
Abstract
We tested whether nanoliposomes containing phosphatidylcholine, cholesterol and phosphatidic acid (NLPA) prevent β-amyloid 1-42 (Aβ42) fibrillation and Aβ42-induced human arteriole endothelial dysfunction. NLPA abolished Aβ42 fibril formation (thioflavin-T fluorescence/electron microscopy). In ex-vivo human adipose and leptomeningeal arterioles, Aβ42 impaired dilator response to acetylcholine that was reversed by NLPA; this protection was abolished by L-NG-nitroarginine methyl ester. Aβ42 reduced human umbilical vein endothelial cell NO production that was restored by NLPA. Nanoliposomes prevented Aβ42 amyloid formation, reversed Aβ42-induced human microvascular endothelial dysfunction and may be useful in Alzheimer’s disease.
Introduction
Alzheimer’s disease (AD) is a chronic degenerative condition without effective treatment and is expected to afflict 80 million people by 2040. 1 It is believed that AD results from the toxic effects of amyloidogenic β-amyloid peptides that form fibrils and are deposited in neural and vascular tissues as amyloid plaques. The role of vascular dysfunction in the etiopathology of AD is gaining recognition as epidemiologic, preclinical and clinical data show strong association between vascular disease/cardiovascular risk factors and the development/progression of AD. 2 Recently, we showed that ex-vivo human leptomeningeal arterioles acutely exposed to soluble β-amyloid peptide 1-42 (Aβ42), a major peptide implicated in AD, demonstrate endothelial dysfunction, and that this effect was similar in human abdominal subcutaneous adipose arterioles. 3 We showed similar induction of human arteriole endothelial dysfunction with exposure to light chain proteins derived from patients with AL amyloidosis,4–6 suggesting common vascular toxicity among amyloidogenic proteins despite differences in amino acid composition. When nanoliposomes of less than 100 nM size composed of phospholipids phosphatidylcholine, cholesterol and phosphatidic acid (NLPA) were co-treated with amyloidogenic light chains, endothelial dysfunction was reversed and the β-sheet structure of the light chain protein was altered. 7 Previous studies showed that nanoliposomes with similar components have very high affinity for Aβ peptides,8,9 which when viewed with our previous results on amyloidogenic light chain proteins, suggest a potential for reversing Aβ-induced vascular dysfunction. The study aim is to test the hypotheses that NLPA inhibits Aβ42 fibril formation and protects against Aβ42-induced human adipose and leptomeningeal arteriole endothelial dysfunction.
Materials and methods
Materials and nanoliposome preparation
Aβ42 peptides were obtained from Anaspec (Fremont CA), Sigma-Aldrich (St. Louis, MO, USA) and from recombinantly expressed methods as previously described. 10 NLPA was prepared according to prior methods. 7 In brief, phosphatidylcholine, cholesterol and phosphatidic acid (Avanti Polar Lipids, Alabaster AL) were mixed at 70:25:5 molar ratios (20 mg lipid/mL). The lipid mixture was dissolved in chloroform followed by removal of the organic solvent by rotary evaporator. To the dry lipid film was added 5 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffering agent (HEPES) followed by probe sonication (Sonic Dismembrator Model 100, Fischer Scientific, 10 W, 30 minutes). Titanium particles were removed by centrifugation (3000 × g, 10 min). Liposome size and zeta potential were assessed by Coulter N4 Submicron Particle size analyzer and were 29 ± 6 nM and −11.1 mV, respectively.
Aβ42 fibril formation
Thioflavin T (ThT) fluorescence assays were carried out on a Flexstation 3 microplate reader (Molecular Devices) using 15 µM Aβ42 alone or with NLPA at 10:1 (NLPA:Aβ42), with 20 µM ThT in 5 mM HEPES, 150 mM NaCl, pH 7.4. Experiments were carried out in triplicate in 96-well black-walled, clear bottomed microplates. The experiments for ThT fluorescence and transmission electron microscopy were performed without cells or tissues. Data was recorded every 5 min using bottom read mode, with excitation/emission at 440/490 nm, at 37℃ with orbital shaking between reads.
Transmission electron microscopy was used to analyze the solutions at endpoint ThT. Peptide suspensions (10 µL) were loaded onto carbon-coated copper grids, negatively stained with 4% uranyl acetate and visualized on a Tecnai 10 electron microscope at 120 kV.
Human subjects and tissue collection
Adipose arterioles
Research volunteers without known vascular disease, diabetes, or AD scheduled to undergo elective abdominal surgery provided written, informed consent for adipose tissue donation (n = 23, 60.6 ± 2.5 years, all males). The study was approved by the Phoenix Veterans Affairs Institutional Review Board and followed ethical guidelines of the Helsinki Declaration of 1975 (revised 1983). Abdominal subcutaneous adipose tissues were collected during surgery and immediately placed in sterile HEPES buffer (4℃, pH 7.4).
Leptomeningeal arterioles
Leptomeningeal arterioles were isolated from cadavers who prior to death provided informed consent for brain donation following death under an existing Brain and Body Donation Program (www.brainandbodydonationprogram.org). 11 The operations of the program have been approved by the Banner Sun Health Research Institute Institutional Review Board. The program involves 24-h on call Pathology technicians performing rapid autopsy of donors within a few hours of declaration of death. Tissues were placed immediately in sterile 4 -(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (4℃, pH 7.4). 3 Of 15 brain donors, seven (47%) were females, age was 86.5 ± 2.8 years, post-mortem interval was 3.17 ± 0.31 h, and clinical neurologic diagnoses were as follows: Alzheimer’s disease (3), cognitively normal (4), cognitively normal with Parkinson’s disease (1) mild cognitive impairment (2), dementia with Lewy bodies (2), Parkinson’s disease (1), frontotemporal dementia (1), and mixed vascular dementia (1).
Arteriole vasoreactivity
The methods for arteriole preparation were previously published. 6 Arterioles (∼80–250 µM diameter) were isolated from adipose tissue (living donors) or leptomeningeal tissue (cadaver donors), cannulated and pressurized until a final pressure of 60 mm Hg. Arteriole diameters were measured using videomicrometers. Following stabilization, arterioles were constricted to ∼60% baseline diameter using increasing doses of endothelin-1. Baseline (control) vasoreactivity was tested by successive administration of acetylcholine (10−9–10−4 M) to test endothelium-mediated dilation and papaverine (10−4 M) to test smooth muscle dependent dilation. After washing, the vessels were then exposed to Aβ42 (2 µM) ± NLPA (1:10 Aβ42: NLPA mass ratio) for 1 h and a second vasoreactivity response was measured. The dose of Aβ42 was chosen because this is less than but close to the reported concentration (∼30,000 ng/g tissue) found in cortical tissue of patients with AD.3,12 The dose of NLPA was chosen because this was similar to the dose that restored endothelial function in adipose arterioles exposed to AL amyloid light chain proteins. 7 In additional arterioles, Aβ42 and NLPA were co-treated with L-NG-nitroarginine methyl ester (L-NAME, 5 mmol), an inhibitor of nitric oxide (NO) synthase (NOS). Some of the data on living subject adipose arteriole response to Aβ42 alone (n = 15) were published in our prior study 3 but additional replicates (n = 6) were added for this study. Some of the data on leptomeningeal arteriole response to Aβ42 (n = 4) were reported in the same previous study 3 with additional replicates (n = 10) added for this manuscript.
Endothelial cell NO production and endothelial nitric oxide synthase (eNOS) protein assay
Human umbilical endothelial cells (HUVECs, Lonza, Walkersville MD) were passaged 24–48 h prior to exposure to vehicle or Aβ42 (2 µM) ± NLPA (1:10 Aβ42: NLPA mass ratio) for 18–20 h. NO head gas was measured using Sievers NO Analyzer (General Electric, Boulder, CO, USA). For eNOS protein assay, HUVECs were lysed in radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldritch), sonicated and DNA pelleted. Bradford protein assay was done to determine protein content. Western blot assay was performed and lanes were loaded with 30 µg of protein. Electrophoresis of proteins was performed in BioRad (Hercules CA) precast MiniProtean TGX gel at 100 mV and then transferred to PVDF low fluorescent membrane over 1 h at 100 mV. Primary antibodies (total eNOS, phosphoeNOS [Thr495] and β-actin, Cell Sigaling, Danvers MA) were used at 1:1000 dilution. A second blocking step was done before detection using goat-raised infrared-fluorescent conjugated secondary antibody (either 680 RD or 800 CW (LI-COR, Lincoln NB). Blots were read with the Li-COR Odyssey Clx infrared imaging system and band density measured using LI-COR Image Studio 4.0 and signal normalized to β-actin content to account for differences in protein loading.
Endothelial cell superoxide and peroxynitrite assay
To measure superoxide production, HUVECs were treated with vehicle or Aβ42 (2 µM) ± NLPA (1:10 Aβ42: NLPA mass ratio) for 18–20 h, and the treatment was then replaced with pre-warmed media containing 25 µM dihydroethidium (Life Technologies, Eugene OR), a fluorescent marker of superoxide production. 13 After 1 h, cells were washed with HEPES buffer and lifted with trypsin (Lonza) and suspended in 500 µL 10% Dulbecco’s modified eagle medium (Invitrogen, Carlsbad CA) and transferred to flow cytometry tubes. Cells were washed twice with HEPES buffer and fluorescent signal read on the Beckman-Coulter (Brea, CA, USA) FC500 flow cytometer excited with 488 nm laser and read on the FL-3 channel. Separately treated HUVECs were washed with cold PBS, stained with 20 µM of coumarin boronic acid pinacolate ester (Cayman Chemical, Ann Arbor MI) for 15 min to assess peroxynitrite 14 and fixed with 4% paraformaldehyde in PS and cold 100% methanol and then washed again. Images were obtained on EVOS FL Auto (Life Technologies) using DAPI light cube (excitation 357/44 nM, emission 447/60 nM). Images were analyzed using ImageJ 1.49 analysis software (National Institutes of Health, Bethesda MD).
Data analyses
Data are expressed as means ± standard errors of means with significant p-value set at p < 0.05 (two-sided). ThT fluorescence at time of peak signal of Aβ42 was compared using paired Student’s t-test. For vasoreactivity, control (baseline) response was compared to post-treatment response in the same arteriole using paired Student’s t-test and responses between two treatments were analyzed using unpaired Student’s t-test. Changes in treatment responses from baseline responses at varying doses of acetylcholine were compared between Aβ42 and Aβ42 + NLPA treatments using repeated measures analysis of variance. HUVEC NO, superoxide and peroxynitrite production as well as eNOS and phospho-eNOS were compared using analysis of variance with post hoc pairwise testing using paired Student’s t-test. Statistical analyses were performed using GraphPad Prism 5.0 (San Diego CA) and Sigmastat 3.5 (Richmond CA).
Results
Co-treatment with NLPA prevented Aβ42 fibril formation as shown by ThT fluorescence (Figure 1A). Lack of fibril/amyloid formation was verified using electron microscopy (Figure 1b). Aβ42-treated adipose arterioles showed impaired dilator response to acetylcholine that was mitigated by NLPA co-treatment (10−4 M acetylcholine: control 91.1 ± 1.7%, Aβ42 59.5 ± 4.7%, Aβ42 + NLPA 77.8 ± 7.8%, p < 0.001 control vs. Aβ42, p < 0.05 Aβ42 vs. Aβ42 + NLPA, p = NS control vs. Aβ42 + NLPA). When NOS inhibitor L-NAME was added to Aβ42 + NLPA, the protective effect of NLPA was abolished (10−4 M acetylcholine: 18 ± 10%, p < 0.01 vs. Aβ and Aβ + NLPA, p < 0.05 vs. baseline control). The magnitude of reduction in dilator responses to acetylcholine from control values was significantly different between arterioles treated with Aβ42 versus Aβ42 + NLPA (Figure 2a). Aβ42-treated arterioles showed more modest impaired dilator response to papaverine compared to baseline control, but there was no significant difference in response with NLPA co-treatment (control 96.5 ± 1.0%, Aβ42 80.7 ± 3.5%, Aβ42 + NLPA 85.7 ± 5%, p < 0.001control vs. Aβ2, p < 0.05 control vs. Aβ42 + NLPA). Response to papaverine when L-NAME was added to Aβ42 + NLPA (78.1 ± 9.6%) was not significantly different from response to Aβ42 + NLPA, Aβ42 or baseline control. To confirm findings in cerebrovascular tissue, similar experiments were performed in leptomeningeal arterioles. In leptomeningeal arterioles, exposure to Aβ42 showed significant reduction in dilator response to acetylcholine that was reversed by NLPA (10−4 M acetylcholine: control 83.3 ± 5.2%, Aβ42 45.5 ± 6.2%, Aβ42 + NLPA 79.8 ± 9.9%, p < 0.001 control vs. Aβ42, p = NS control vs. Aβ42 + NLPA p = 0.01 Aβ42 vs. Aβ42 + NLPA). The magnitude of reduction in dilator responses to acetylcholine from control values was significantly different between leptomeningeal arterioles treated with Aβ42 versus Aβ42 + NLPA (Figure 2B). Unlike adipose arterioles, there was no significant reduction in dilator response to papaverine in leptomeningeal arterioles exposed to Aβ42 when compared to control and Aβ42 + NLPA (control 95.3 ± 1.6%, Aβ42 87.0 ± 4.2%, Aβ42 + NLPA 90.3 ± 6.5%, p = NS). However, the change in dilator response to papaverine following Aβ42 treatment versus baseline control in adipose arterioles (−15.2 ± 3.9%) was not significantly different when compared to leptomeningeal arterioles (−8.3 ± 5.2%).
(a) Thioflavin T fluorescence. In-vitro experiment showing Aβ42 fibril formation as measured by thioflavin T fluorescence. With time, there was increased fluorescence signal with Aβ42 alone. Fluorescence signal was not changed when Aβ42 was co-treated with NLPA (10:1 NLPA: Aβ42), demonstrating lack of fibril formation with NLPA administration. (b) Electron microscopy. In-vitro EM images confirms amyloid formation in Aβ42 containing preparations that showed increased thioflavin-T fluorescence. Co-treatment with NLPA resulted in absence of amyloid formation. Scale bar is 500 nm. (a) Changes in dilator responses (treatment minus baseline control) in adipose arterioles. Adipose arterioles exposed to Aβ42 showed greater degree of reduction from control dilator response to acetylcholine. When Aβ42 was co-treated with NLPA, there was significant restoration of dilator response (ANOVA p = 0.009 Aβ42 versus Aβ42 + NLPA). (b) In leptomeningeal arterioles, Aβ42 also showed greater degree of reduction from control dilator response to acetylcholine whereas co-treatment with NLPA showed full restoration of dilator response to acetylcholine (ANOVA p = 0.004 Aβ42 versus Aβ42 + NLPA). (c) NO production. HUVECs (n = 6 each) treated for 18–20 h with Aβ42 showed reduced NO production versus vehicle control (veh) (ANOVA p < 0.05). NO production was restored by co-treatment with NLPA. (d) Total endothelial nitric oxide synthase (eNOS) and phosphorylated eNOS (peNOS) protein in HUVECs by Western blot. There was no significant difference in eNOS, peNOS and peNOS/eNOS ratio in HUVECs treated for 18–20 h with vehicle, Aβ42 or Aβ42 + NLPA (n = 3 each). (e, f) HUVEC superoxide and peroxynitrite production. Superoxide and peroxynitrite production were increased following 18–20 h exposure to Aβ42, and was reduced by co-treatment with NLPA (n = 7–8 per treatment for superoxide assay, n = 5 per treatment for peroxynitrite assay).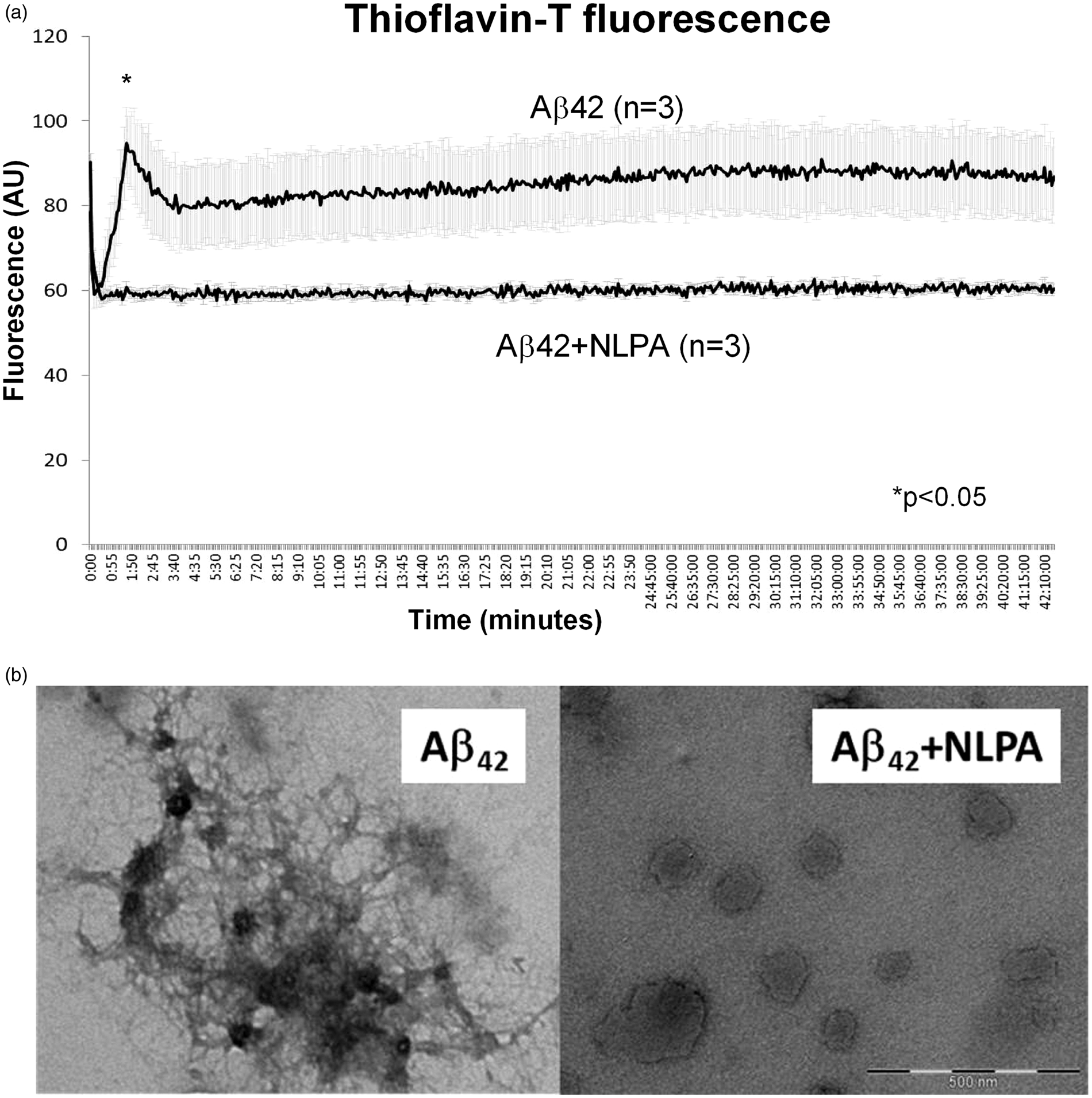
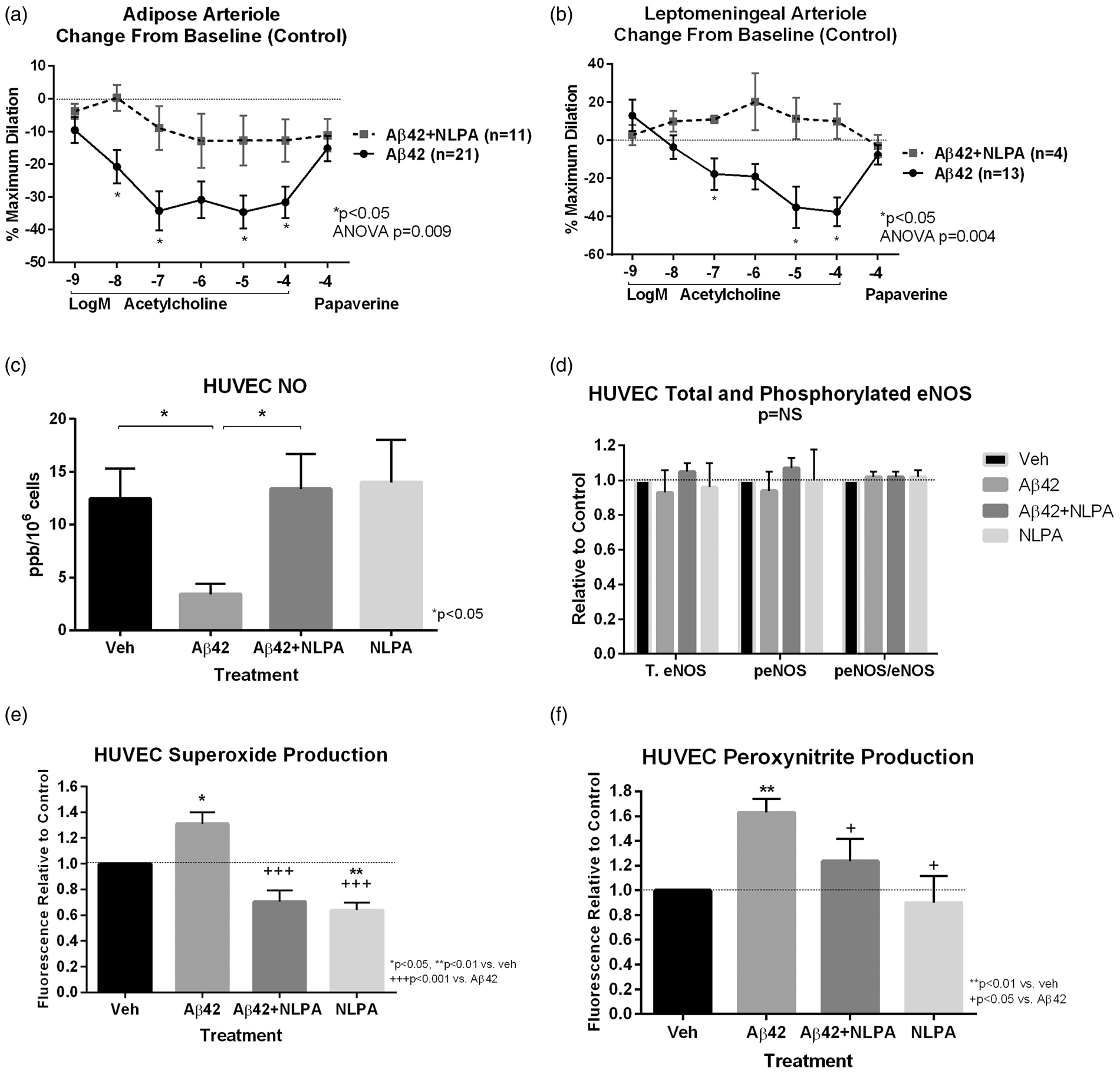
HUVECs treated with Aβ42 (18–20 h) showed reduced NO production that was restored with co-treatment with NLPA (ANOVA p < 0.05, Figure 2c). There was no significant difference in total eNOS, phospho-eNOS protein content, and peNOS/eNOS ratio in HUVECs among the treatment groups (Figure 2D). Aβ42-treated cells showed increased superoxide and peroxynitrite production that was attenuated by co-treatment with NLPA (Figure 2(e) and (f)).
Discussion
Our results demonstrate two novel findings: (1) nanoliposomes composed of phosphatidylcholine, cholesterol and phosphatidic acid prevent Aβ42 fibril formation and (2) the endothelial dysfunction induced by Aβ42 is mitigated by co-administration of NLPA through increased NO bioavailability. These findings point to the potential utility of nanoliposomes as novel treatment class for Alzheimer’s disease.
There is increasing recognition of the important role of vascular dysfunction in AD pathology, especially during the early preclinical phase. Brain hypoperfusion is a prominent and early feature of AD. 15 Endothelial dysfunction and cerebral hypoperfusion led to loss of spatial memory even before Aβ amyloid accumulation in rat hippocampus. 16 Other investigators and our group showed that Aβ caused cerebrovascular endothelial dysfunction in both transgenic mouse models and human tissues3,17 pointing to its potential role in chronic ischemic brain injury in AD. Reversing the adverse effects of Aβ on human microvascular endothelial function may represent an important early therapeutic approach.
Nanoliposomes composed of varying phospholipids may be potential treatment agents alone (“naked”) or as carriers of drugs because, unlike other nanoparticles, they are considered non-toxic, non-immunogenic, fully biodegradable and structurally versatile. 9 Nanoliposomes (including those containing negatively charged phosphatidic acid) were shown to bind to Aβ42 peptides, and when conjugated with apolipoprotein E, allowed enhanced interaction with brain capillary endothelial cells.8,9 Our group previously demonstrated the potential utility of nanoliposomes in mitigating amyloid protein vascular injury when we showed restoration of endothelial function in human adipose arterioles treated with AL amyloid light chain proteins; 7 these proteins share common features with Aβ peptides in causing endothelial dysfunction, downstream ischemic tissue injury and vascular amyloid deposition although they involve coronary and peripheral vessels. We demonstrate similar endothelial dysfunction responses to Aβ42 and protective NLPA effect in both peripheral adipose and leptomeningeal arterioles, suggesting that the easily accessible adipose arterioles may be acceptable surrogates to study cerebrovascular microvascular function (inaccessible in living subjects). There is a note of caution in that there may be a subtle difference in smooth muscle-dependent dilation between adipose arterioles (where Aβ42 showed significant reduction versus control, n = 21) and leptomeningeal arterioles (where Aβ42 showed no significant reduction versus control, n = 13), although no difference was noted between adipose and leptomeningeal arterioles when compared as to change in dilator response to papaverine versus baseline control. Whether this represents true variance in Aβ42 effects on smooth muscle-dependent vasodilation between peripheral and cerebrovascular microvasculature, or similar responses but degrees of statistical significance were affected by difference in sample sizes, remain to be elucidated.
Our group previously showed that NLPA altered the properties of AL amyloid light chain proteins by reducing the amount of partially folded states and shifting the equilibrium towards the folded state, 7 and atomic force microscopy imaging demonstrated physicochemical interaction between nanoliposome and light chain proteins (unpublished data). Our current data suggest that NLPA similarly alters the properties of Aβ42 leading to inhibition of fibril and amyloid formation, potentially useful as a strategy to reduce amyloid deposition that is the hallmark of advanced and irreversible AD. This preliminary report cannot yet fully and definitely ascertain the underlying signaling mechanisms linking the NLPA effects on altering the structural behavior of Aβ42 (as demonstrated by fibrillation assays) and the beneficial effects on improving endothelial function. However, our data suggest a potential mechanism underlying the beneficial effect. Aβ42 caused increased superoxide and peroxynitrite production; peroxynitrite is a reactive nitrogen species formed by the reaction between superoxide and nitric oxide and is one of the most potent mediators of DNA and protein damage. 6 Our findings show that the restoration of NO production by NLPA in Aβ42-treated cells was accompanied by reduction in superoxide and peroxynitrite production, suggesting that NLPA improves endothelial cell NO bioavailability by reducing superoxide that can rapidly react with NO to form peroxynitrite 14 thereby also reducing oxidative and nitrative stress. Aβ42 reduced NO without changing total eNOS, peNOS and peNOS/eNOS ratio, suggesting that one possible mechanism of increased superoxide production and reduced NO bioavailability is eNOS uncoupling, 18 and the increased NO, reduced superoxide and peroxynitrite with NLPA co-treatment may point to the ability of NLPA to restore eNOS coupling.
Limitations
The results represent an early proof-of-concept on potential use of nanoliposomes to mitigate vascular injury induced by Aβ peptide. Although use of human ex-vivo model enhances the translational relevance to human disease, in-vivo testing on appropriate preclinical models (e.g. transgenic AD mouse models) will need to be done to assess efficacy in reversing some or all of the tissue and endothelial pathologies in AD and preventing Aβ amyloid tissue formation, which are goals for future follow-up studies. Because nanoliposomes of various types are already being used in the clinical setting as delivery agents for therapeutic cargo (such as chemotherapeutic agents) for specific organ targeting, testing our nanoliposomes from our ex-vivo model to in vivo experiments should be an easy transition. Our study was also limited in using one formulation of nanoliposomes composed of phosphatidylcholine, cholesterol and phosphatidic acid. Although this was chosen because of previous demonstration of efficacy in vascular protection in amyloidogenic protein-induced microvascular dysfunction 3 and high affinity for Aβ,8,9 various other compositions may provide superior effects but were not tested in our study. Although human umbilical vein endothelial cells are not sourced from brain tissue, nevertheless this human cell line is an accepted cell model to study effects of Aβ on vascular tissue.19,20 The study was limited in using leptomeningeal arterioles from an elderly cohort with existing comorbidities as this was the only available source of donated tissue for this study.
Footnotes
Funding
National Institutes Health (NIA R21AG044723, NINDS U24NS072026, NIA P30AG19610, NIA RO1AG019795), Veterans Affairs Merit grant (I01BX007080), the Arizona Department of Health Services (contrast 211002), the Arizona Biomedical Research Commission (4001, 0011, 05-901 and 1001), Michael J. Fox Foundation for Parkinson’s Research and Amyloidosis Foundation. Support was provided by the Midwestern University College of Pharmacy-Glendale and Carl T. Hayden Medical Research Foundation. The study was based on work supported by the Department of Veterans Affairs and VA employment.
Acknowledgement
We would like to thank our research volunteers, John Hatfield, Sara Schwab, the surgeons and staff of the Surgery Service of the Phoenix VA. The contents of the manuscript do not represent the views of the Department of Veterans Affairs or the United States government.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
RQM, ST, VW, JM, DAF took part in conception and design; ST, VW, JM, HAD, DGV, DAF, CB, NK, TGB, GS and RQM took part in acquisition, analysis or interpretation of data; RQM contributed to drafting the article; and ST, VW, JM, HAD, DGV, DAF, CB, NK, GS, TGB and RQM contributed to reviewing critically for intellectual content and final approval of article.