a presentation typically accompanied by a “soft” appearing brain.28,29 Some observers speculated that the brain itself held the excess fluid.
Soon after these observations, writings and experiments by Alexander Monro (1733–1817), George Kellie (1720–1779), and John Abercrombie (1780–1844) led to the formation and rise in popularity of the Monro-Kellie axiom, which states that during health, the volume occupied by the contents of the cranium must remain in dynamic equilibrium, the implication being that the fluid influx rate must equal the efflux rate.30,31 This axiom was an important precursor to models of brain swelling. Unfortunately, excepting a few articles describing brains with a soggy gross appearance, interest in cerebral edema waned until the late 1800s. 32
Interest in cerebral edema was renewed due to Paul Ehrlich’s (1854–1915) identification of the BBB with aniline dyes in 1886, and the revival of the Monro-Kellie axiom. The latter was partially driven by the writings of Harvey Cushing (1869–1939). 33 The growing popularity of these concepts drew attention to the unique anatomy and physiology of cerebral circulation and indicated to contemporaneous researchers that mechanisms of cerebral edema formation were unique from those that drive edema formation in other body regions.
Based upon the gross appearance of brain tissue and data from a newly developed technique to quantify brain swelling, in the late 1910s and early 1920s cerebral edema was subdivided into “brain swelling” and “cerebral edema”, characterized by wet and dry tissue, respectively.34,35 While debate arose regarding the exclusivity of these subtypes, the controversy remained unresolved until the advent and optimization of electron microscopy techniques.
In 1965, Bakay and Lee applied electron microscopy to describe two different types of cerebral edema in their text Cerebral Edema. 36 In 1967, Igor Klatzo (1916–2007) termed these subtypes “cytotoxic” and “vasogenic”. 37 Vasogenic edema was defined by extravasation of fluid that contained plasma proteins and was attributed to vascular injury. In contrast, cytotoxic edema was characterized by cellular swelling and was attributed to inhibited cell volume regulation. While the latter edema subtype was termed cytotoxic to reflect its occurrence following toxicant exposure, such as triethyltin (TET) poisoning, water intoxication, or cyanide poisoning, cytotoxic edema was also known to occur following ischemia. Importantly, in Klatzo’s schema, cytotoxic edema could include extravasation of ions and water, but by definition did not include extravasation of plasma protein.
While reviewing these historical models, it is important to note that they are purely phenomenological and offer little in terms of mechanistic explanation. The development of molecular biology in the 1950s and 1960s allowed researchers to probe the molecular drivers of edema formation. Findings from studies utilizing these techniques indicated that all subtypes of cerebral edema, as well as hemorrhagic transformation, share common molecular antecedents. 38 Thus, subtypes of cerebral edema are best viewed as the manifestations of a program of pre- and post-transcriptional molecular events that is ultimately triggered by a brain insult. 38
Historical approaches to post-ischemic therapeutic intervention
Excepting neurons in specialized regions, neurons in the adult mammalian brain are arrested in the G0-phase of cell-cycle and can be considered to be essentially irreplaceable. Therefore, over the past few decades, acute CNS research has attempted to mediate direct neuroprotection through strategies such as attenuation of excitotoxicity, apoptosis, or oxidative stress. During this time, preclinical work in animal models of acute CNS injury led to the identification of over 1000 new potential neuroprotectants.39,40 However, this great expenditure of effort, time, and money has essentially failed, as none of these agents has shown effectiveness in clinical trials. 39
Possible explanations have been offered for the failure to translate promising preclinical findings into the clinic. Some have criticized the commonly used animal models of acute CNS injury, arguing that they do not accurately reflect human disease. 41 Others find fault with the experimental design used in many preclinical studies, arguing that methods like blinding would have prevented many of said false positives. 42 Yet others point out that clinical trials often do not replicate the experimental preclinical studies that appeared so promising.
While model validity and experimental design are clearly important, a more fundamental issue might be that agents designed to specifically salvage neurons may not abort the death or dysregulation of other components of the neurovascular unit. Neurons are fragile cells and cannot survive without the support of other cell types. Therefore, in addition to direct neuroprotection, a new goal for acute brain injury research is to investigate and attenuate mechanisms of endothelial, astrocytic, and microglial dysfunction and, thereby, create an environment permissible to neuronal survival. It follows that cerebral edema, a phenomenon that arises from dysfunction of astrocytes and endothelium, represents an important target for basic research and therapeutic intervention.
Core concepts of cerebral edema
Cerebral edema and swelling
The cranial contents are divided into a series of fluid compartments, which are spaces separated by barriers that are relatively impermeable to water and are maintained at homeostatic volumes. Examples of fluid compartments include the vasculature (∼100 mL), CSF (∼100 mL), brain interstitial space (∼100 mL), and brain intracellular space (∼1.1 L) (volumes refer to the human brain). 43 The water masses contained by these compartments are dynamic during health; for example, neuronal activity precipitates an increase in the intracellular water mass of local astrocytes.44,45
Cerebral edema is a pathological increase in the water mass contained by the brain interstitial space. Incidentally, although cytotoxic edema (oncotic cell swelling) is referred to as “edema” for purely historical reasons, it results in intracellular, rather than extracellular, fluid accumulation, it does not include a swelling component, and it is best regarded as a premorbid precursor to extracellular ionic edema. Transvascular cerebral edema (ionic edema and vasogenic edema) is detrimental because it manifests as brain tissue swelling. Swelling refers to a volumetric expansion of a given mass of tissue and can be generated by the accumulation of tumor, edema, or blood, although here, the focus is on edema.
Brain swelling causes a mass effect that exerts pressure on the surrounding shell of tissue. This pressure increase is magnified by the rigid enclosure of the skull, which places an upper limit on the volume that the brain might expand to. As the brain swells, it exerts mechanical forces on the skull interior, thereby increasing intracranial tissue pressure. When tissue pressure exceeds capillary pressure, capillary lumens collapse, precipitating a feedforward process wherein ischemia of the surrounding shell triggers further edema formation and further swelling in the next shell. 46
Cerebral edema requires perfusion
For cerebral edema and swelling to occur, the brain tissue must be perfused by an external fluid source. To illustrate this concept, imagine that a fresh biopsy of brain tissue is placed upon a dry surface. As the tissue is completely ischemic, cytotoxic edema will form and water will redistributed from the interstitium to the intracellular compartment. However, as the tissue is isolated from any possible source of new ions or water mass, the tissue will not become heavier and will not swell. For in vivo brain tissue, blood or CSF might serve as the source of this new water, although the relative contributions of these sources are in debate, as is described in the next section.
Water sources of ionic cerebral edema
While the water that drives ionic edema originates ultimately from the vasculature, there exist two major hypotheses regarding the immediate source of the new water mass that is required for brain swelling. In one hypothesis, water moves from the capillary lumen into the parenchyma, driven by osmotic forces, and is conveyed across capillary endothelial cells by mechanisms discussed in later sections of this review. In the context of ischemic stroke, the requirement for perfusion by an outside water source is fulfilled by post-ischemic reperfusion of the core and/or the ischemic penumbra.
The recent description of the glymphatic system led to the formulation of a second hypothesis, whereby CSF serves as the immediate source of ions and water. In this hypothesis, swelling occurs when influx of CSF into the parenchyma is enhanced and/or efflux of ISF is impaired, a situation that precipitates relative accumulation of ISF within the parenchyma. 47 Interestingly, both of these major hypotheses were alluded to in an essay on cerebral edema published in 1894. 32
In support of the first hypothesis, ionic edema formation is intimately associated with the local blood perfusion status. The post-ischemic reperfusion flow magnitude is positively correlated with edema load. 48 Furthermore, regional brain perfusion is correlated with the spatial magnitude of edema influx: In a rat model of malignant cerebral edema 8 h after permanent middle cerebral artery occlusion, edema fluid is located mostly in peri-infarct regions, with minimal edema fluid in the poorly-perfused core. 49 In human stroke, magnetic resonance imaging (MRI) shows that edema is first found in peri-infarct regions that are actively perfused. 50 In addition, following acute liver failure, cerebral blood flow is positively associated with edema load. 51 However, acceptance of this hypothesis is not universal, as there are uncertainties regarding the expression level of commonly cited molecular mechanisms for ion and water influx across brain endothelium. 47
As the second hypothesis was only recently formulated, relatively little literature exists to support or refute it. However, as acute CNS injury does appear to trigger chronic dysregulation of glymphatic clearance of interstitial solutes, this hypothesis merits additional scrutiny. 52
Two salient features of the BBB indicate that these hypotheses need not be mutually exclusive. Firstly, since brain endothelial cells in the healthy brain mediate high rates of water flux between the vascular compartment and interstitium without net water movements, 53 and thus exhibit high water permeability, the two aforementioned hypotheses could represent steps in a sequential process of water movements from blood to parenchyma. Secondly, the absence of a perivascular CSF-filled space surrounding brain capillaries suggests that their contribution to the glymphatic system may be minimal.14,15 Thus, these two hypotheses could explain ionic edema formation at different levels of the vascular tree.
Interestingly, the absence of a perivascular CSF-filled space surrounding brain capillaries does not prevent intracisternally-delivered fluorescent tracers from appearing in the capillary basement membrane. 18 In the glymphatic hypothesis, this presumably occurs due to the marker migrating first along the periarteriolar Virchow Robin spaces and then ostensibly by migrating longitudinally through the capillary basement membrane itself. However, it is unclear how the relatively dense pericapillary basement membrane is able to serve as a high-capacity channel for CSF flux, especially since the basement membrane is usually thought to serve as a physical barrier to solute movements. 54 It has been suggested that nonspecific binding of the fluorescent dextran tracer to the capillary basement membrane could underlie the pericapillary fluorescence reported in the abovementioned study. 21
Importantly, although the aforementioned points accurately reflect our current understanding of the formation of ionic edema, the two hypotheses do not account as equally well for the formation of vasogenic edema, which is best understood as reflecting the transcapillary flow of plasma proteins.
Diffusion versus bulk flow
The phenomenon of diffusion, namely, the tendency for molecules to spread from a point of high concentration to surrounding points of lower concentration, arises from random Brownian motions exhibited by particles when suspended in fluid. One property of diffusion is that the probability of finding a molecule at a given distance from its starting position within a given period of time, essentially the molecule’s speed of diffusion, is inversely proportional to the hydrodynamic radius of the molecule. Larger molecules will diffuse more slowly than smaller molecules.
Solutes may also move by bulk flow. In contrast to diffusion, where molecular migration is driven by movement of the molecule within a static substrate, bulk flow migration is driven by the movement of the fluid substrate itself, which in turn is driven by hydrodynamic or osmotic forces. Essentially, suspended particles are “swept along” by the fluid substrate. An important property of bulk flow is that the speed of migration of a suspended particle is dictated solely by the forces driving the migration of the fluid substrate and is therefore independent of the solute’s hydrodynamic radius. This property can be exploited to probe whether observed movements are due to diffusion or bulk flow. Parenthetically, bulk flow is utilized in convection-enhanced delivery, an intraparenchymal drug delivery technique that allows for much wider distribution of a given agent than diffusion-driven delivery. 55
Cerebral edema moves by bulk flow
The movement of ISF through the brain parenchyma was recognized as early as 1865, when His observed that material injected into the brain parenchyma spreads from the initial point of delivery. 56 In the early 1980s, Cserr et al. observed that radiotracers of varying molecular weights were cleared from the parenchyma with identical rates, indicating that brain ISF in the healthy brain moves by bulk flow rather than diffusion, with white matter tracts exhibiting higher fluid transport.57,58 Experiments using a cold lesion model of extracellular edema indicated that edema fluid also spreads through the parenchyma through bulk flow rather than diffusion. 59 Interestingly, extracellular fluid appears to take both pericellular and transcellular routes through the parenchyma. 60 Bulk flow of edema fluid is driven by hydrostatic and osmotic forces, produced by mass effect and derangements in ion transport, respectively. Better models of how these forces are generated in vivo are needed to improve our understanding of directional flux of ISF and cerebral edema in vivo.
Clearance of cerebral edema
The clearance routes of extracellular edema are incompletely understood. Studies examining clearance of ISF indicate that a sizable portion is removed from the parenchyma along perivascular spaces and is deposited into the subarachnoid space, whereupon it is cleared by mechanisms of CSF absorption, a topic that itself is not without controversy.18,21,61,62 Historically, the arachnoid villi were considered to mediate most of the absorption of CSF. However, a significant portion of CSF might be cleared through the cervical lymphatics, either through the perineuronal subarachnoid spaces that surround cranial nerves or by passage through the olfactory submucosa. 63 In addition to efflux into the subarachnoid space, a portion of brain ISF is cleared by cerebral capillaries, though their precise contribution remains controversial.
The clearance pathway taken by ISF depends upon its location in the brain: In rats, CSF in the cisterna magna accounts for efflux of 60–70% of midbrain ISF but only 10–15% of caudate nucleus ISF. 64
The transcapillary mechanism and Starling’s principle
In the late 1800s, Starling formulated the basic model for the transcapillary driving forces during edema formation.
65
In his model, edema formation requires two factors: (i) a driving force that either “pushes” or “pulls” substances into or out of tissues; (ii) a “permeability pore” that mediates the transcapillary flux of these substances. In its original formulation, Starling’s principle stated that the transendothelial flux of fluid depends upon the net osmotic and hydrostatic pressure and a single permeability coefficient, K. To reflect mechanisms of cerebral edema formation, Starling’s principle was reformulated in 2007, and the permeability coefficient K, was separated into two constants, the net hydraulic conductivity, KO, and the net osmotic conductivity, KH, to account for the transcapillary efflux of water (Jv).
38
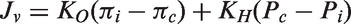
Starling’s principle states that the driving force is the sum of the hydrostatic and osmotic pressure gradients. Capillary hydrostatic pressure (Pc) is dictated by the precapillary arteriolar pressure, the postcapillary venular pressure, and the capillary resistance, while the tissue pressure (Pi) is a function of the volume of the ISF and the tissue compliance. The osmotic pressures of blood (
In healthy tissues, both the osmotic term [
Driving forces for cerebral edema formation
Cytotoxic edema
Cytotoxic edema (oncotic cell swelling) is a premorbid process whereby cells swell due to influx of osmolites (mainly Na+ and Cl–) and water from the interstitial spaces into the intracellular compartment. This process takes place in all CNS cell types following CNS injury, and is particularly prominent in astrocytes. Astrocyte swelling appears to be a general response of astrocytes to injury and occurs quickly following a variety of types of CNS injury, including ischemia, trauma, hypoglycemia, status epilepticus, and fulminant hepatic failure, though importantly, the mechanisms driving the swelling may differ among etiologies. 67 Importantly, cytotoxic (cellular) edema does not generate tissue swelling, as it simply represents a rearrangement of parenchymal water mass and does not involve the addition of new water mass to the brain. Nevertheless, cytotoxic edema is an important initial step in the formation of cerebral edema and swelling, as it generates the driving force for influx of ionic and vasogenic edema, which do cause swelling.
Cellular influx of osmolites may occur due to primary active transport or secondary active transport. Primary active transport requires a continuous supply of adenosine triphosphate (ATP) to provide energy for “pumps” such as the Na+/K+-ATPase and Ca2+-ATPase. Secondary active transport harnesses the potential energy stored in transmembrane ionic gradients previously generated through primary active transport. Examples of secondary active transporters include ion channels and cotransporters such as the Na+/ K+/ Cl– co-transporter (NKCC1) and the Na+/Ca2+ exchanger. Following many types of CNS injury, intracellular ATP becomes depleted and thus, mechanisms that are independent of intracellular ATP, like secondary active transport, are more likely to be relevant to the formation of ionic edema.
Ions involved in cytotoxic edema can be conceptually divided into primary drivers and secondary participants. Primary drivers are substances that, through extrusion by primary active transport, are more concentrated outside of the cell than inside. Secondary participants exhibit no pre-existing electrochemical gradient. However, rearrangement of primary drivers stimulates secondary participants to flux. For example, if Na+ is the primary driver, Cl– and water are secondary participants that move in order to maintain electrical and osmotic neutrality.
Per its namesake, astrocytic cytotoxic edema is usually triggered by exposure to endogenous toxins (K+, glutamate, H+), exogenous toxins (ammonia, TET, cyanide)68,69 or, unique to the Sur1-Trpm4 (sulfonylurea receptor 1 – transient receptor potential melastatin 4) channel, intracellular ATP depletion. Maladaptive ion influx may ensue, generating a transmembrane osmotic gradient that drives water influx and causes cell swelling. Cytotoxic edema is one instance of the more general category of astrocyte swelling; the latter includes forms of astrocyte swelling that are not strictly pathological. For example, while hypotonicity, i.e. water intoxication, may trigger astrocyte swelling, this swelling is a purely osmotic phenomenon and lacks the maladaptive ion transport characteristic of cytotoxic edema.
Constitutively expressed drivers of cytotoxic edema
Swelling due to endogenous brain toxin exposure occurs because astrocytes possess strong homeostatic mechanisms that evolved to maintain the extracellular fluid composition within a range of acceptable values. Following injury, certain molecules normally present in ISF greatly increase in concentration, whereupon astrocytes attempt to maintain ISF homeostasis by activating a variety of normally beneficial secondary active transporters that drive solute transport. In extreme conditions, excessive activation of these secondary active transport mechanisms occurs, leading to massive Na+ and water influx and cytotoxic edema (Figure 2).
70
All mechanisms of cytotoxic edema involve Na+ overload which, interestingly, is sufficient to impair astrocyte volume regulation
71
perhaps indicative of why astrocytes normally exhibit relatively strong volume regulation,47,72–74 yet swell so markedly following injury.
Major routes for influx of ions and water in astrocytic cytotoxic edema. Schematic depiction of the major astrocytic transporters and channels that are implicated in the formation of cytotoxic edema; in regards to water transport, single-headed arrows denote water co-transport, while double-headed arrows denote passive water transport.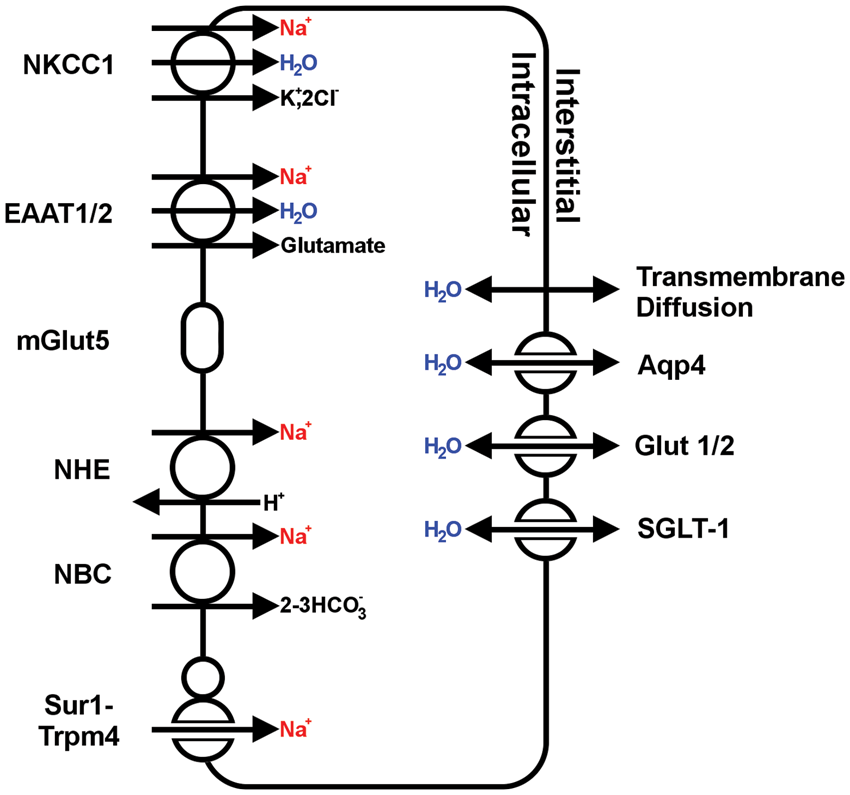
The normal ISF K+ concentration ranges between 2.7 and 3.5 mM and is maintained by astrocytic Na+/K+-ATPase, NKCC1, and Kir4.1.44,75,76 Following many types of CNS injury, extracellular K+ accumulates to dangerous levels, sometimes in excess of 60 mM, due to energy depletion and Na+/K+-ATPase failure, cell membrane rupture, or as a byproduct of glutamate excitotoxicity. 77 In the healthy brain, astrocytes function to clear excess extracellular K+, a function that is associated with benign astrocyte swelling.44,45,78 However, in conditions of greatly increased extracellular K+, K+ clearance triggers cytotoxic edema formation. In the range of 25–117 mM, the magnitude of astrocyte swelling becomes linearly related to extracellular [K+]. 79 The bumetanide-sensitive electroneutral NKCC1 transporter, a member of the Na+/K+/2Cl– transporter family, is particularly important to K+-induced astrocyte swelling. NKCC1 is constitutively expressed by astrocytes in all regions of the adult brain and its activity is enhanced after ischemia and acute liver failure due to increased protein expression and phosphorylation.80–82 NKCC1 carries a net of four osmolites inward per turnover and is capable of water co-transport. 83 In vitro experiments using cultured primary astrocytes demonstrated that NKCC1 contributes to cell swelling in conditions of high extracellular potassium.84–86 In vivo, swelling is attenuated with bumetanide, an NKCC1 inhibitor, following trauma and ischemia.87–90
ISF glutamate in the healthy brain is typically maintained around 10 μM, depending on the region sampled. Following CNS injury such as stroke or trauma, extracellular glutamate can accumulate to greater than 200 μM due to spreading depolarization, synaptic release, or neuronal lysis.91–93 Astrocyte swelling occurs when extracellular glutamate ranges between 50 μM and 5 mM. 94 Parenthetically, while brain extracellular glutamate is increased after acute liver failure, 95 glutamate is thought to play a minor role in astrocyte swelling after acute liver failure, a process that is driven primarily by ammonia. Glutamate can induce astrocyte swelling through two mechanisms. Firstly, the human excitatory amino acid transporter (EAAT) glia-specific family members EAAT1 (a.k.a. GLAST in rat) and EAAT2 (a.k.a. Glt-1 in rat) mediate influx of glutamate with co-transport of Na+ and water. 96 Both transporters are constitutively expressed by astrocytes in the adult brain. 97 EAAT2 is the major (>90%) contributor to CNS glutamate uptake and homeostasis during health, and forms a multiprotein complex with aquaporin-4.98–100 Secondly, the metabotropic glutamate receptor 5 (mGluR5), 101 which forms a multiprotein complex with Na+/K+-ATPase and aquaporin-4, also has been implicated in glutamate-induced astrocyte swelling.102,103 Notably, mGluR5 is minimally expressed by resting (nonreactive) adult astrocytes. 104 In line with the notion that many of the mechanisms that drive cytotoxic edema were evolved due to their beneficial actions, if glutamate uptake and glial swelling are inhibited, glutamate mediated neurotoxicity is worsened. 105
ISF pH is also tightly regulated by astrocytes. Many types of CNS injury interrupt oxygen delivery and lead to ATP depletion, triggering anaerobic metabolism and lactic acid generation that can precipitate a drop in extracellular pH. If ISF pH drops below 6.8, compensatory ion fluxes in astrocytes are sufficient to induce cytotoxic edema.106,107 Two general classes of astrocyte transporters are involved in pH homeostasis and acidosis-induced astrocyte swelling. Firstly, the constitutively expressed bicarbonate-independent and amiloride sensitive Na+/H+ exchanger (NHE), 108 which facilitates a 1:1 exchange of intracellular H+ for extracellular Na+, mediates acidosis-induced astrocyte swelling in vitro and in vivo.109–116 Secondly, the constitutively expressed bicarbonate dependent Na+/HCO3– transporter family (NBC) may also contribute to acid-induced swelling.107,108,117 However, compared to the NHE family, the contribution of the NBC family to cytotoxic edema in vivo is less well understood.
Brain interstitial ammonia is normally ∼100 μM, 118 and can rise above 5 mM during acute liver failure. 119 Astrocyte swelling occurs when cells are exposed to millimolar concentrations of ammonia. 120 Ammonia is internalized by astrocytes and converted to glutamine via glutamine synthetase. It has been hypothesized that glutamine is shuttled to the mitochondria, whereupon mitochondrial phosphate-activated glutaminase (PAG) converts glutamine to glutamate and ammonia, which can trigger production of reactive oxygen species (ROS) and activation of the mitochondrial permeability transition, culminating in astrocyte swelling, i.e. the “Trojan horse hypothesis”. 121 Treatment of astrocytes with ammonia also triggers upregulation and phosphorylation of NKCC1, which mediates ammonia-induced osmolite influx and astrocyte swelling. 122
De novo expressed drivers of cytotoxic edema
In contrast to constitutively expressed mechanisms of cytotoxic edema, which can be viewed as maladaptive versions of normally beneficial processes, the Sur1-Trpm4 channel is expressed in the CNS only following injury, and is absent from healthy brain. Trpm4, a constitutively expressed monovalent cation channel that opens in response to intracellular Ca2+, is the pore forming subunit of Sur1-Trpm4.123–125 In all CNS cells, CNS injury triggers activation of the hypoxia-inducible factor 1 (HIF1) transcription factor, which induces de novo expression of Sur1, 126 an ATP-binding cassette, which associates with Trpm4 and doubles its Ca2+ sensitivity and sensitizes the channel to ATP depletion.123,125,127 It is hypothesized that this channel evolved to protect cells against Ca2+influx in less extreme types of CNS injury by allowing Na+ influx, membrane depolarization, and thus a reduction of the inward driving force for Ca2+. However, in the context of severe CNS injury and ATP depletion, excessive influx of Na+ through Sur1-Trpm4 drives maladaptive cell swelling and cytotoxic edema (Figure 2). Inhibition of Sur1-Trpm4 with sulfonylurea drugs prevents cytotoxic edema in vitro and reduces edema in vivo following ischemia and trauma.49,123,128,129 Evidence also suggests that the Sur1-Trpm4 channel contributes to ammonia-induced astrocyte swelling in vitro and in vivo. 130
Routes for transmembrane water flow during cytotoxic edema
The influx of ions generates a transmembrane osmotic gradient that favors the influx of water. Water might flow into astrocytes through three routes. Firstly, simple diffusion through the lipid bilayer can account for significant water influx; however, this route is low capacity and not thermodynamically favored. Secondly, transmembrane water channels, including the aquaporin family as well as certain astrocytic transporters such as SGLT1, GLUT1, and GLUT2 possess passive water permeable pores, where water flux is driven by an osmotic gradient. 83 Thirdly, certain ion transporters expressed by astrocytes that drive ion fluxes during cytotoxic edema also mediate secondary water co-transport by carrying a fixed number of water molecules with their ionic cargo per turnover. Examples of these transporters include NKCC1 and the glutamate transporters EAAT1 and EAAT2. 83
Aquaporin channels in the CNS
Recently, aquaporin channels have been recognized as important mediators of plasmalemmal water fluxes in cerebral edema. In 1992, Peter Agre described a novel molecular conduit for bulk water flow through the plasma membrane, a protein later christened aquaporin-1. 131 Aquaporin monomers constitute the functional water channel subunit of aquaporin channels and are exquisitely selective to water by virtue of a dumbbell-shaped pore with an amphipathic bottleneck. 132 Aquaporin water transport is passive and bidirectional; the rate and directionality of aquaporin mediated water flux is determined exclusively by the transmembrane osmotic pressure gradient.
While there are 14 known aquaporin channels, only aquaporin-1, aquaporin-4, aquaporin-9, and aquaporin-11 are expressed in the CNS.133,134 Of these, aquaporin-4 is the major aquaporin expressed by astrocytes, and is the dominant contributor to cerebral edema formation and clearance. In the prefrontal cerebrum under normal conditions, aquaporin-4 expression is strongly localized to the perivascular astrocyte endfoot; astrocyte soma and main processes do not exhibit notable aquaporin-4 immunoreactivity. 135 Aquaporin-4 also is localized to the submeningeal astrocyte endfeet and in the leading lamellipodia of migrating astrocytes. 136 While aquaporin-4 is mostly astrocyte-specific, it has been reported to be upregulated in microglia following LPS injection. 137
Aquaporin-4 is expressed in two major N-terminal splice variants, the M1 (323 amino acids) and M23 (301 amino acids), as well as four other alternative isoforms whose functional significance is still being investigated.138–141 The M23 isoform is able to multimerize on the plasmalemma into large complexes called orthogonal arrays of intramembraneous particles (OAPs) that, on the luminal face of the astrocyte endfoot, exhibit a density of 500–600/µm2 and occupy approximately 50% of its surface area.135,142,143
The precise contribution of the astrocyte endfeet and aquaporin-4 to brain ISF during health is currently being investigated. Aquaporin-4 represents a high capacity route of water flux past the astrocyte endfoot layer of the BBB; other routes include paracellular water flux, co-transport of water, or simple diffusion. 70 Aquaporin-4 may then be necessary to rapidly neutralize osmotic gradients that are generated during ion transport, akin to its role in skeletal muscle during contraction. 144 This might explain the dependence of the aforementioned glymphatic system on aquaporin-4. 18 In addition, aquaporin-4 knockout animals exhibit CNS abnormalities linked with altered ion transport, such as expansion of the extracellular space, increased brain water content, cochlear deafness, and increased seizure threshold.145–148
Aquaporin channels rely on ion transport
As passive channels, aquaporins are completely dependent upon the activity of ion transporters for water flux. Therefore, the study of aquaporin-dependent cerebral edema is essentially the study of ion transport. 70 While aquaporins alone are undoubtedly important in the generation of cerebral edema, future work will address the interaction between ion transport and aquaporin water flux. 149
Role of aquaporin-4 in cytotoxic edema
Aquaporin-4 worsens subtypes of cerebral edema that form in the context of an intact BBB.150–154 Mice with aquaporin-4 knockout or mislocalized aquaporin-4 exhibit reduced astrocyte swelling during water intoxication, suggesting a role for aquaporin-4 in cytotoxic edema of astrocytes.153,155,156 Notably, the alternative routes of astrocyte plasmalemma water influx are physiologically and pathologically important, as even with knockout of aquaporin-4, astrocytes still swell quickly in response to hypoosmotic challenge. 157
Cytotoxic edema generates driving forces
The influx of primary drivers like Na+ and secondary participants like Cl– and water into cells during cytotoxic edema depletes these constituents from the extracellular space.158,159 Sequestration of these constituents is possible because the intracellular compartment is much larger than the extracellular compartment, which only comprises 12–19% of total brain volume. 160 Cytotoxic edema thereby generates a new Na+ gradient across the BBB, where Na+ concentration becomes higher in the vascular compartment compared to the interstitial compartment. For example, in one study that used a rat model of global ischemia, extracellular [Na+] declined from a baseline of 141 mM to 74 mM; as plasma [Na+] was 134 mM, cytotoxic edema generated a transendothelial Na+ concentration differential of approximately 60 mM. 158
This newly formed Na+ gradient is preserved even if cells lyse and release their intracellular contents because the extracellular space is much smaller than the intracellular space and because K+ will remain mostly bound to intracellular proteins and macromolecules. 161 The Na+ gradient generated by cytotoxic edema serves as a source of potential energy that drives subsequent influx of ionic edema fluid.
Endothelial dysfunction
The endothelial permeability pore
Acute CNS injury triggers a program of pre- and post-transcriptional molecular changes in the neurovascular unit that results in the formation of endothelial “permeability pores” and subsequent loss of BBB integrity. Progressive endothelial dysfunction can be organized into three phases (ionic edema, vasogenic edema, and hemorrhagic transformation), based upon the principle substances that undergo transcapillary movement (Figure 3).
Phases and select mechanisms of endothelial dysfunction. In ionic edema, water flux (blue arrows) and ion flux (grey arrows) are mediated by plasmalemma channels and transporters; vasogenic edema, which includes extravasation of plasma proteins, but not erythrocytes, is mediated by transcellular channels, MMP degradation of tight junctions, and endothelial retraction, phenomena that are, in part, triggered by VEGF, Ang2, and CCL2 signaling; hemorrhagic transformation occurs due to structural failure of the vessel, driven by either complete degradation of tight junctions or Sur1-Trpm4-mediated oncotic cell death of endothelial cells.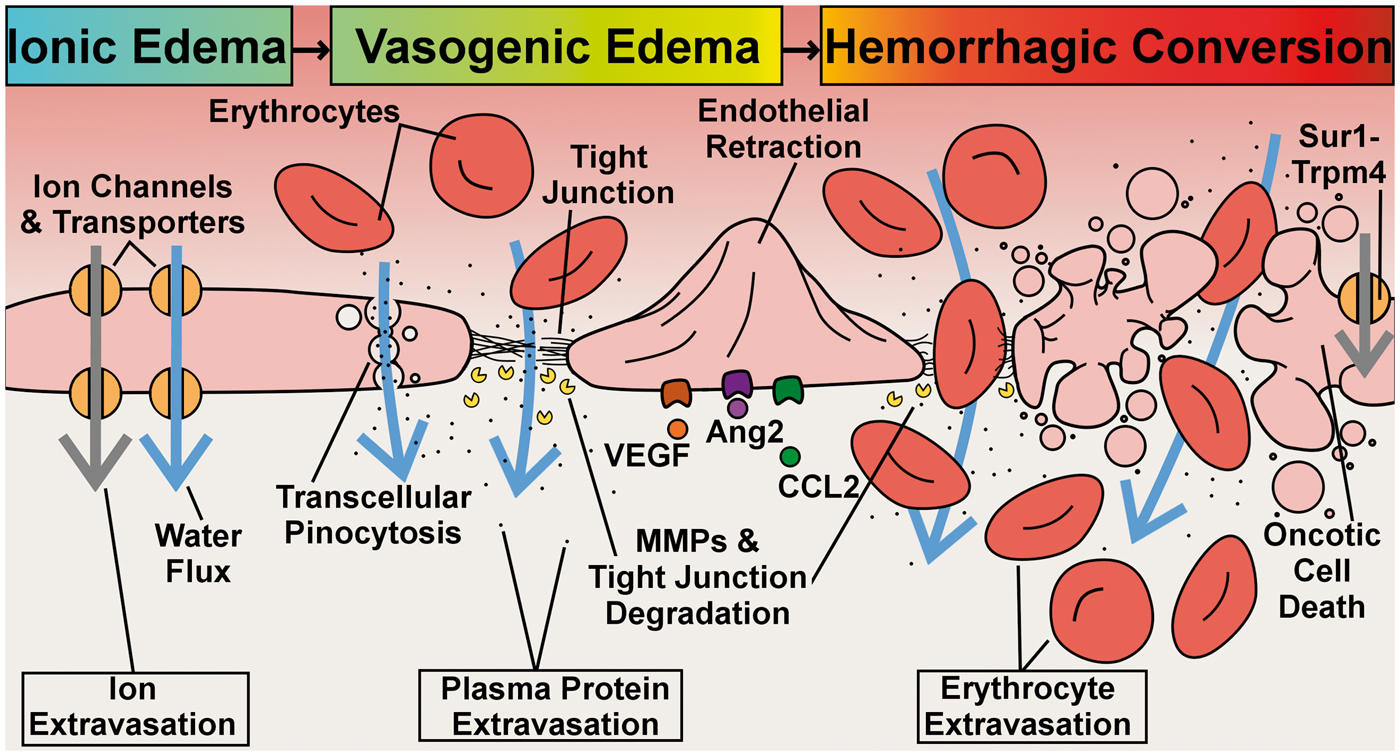
The three phases of endothelial dysregulation are thought to occur sequentially, although the rapidity of transition between phases probably depends on injury type and severity. Furthermore, as many etiologies of brain endothelial dysregulation and cerebral edema are focal in nature, brain tissue usually exhibits a complex spatiotemporal pattern of the different phases of endothelial dysregulation.
First phase: Ionic edema
During ionic edema, the potential energy contained in the transendothelial Na+ gradient generated by cytotoxic edema drives extravasation of osmolites and water. Na+ is transported inward along its concentration gradient by brain endothelial cells, resulting in the accumulation of Na+ in the brain parenchyma and the generation of a nonzero osmotic driving force (the
Ionic edema and vasogenic edema are forms of extracellular edema that differ in an important way. Vasogenic edema, but not ionic edema, includes extravasated serum proteins. The preservation of BBB integrity has two important implications for mechanisms of ionic edema. Firstly, ion influx during ionic edema is exclusively mediated by endothelial ion channels and transporters; physical disruptions to the BBB, such as occurs with reverse pinocytosis or degradation of tight junctions, do not represent viable pathways for ion flow, as they would inevitably include plasma proteins. Secondly, preservation of the BBB integrity implies that KH remains zero. The dynamics of ionic edema are governed only by the osmotic term in Starling’s principle.
Parenthetically, while a subtype of extracellular edema that lacked serum protein extravasation has long been recognized, ionic edema was only recently defined as a distinct cerebral edema subtype. 38 Prior to this distinction, all edema subtypes not involving serum protein extravasation were grouped under the common term “cytotoxic edema”, which therefore encompassed cytotoxic edema, as the term is used presently, as well as ionic edema.
For ionic edema to form, Na+, Cl–, and water must first be transported inward through the luminal membrane and then transported through the abluminal membrane of brain capillary endothelial cells (Figure 4). Therefore, ionic edema is essentially a two-step transport process. Given that many brain endothelial channels and transporters exhibit a polarized distribution at these membrane faces, the transmembrane routes taken by ions and water differ between the luminal and abluminal membrane. Similar to cytotoxic edema, ion flux is driven by either primary active transport or secondary active transport.
Major routes for influx of ions and water in ionic edema. Schematic depiction of the major endothelial transporters and channels that have been implicated in the formation of ionic edema; in regards to water transport, single-headed arrows denote water co-transport, while double-headed arrows denote passive water transport.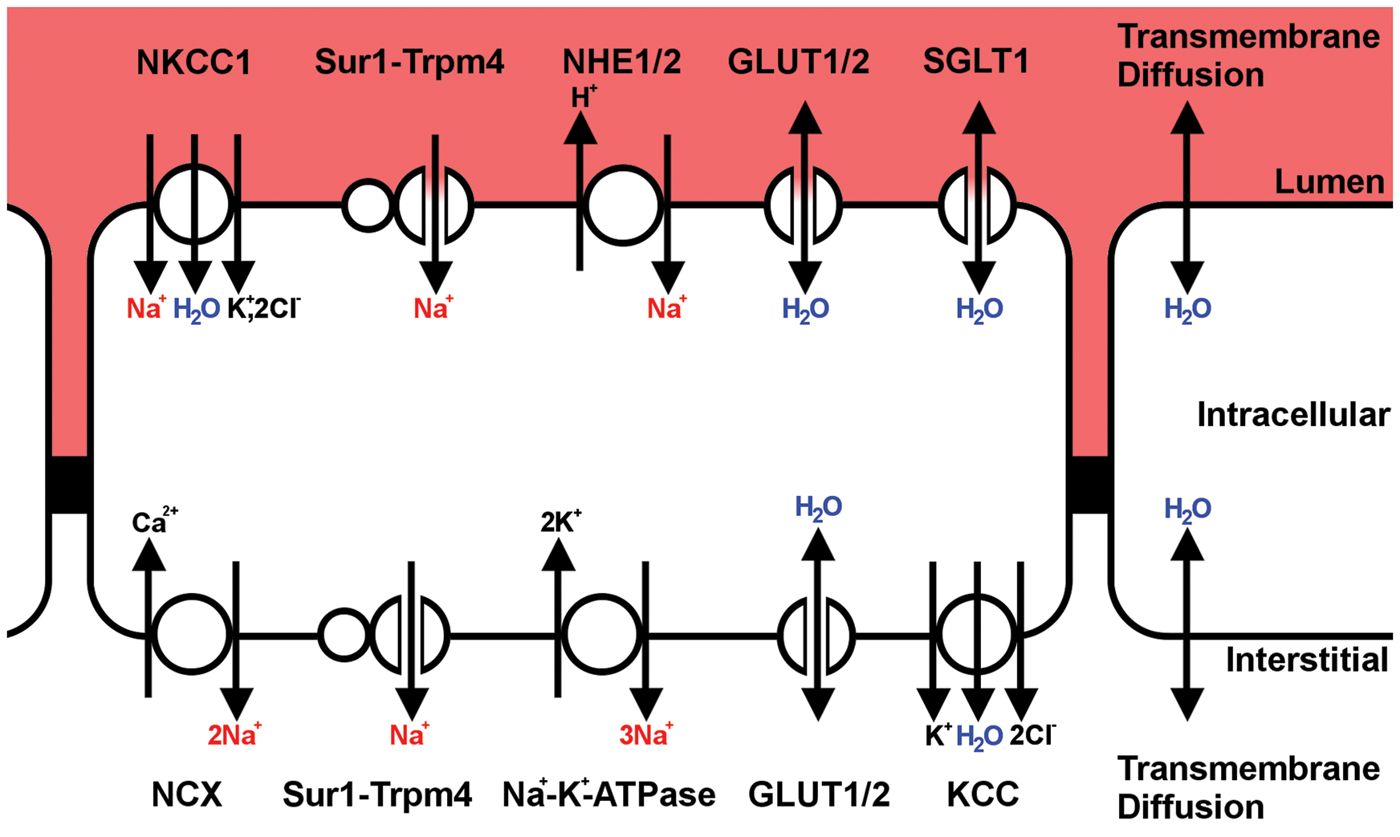
As ionic edema involves, in part, the uptake of ions and water by brain endothelial cells, it is analogous to cytotoxic edema, but differs in that the ion and water fluxes are polarized. Specifically, channels on the luminal membrane of brain endothelial cells drive the cellular uptake of vascular ions and water, which may manifest as endothelial cell swelling. Endothelial swelling is then “relieved” by channels on the abluminal membrane, which permit efflux of ions and water into the brain interstitium, thereby completing the transcapillary flux of ions and water.
Constitutively expressed drivers of ionic edema
The sodium-hydrogen antiporter (NHE) family members, NHE1 and NHE2, are constitutively expressed on both luminal and abluminal membranes of brain endothelium. During ionic edema, NHE family members contribute to Na+ influx across the luminal membrane. 165 Na+/H+ exchange in vitro is stimulated by hypoxia and hypoglycemia, conditions that occur following cerebral ischemia. 165 Intravenous delivery of Na+/H+ exchange inhibitors reduce cerebral edema formation in a rat stroke model, putatively by attenuating luminal NHE activity.113,166,167
The cation-chloride transport family member, NKCC1, is constitutively expressed on the luminal membrane of brain endothelium, is upregulated and activated via phosphorylation in response to ischemia, and contributes to Na+ influx across the luminal membrane during ionic edema formation.90,168 In addition to mediating solute influx that osmotically drives water influx, NKCC1 also mediates secondary active transport of water at 590 molecules of H2O per transporter turnover, thereby allowing it to pump water up an osmotic gradient. 83 Influx of ionic edema is attenuated with intravenous delivery of bumetanide, an NKCC1 inhibitor, thus supporting the role of NKCC1 in ionic edema formation.87,88
Under conditions of adequate energy, Na+ efflux across the abluminal membrane is primarily mediated by the Na+-K+-ATPase, a primary active transporter that is selectively expressed on the abluminal membrane of brain endothelial cells. 9 Notably, as this efflux route depends on ATP, its contribution is likely minimal during severe energy depletion such as during ischemia, although it may become relevant with timely reperfusion. In addition, the Na+/Ca2+ exchanger exists on the abluminal membrane and might contribute to Na+ efflux by virtue of its ability to operate in reverse-mode where Na+ is expelled in exchange for Ca2+ influx.169,170 As this efflux route is ATP-independent, Na+/Ca2+ mediated Na+ efflux might be particularly relevant during total ischemia.
De novo expressed drivers of ionic edema
The Sur1-Trpm4 channel is upregulated by capillary, arteriole, and venule endothelial cells in response to CNS injury, 127 and contributes to the formation of ionic edema by mediating Na+ influx at the luminal membrane and Na+ efflux at the abluminal membrane. Post-ischemic blockade of the channel by glibenclamide, a potent Sur1 antagonist,171–173 reduces edema formation by 50%, indicating that Sur-Trpm4 plays a key role in transcapillary Na+ influx that occurs during ionic edema. Parenthetically, as glybenclamide penetrates the BBB in ischemic brain tissue, 49 and Sur1 is expressed after injury by all CNS cells, its effects upon edema formation may not be solely due to inhibition of luminal endothelial Sur1. 174 As Sur1 is not constitutively expressed, this mechanism is injury-specific. Furthermore, as this mechanism relies on transcriptional gene expression and hence ATP, it is most relevant in ischemic, but still perfused vessels.
A disease-specific molecular driver of cytotoxic and ionic edema, the Sur1-Trpm4 channel is a highly promising target for pharmacological inhibition. Notably, the glyburide advantage in malignant edema and stroke (GAMES-RP) trial, a randomized phase II trial that evaluated the efficacy of an intravenous formulation of glybenclamide (CIRARA or RP-1127) in patients with large territory ischemic stroke, 175 was recently concluded. Adverse drug reactions are most commonly due to unwanted effects upon normal function that are mediated by the drug’s primary pharmacological mechanism. For example, inhibition of the N-methyl-D-aspartate receptor (NMDAR), a protein that is constitutively expressed throughout the brain, is associated with adverse CNS drug effects such as dizziness, sedation, agitation, hallucination, and confusion. 176 These adverse effects have led many researchers to abandon NMDAR antagonism as a strategy to combat excitotoxic injury following ischemia. However, as Sur1-Trpm4 is only expressed in the injured brain, CNS effects resulting from the drug’s primary pharmacology will be specific to injured brain tissue, and adverse drug effects will be minimized.
Transendothelial routes for Cl– and water during ionic edema
Na+, the primary driver of ionic edema, drives the influx of secondary participants like Cl– and water in order to equalize electrical and osmotic gradients. Transendothelial Cl– flux is likely mediated by Cl– channels and Cl– co-transporters such as NKCC1 and KCC. 177
Routes for transendothelial water influx during cerebral edema formation.a

bExpressed as the hydraulic permeability coefficient (Lp with units of cm3/s).
cExpressed as the diffusive water permeability coefficient (Pf with units of cm sec-1) of unmodified Xenopus oocytes (first row), or Xenopus oocytes expressing a particular water-permeable channel. Note that Pf is dependent upon the abundance of the expressed water channel and thus measurements of single-channel Lp is not directly translatable to plasmalemma Pf measurements. In addition, note that Pf is surface-area independent and is related to Lp through the equation:
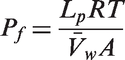
where A is the membrane area,
dExpressed as number of water molecules co-transported per transporter turnover.
eNot applicable (–).
fAquaporin channels are minimally expressed by brain endothelium.
gNot determined (ND).
Role of the astrocyte endfoot and aquaporin-4 in ionic edema
Aquaporin-4 worsens ionic edema, a subtype of cerebral edema that occurs in the context of an intact BBB.150–154 Conversely, overexpression of aquaporin-4 enhances ionic edema formation. 183 Interestingly, following cerebral ischemia, aquaporin-4 is upregulated primarily by white matter astrocytes. 184 Given that white matter can exhibit greater swelling after ischemia than grey matter,184–186 these data indicate that white matter may play an underappreciated role in ionic edema formation and brain swelling.
Recently, it was postulated that a primary function of the astrocyte endfoot syncytium, and of aquaporin-4, is to mediate transglial water flux. 18 It was suggested that dysregulation of this function occurs following CNS injury, and results in reduced glymphatic flow, which is thought to represent a primary mechanism driving ionic edema formation. 47 While this hypothesis might account in part for ionic edema, a strict adherence to this model would imply that the brain endothelium has little, if any, contribution to cerebral edema formation. Omitting a key role for endothelium is implausible, given the many endothelial transporters and channels implicated in the formation of edema. It is more likely that, in addition to its hypothesized role in glymphatic-mediated edema, aquaporin-4 interacts with and potentiates the endothelial water fluxes that drive ionic edema formation. Unfortunately, without a greater understanding of how aquaporin-4 controls water and solute flux through the endfoot layer of the BBB, it is unclear how aquaporin-4 water transport might affect endothelial ion transport.
Second phase: Vasogenic edema
Vasogenic edema is a form of extracellular edema characterized by breakdown of the BBB, wherein a transendothelial permeability pore forms that permits extravasation of water and plasma proteins such as albumin and IgG into the brain interstitial compartment. Unlike hemorrhage, capillary structural integrity is maintained during vasogenic edema such that passage of erythrocytes is prohibited. Therefore, vasogenic edema is best viewed as a cell-free blood ultrafiltrate, i.e. plasma.37,187 The permeability pore that allows the passage of solutes during vasogenic edema likely has contributions from more than one mechanism (see below). While all levels of the vascular tree contribute to vasogenic edema formation, brain capillaries are a particularly major contributor. 188
Once physical communication between the vascular and interstitial compartments is established, microvessels behave like fenestrated capillaries and therefore both hydrostatic and osmotic pressure gradients can affect edema formation, although hydrostatic pressure represents the primary driving force for vasogenic edema formation. 189 Determinants of the hydrostatic pressure gradient, such as intracranial pressure, systemic blood pressure, capillary occlusion, and vasospasm, are important to vasogenic edema dynamics. Determinants of the osmotic pressure gradient, which now include all osmotically active molecules such as Na+ and proteins, also can influence water flux.
The influence of hydrostatic pressure on vasogenic edema has direct clinical implications. For example, systemic blood pressure must be kept high enough to maintain brain perfusion, but in excess will promote edema formation.189,190 In addition, intracranial pressure must be kept low enough to maintain tissue perfusion, but high enough to counteract edema influx. 189 Optimization of these parameters is a difficult, multifactorial problem.
The concept that only the osmotic forces influence ionic edema, while both osmotic and hydrostatic gradients influence vasogenic edema may help to explain the mixed outcomes that occur following decompressive craniectomy, a procedure that abruptly lowers intraparenchymal pressure.191,192 Decompression is safe if done early, during the ionic edema stage, as it aids in the restoration of tissue perfusion. However, if done later during vasogenic edema, decompression will decrease tissue pressure, thereby increasing the hydrostatic gradient and driving edema influx.193,194
Mechanisms of vasogenic edema
Protein and water may passage from the vascular compartment to the interstitial compart through transendothelial channels formed by dysregulation of pinocytosis. Pinocytosis, a process whereby blood solutes are enveloped by luminal endothelial membrane ruffles, 195 are transported across the cytoplasm, and are released at the abluminal membrane. 196 Following CNS injury, pinocytic vesicles that are capable of carrying solutes and water have been observed to fuse and form transendothelial channels that span from the luminal to the abluminal membrane.197,198 Controversy still exists regarding this mechanism. 199
It is generally agreed that vasogenic edema can form via paracellular transport past endothelial cells. Inflammation and cerebral ischemia can trigger actin-dependent endothelial cell rounding or retraction and increased endothelial permeability. 200 Endothelial retraction is an ATP-dependent process that can be triggered by thrombin,200–203 a protease that is upregulated following ischemia and is highly abundant in the brain parenchyma following intracerebral hemorrhage. However, as some evidence suggests that in lieu of tight junction disruption, endothelial retraction is not sufficient to impair barrier resistance, 204 this mechanism might serve to enhance rather than initiate vasogenic edema formation. It has been speculated that endothelial cell retraction might have been evolved to facilitate transmigration of leukocytes that contribute to the beneficial clearance of necrotic debris produced by many types of CNS injury. 205
Paracellular permeability pores also can be generated by vascular endothelial growth factor (VEGF) signaling. Brain injury triggers expression of VEGF,206–208 which triggers decreased expression of tight junction proteins,209,210 uncoupling of interendothelial tight junctions, increased hydraulic permeability of vessels 211 and promotes edema formation. 212 Inhibition of VEGF reduces edema associated with post-ischemia reperfusion and brain tumors.213,214 Like many mechanisms that produce cerebral edema, VEGF signaling is not purely maladaptive, but rather is linked to angiogenesis. If administered early following experimental stroke in rats, recombinant VEGF increases edema formation, but if given at later times, VEGF stimulates angiogenesis in the penumbra and improves neurological recovery. 215 As some studies have shown that the extent of angiogenesis is correlated with survival following ischemic stroke, angiogenesis might be an important factor in recovery following ischemia. 216 In addition to VEGF, a host of signaling molecules including CCL2, angiopoietin 2 (Ang2), and nitric oxide are released following injury, and can inhibit expression of tight junction proteins and thereby exacerbate vasogenic edema formation.217–221
Endothelial basement membrane proteins and tight junction proteins also can be lost following CNS injury through protease degradation. Following injury, matrix metalloproteinase (MMP) activity increases through de novo expression and activation of latent MMPs, resulting in degradation of basement membrane and tight junction proteins.222–227 MMP inhibitors reduce ischemia- and reperfusion-associated cerebral edema,36,228,229 partially by preventing degradation of tight junction proteins. 230
Role of the astrocyte endfoot and aquaporin-4 in vasogenic edema
In contrast to its role in cytotoxic edema or ionic edema, knockout of aquaporin-4 is associated with worsened edema following injuries that precipitate vasogenic edema formation, such as trauma or cold lesion. 148 These data suggest that the astrocyte endfoot and aquaporin-4 contributes to the clearance of vasogenic edema, and likely mediates the clearance of other forms of extracellular edema, such as ionic edema.
Third phase: Intracerebral hemorrhage
Intracerebral hemorrhage can occur as a primary injury, as in the context of traumatic brain injury, or as secondary injury, where its formation is due to downstream injury-related mechanisms. The latter, also referred to as hemorrhagic conversion or hemorrhagic transformation, represents the third and final phase of endothelial dysfunction, where the structural integrity of the capillary is lost, allowing extravasation of all constituents of blood, including erythrocytes and other cells. Up to 30–40% of ischemic strokes undergo hemorrhagic transformation, a phenomenon that accounts for approximately 26–154 additional deaths per 1000 patients.231–233
Like vasogenic edema, hydrostatic pressure is the primary driving force for hemorrhagic transformation. Interestingly, extravasated blood increases the local tissue hydrostatic pressure and should thus impair further hemorrhage. However, this meager benefit is outweighed by the mass effect and tissue distortion created, as well as the robust inflammatory response triggered by blood products such as methemoglobin. Implications for clinical management of hemorrhage are similar to vasogenic edema, but this challenge is much more difficult, given the inflammatory milieu created by hemorrhage.
Mechanisms of hemorrhagic transformation
Mechanisms that drive hemorrhagic transformation are complicated, multifactorial, and incompletely understood. The aforementioned mechanisms of vasogenic edema formation are likely also relevant to hemorrhagic transformation. For example, exogenous VEGF administration following reperfusion worsens hemorrhagic transformation. MMP-driven extracellular proteolysis appears to play a major role in hemorrhagic transformation as its inhibition reduces hemorrhage.224,234–237 In addition to these shared mechanisms, there exist several mechanisms that are specific to hemorrhagic transformation. Oncotic death of endothelial cells mediated by Sur1-Trpm4 is likely an important factor in hemorrhagic transformation after a variety of CNS injuries.49,238,239 Other mechanisms might include damage mediated by ROS, basement membrane degradation, endothelial cell activation, and transmigration of leukocytes.231,235
Perihematomal cerebral edema
As blood is exquisitely toxic to brain tissue, hemorrhage by itself is a form of focal CNS injury that triggers formation of cerebral edema in the shell of tissue immediately surrounding the hemorrhage, i.e. the perihematomal space, a phenomenon referred to as perihematomal edema. 240 Perihematomal edema occurs in three stages: ionic edema, vasogenic edema, and delayed vasogenic edema. While the aforementioned core concepts that govern the formation of edema during phases of endothelial dysregulation (e.g. Starling’s principle) apply also to perihematomal edema, some mechanistic details are unique to the latter.
Perihematomal ionic edema, the first stage of perihematomal edema, is driven by transendothelial osmotic forces generated by two processes. Firstly, cytotoxic edema forms in the perihematoma space, putatively because glutamate tends to accumulate in this region.241,242 As described above, cytotoxic edema generates a strong driving force for the influx of ionic edema. Secondly, a hemorrhage-specific phenomenon called clot retraction, where activation of the coagulation cascade in the hematoma results in exudation of serum proteins and increased colloidal pressure of the perihematomal space, drives influx of water.243,244
Perihematomal vasogenic edema, the second stage of perihematomal edema, occurs when extravasation of blood products triggers changes in brain endothelium that manifest as extravasation of serum proteins without extravasation of erythrocytes. Thrombin, a protein that is extravasated with hemorrhaged blood and is produced at the site of injury, is a major contributor to the formation of perihematomal vasogenic edema. 240 Thrombin activates microglia primarily through PAR-1 receptors,245–247 resulting in secretion of tumor necrosis factor (TNF) and IL-1β,246,248,249 cytokines that elicits downregulation of tight-junction proteins in endothelial cells and BBB opening.250,251 In addition, thrombin enables the transmigration of circulating leukocytes by triggering endothelial retraction (as discussed above) and endothelial upregulation of chemokines and adhesion molecules,246,252 Infiltrating leukocytes contribute to perihematomal edema through the secretion of mediators such as ROS. In addition to thrombin, the compliment cascade is an important mediator of perihematomal vasogenic edema. Activation of the compliment cascade results in the production of anaphylatoxins, membrane attack complex (MAC)-mediated lysis of red blood cells and iron-induced edema, as well as infiltration of neutrophils. 253
The third and final stage of perihematomal edema, delayed vasogenic edema, is principally mediated by hemoglobin degradation products that originate from extravasated and lysed erythrocytes. In the interstitium, hemoglobin is quickly oxidized to methemoglobin; the latter can spontaneously release its heme moiety, which may be further degraded by heme oxygenase enzymes to free iron. 254 Erythrocyte lysis and hemoglobin degradation is apparently a relatively slow process, as free iron reaches maximal tissue levels at approximately 3 days following a hemorrhage event.246,255 Perihematomal delayed vasogenic edema exhibits similar temporality: Infusion of free iron increases brain water content by 24 h, whereas infusion of packed red blood cells increases brain water content only after 3 days.255,256 Many hemoglobin degradation products can independently contribute to perihematomal delayed vasogenic edema. Free iron triggers ROS generation, MMP-9 activation, and BBB breakdown, while iron chelation reduces edema influx in models of intracerebral hemorrhage.257–259 In addition, extracellular methemoglobin is a potent TLR-4 ligand that can trigger microglial TNF secretion and neuroinflammation. 260
Conclusions
While historical models have focused on the gross or ultrastructural appearance of edematous brain tissue, cerebral edema is better understood in a cellular and molecular context. The water movements involved in cerebral edema are dependent upon ionic fluxes, which are ultimately mediated by individual channels and transporters. The study of cerebral edema is essentially the study of maladaptive ion transport. While significant gaps still remain in our understanding of how specific proteins contribute to cerebral edema, the fields of cerebral edema and brain ISF dynamics are robust and productive. Doubtlessly, the next few years will yield new knowledge of how particular proteins drive edema influx, paving the way for rationally designed therapeutics that directly target key steps in cerebral edema formation, thereby achieving what currently approved therapies do not.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants to JMS from the National Institute of Neurological Disorders and Stroke (NINDS) (NS060801; NS061808) and the National Heart, Lung and Blood Institute (HL082517), and to VG from NINDS (NS061934; NS072501).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author’s contributions
JS conceived of and wrote the manuscript. JMS and VG contributed intellectually and provided critical feedback on the manuscript.