
Abstract
Cerebral small vessel disease (SVD) gives rise to one in five strokes worldwide and constitutes a major source of cognitive decline in the elderly. SVD is known to occur in relation to hypertension, diabetes, smoking, radiation therapy and in a range of inherited and genetic disorders, autoimmune disorders, connective tissue disorders, and infections. Until recently, changes in capillary patency and blood viscosity have received little attention in the aetiopathogenesis of SVD and the high risk of subsequent stroke and cognitive decline. Capillary flow patterns were, however, recently shown to limit the extraction efficacy of oxygen in tissue and capillary dysfunction therefore proposed as a source of stroke-like symptoms and neurodegeneration, even in the absence of physical flow-limiting vascular pathology. In this review, we examine whether capillary flow disturbances may be a shared feature of conditions that represent risk factors for SVD. We then discuss aspects of capillary dysfunction that could be prevented or alleviated and therefore might be of general benefit to patients at risk of SVD, stroke or cognitive decline.
Introduction
Cerebral small vessel disease (SVD) denotes a range of pathological processes, which affect the small arteries, arterioles, capillaries and small veins of the brain. 1 SVD is associated with small subcortical infarcts, lacunes, white matter hyperintensities, enlarged perivascular spaces, microbleeds, and cortical atrophy, 2 gives rise to one in five strokes worldwide, and constitutes a major source of cognitive decline, particularly in the elderly. 1
Until recently, changes in capillary morphology and blood–brain barrier (BBB) function have received little attention in a etiopathogenesis of SVD and associated stroke and cognitive decline.3,4 In addition, capillary flow patterns have now been shown to limit the extraction efficacy of oxygen in tissue, 5 and capillary dysfunction proposed as a source of stroke-like symptoms 6 and neurodegeneration, 7 even in the absence of flow-limiting vascular pathology.
Here, we briefly review the properties of capillary dysfunction and the evidence for capillary involvement in SVD and in conditions that impose risk of SVD. We then examine whether capillary dysfunction may play a role in the aetiopathogenesis of SVD and the subsequent development of stroke or cognitive decline. Finally, we discuss whether capillary dysfunction may serve as a common therapeutic target in efforts to prevent or ameliorate stroke and cognitive decline across the diverse range of conditions associated with SVD.
Capillary dysfunction
In normal brain, only 30–40% of blood’s oxygen passes into the brain parenchyma from small arteries, arterioles and capillaries to fuel cerebral metabolism. 8 Oxygen extraction is known to be inefficient from capillaries with high flow velocities. 9 The heterogeneity of flow velocities across the capillary bed of normal, resting brain tissue is extremely high10–12 and therefore limits net oxygen extraction5,13,14 – a biophysical property referred to as functional shunting. In the normal brain, capillary flow patterns homogenize when cerebral blood flow (CBF) increases in relation to cortical activation10,12,15,16 and thereby facilitate more efficient oxygen extraction.5,14 This homogenization is partly a passive property of normal microvascular networks: as CBF increases, the blood tends to distribute in a more homogenous way across ‘ideal’ capillary networks. 17 In a later section, we discuss how cerebral pericytes regulate capillary diameter during functional activation 18 and possibly provide active regulation of capillary flow patterns. 19
Capillary dysfunction refers to conditions in which changes in capillary function and/or patency, or in blood rheology, disturb either capillary flow patterns, their homogenization during hyperaemia or both. To determine the effects of capillary dysfunction on oxygen extraction, the distribution of erythrocyte velocities or transit times across the capillary bed must be known.5,14,17,20,21 For convenience, we quantify capillary dysfunction by the accompanying capillary transit time heterogeneity (CTH) and use accepted transit time distributions for which the standard deviation describes CTH by a single parameter.5,13,14,17,21 Figure 1 illustrates how CBF and its microvascular distribution (CTH), combined, affect oxygen uptake in brain tissue.
The green isocontour surface corresponds to all combinations of CBF, CTH, and P
t
O2 for which brain oxygenation – according to our model
5
– matches the metabolic rate of oxygen in resting brain.
187
Transitions to combinations of CBF, CTH and P
t
O2 that correspond to points located outside the resulting, bell-shaped surface are therefore predicted to result in immediate neurological symptoms, and tissue damage if they persist. The red plane marks the boundary, left of which vasodilation reduces tissue oxygen availability (dubbed malignant CTH). The maximum value that CTH can attain at a P
t
O2 of 25 mmHg, if oxygen availability is to support the metabolic needs of resting brain tissue, is indicated by the label A. As CTH increases further (progressive capillary dysfunction), CBF must be attenuated in order to reduce the level of ‘physiological shunting’. Importantly, continued tissue oxygen metabolism reduces tissue oxygen tension, and thereby improves blood-tissue concentration gradients and net extraction. As a result, the bell-shaped surface widens towards its base, reflecting that higher levels of CTH (more severe capillary dysfunction) can be accommodated by attenuating CBF and CBF responses. A critical limit is reached, however, as P
t
O2 approaches zero – label B. At this point, the metabolic needs of tissue are met by ‘delaying’ mean transit time (MTT) to a threshold of approximately 4 s, corresponding to CBF = 21 ml/100ml/min. As a result, slight increases in CTH (e.g. caused by an infection or dehydration) or a slight change in CBF (small flow reductions as well as flow increases) can trigger a critical reduction in tissue oxygen availability, and thereby stroke-like symptoms. The blue arrow indicates progressive capillary flow disturbances, which cause CTH to increase and tissue oxygen availability to approach the metabolic requirements of resting brain tissue (the green iso-contour). Note that the traditional notion of ischemia (which disregards capillary flow patterns) considers only a reduction in CBF (increase in MTT), that is, a transition along the x-axis in the three-dimensional plot. Source: Reproduced and modified from the literature.
6
CBF: cerebral blood flow; CTH: capillary transit time heterogeneity; P
t
O2: tissue oxygen tension.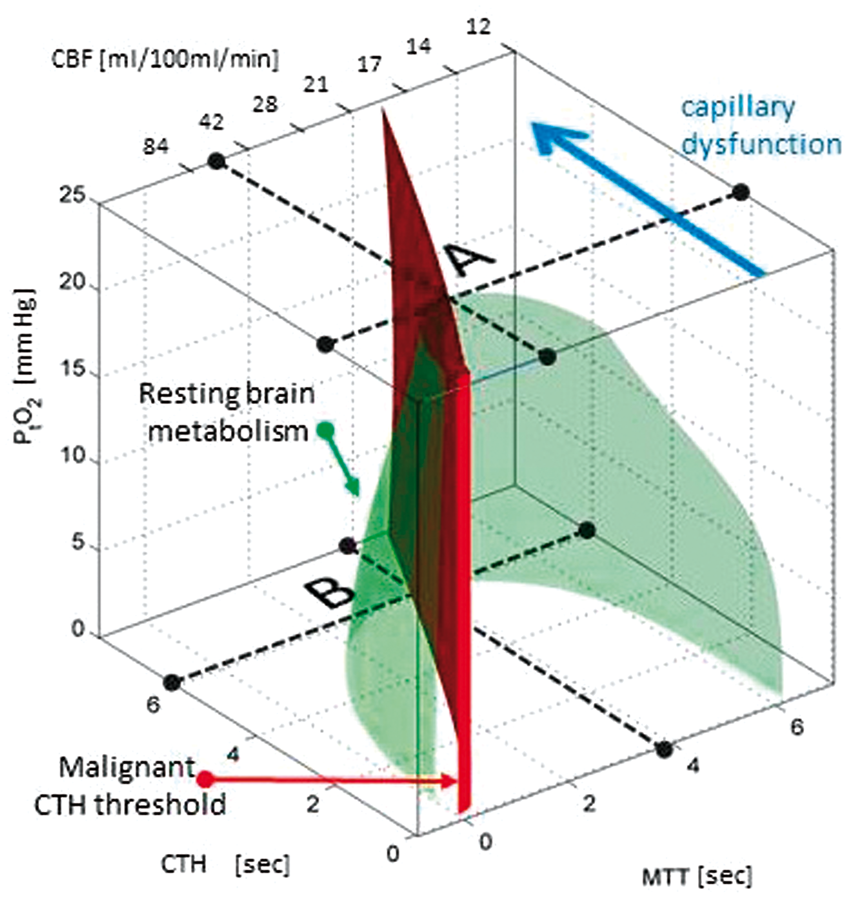
If changes in capillary patency or blood rheology become severe, our analyses predict that the flow-metabolism coupling, which is crucially required for tissue function and survival, modifies CBF in a counterintuitive direction. 6 While homogenization of capillary flows (CTH reduction) maintains efficient oxygen extraction during hyperaemia in normal brain, 5 only the suppression of CBF can reduce functional shunting if CTH can no longer be reduced. Attenuated CBF responses (reduced cerebrovascular reserve capacity) are therefore expected in both capillary dysfunction and flow-limiting conditions – but to represent the maintenance of flow-metabolism coupling in the former and flow limitations at the level of resistance vessels in the latter.
In cerebral ischemia, tissue function and survival are threatened by tissue hypoxia as a result of limited blood supply, whereas in capillary dysfunction, the source of hypoxia is inefficient oxygen extraction from the microcirculation. Notably, stroke-like symptoms and hypoxic tissue injury can therefore, in principle, be caused by capillary flow disturbances, in the absence of a flow-limiting condition.6,7 We have shown that CTH can reach a critical biophysical threshold CTHmax, above which net oxygen extraction can no longer meet the metabolic demands of resting brain tissue, although CBF is suppressed to minimize net functional shunting. 6 Notably, the CBF value that optimizes net oxygen extraction for CTH = CTHmax seems almost identical to the classical ischemic threshold of approximately 20 ml/100ml/min, 6 making critical capillary dysfunction and cerebral ischemia 22 indistinguishable in terms of their low CBF and elevated oxygen extraction fraction (OEF).
If CBF suppression fails to compensate for capillary dysfunction, tissue hypoxia and injury can occur at CBF values above the ischemic threshold and even above normal brain perfusion. The ‘luxury perfusion syndrome’, 23 which is sometimes observed upon reperfusion of tissue after prolonged or severe ischemia, may represent an instance of tissue damage caused by excessive functional shunting, keeping in mind that capillary constrictions can be observed after ischemia.19,24
Reductions of CBF to levels below the ischemic threshold can cause neurological symptoms and tissue injury irrespective of whether vascular patency is reduced at the arterial or the capillary level, but some vascular causes of neurological symptoms and tissue injury are specific to capillary dysfunction: capillary dysfunction can thus cause neurological deterioration and hypoxic tissue injury (a) in the absence of primary, flow-limiting pathology (e.g. no severe stenosis, thrombosis, embolism) (b) under conditions of augmented CBF (iatrogenic or spontaneous) and (c) due to increased blood viscosity if this increases CTH beyond CTHmax.
We discuss additional signatures of capillary dysfunction below.
Sources of capillary dysfunction in conditions that represent risk factors for SVD
Abbreviations.
Type 1: Arteriolosclerosis (age- and vascular risk-factor-related SVD). Type 2: CAA, sporadic or hereditary.

AD: Alzheimer’s disease; APOE: apolipoprotein; Aβ: amyloid β; BOLD: blood oxygen level dependent; CAA: cerebral amyloid angiopathy; CBF: cerebral blood flow; STZ: streptozotocin; SVD: small vessel disease.
Type 3: Inherited or genetic SVD other than CAA.

CADASIL/CARASIL: erebral autosomal dominant/recessive arteriopathy with subcortical ischemic strokes and leuko-encephalopathy; GOM: granular osmophilic material; HERNS: Hereditary endotheliopathy with retinopathy, nephropathy and stroke; MELAS: Mitochondrial encephalo-pathy with lactic acidosis and stroke-like episodes; N3ECD: NOTCH3 extracellular domains; PADMAL: Pontine autosomal dominant angiopathy and leukoencephalopathy; SVD: small vessel disease.
Type 4: Inflammatory and immunologically mediated SVD. Vasculitis caused by infection.

BOLD: blood oxygen level dependent; CBF: cerebral blood flow; CMRO2: cerebral metabolic rate of oxygen; HAND: HIV associated neurocognitive disorder; HCV: hepatitis C virus; HIV: human immunodeficiency virus; SVD: small vessel disease.
Type 4: Inflammatory and immunologically mediated SVD.

ANCA: antineutrophil cytoplasmic antibodies; Aβ: amyloid β; BOLD: blood oxygen level dependent; CNS: central nervous system; SVD: small vessel disease.
Type 4: Inflammatory and immunologically mediated SVD. Vasculitis caused by connective tissue disorders.

BOLD: blood oxygen level dependent; MRI: magnetic resonance imaging; NCM: nailfold capillary microscopy; SLE: systemic lupus erythematosus; SVD: small vessel disease.
Type 5: Venous collagenosis, and type 6: Other SVDs.

CTH: capillary transit time heterogeneity; OEF: oxygen extraction fraction; SVD: small vessel disease.
Pericyte dysfunction
Pericytes are embedded in layers of the basement membrane that surround the capillary endothelium. 27 Pericytes are thought to cover most capillaries in the central nervous system where they regulate BBB function 28 and aspects of the brain’s immune response. 29 Pericytes are involved in the regulation of capillary development (angiogenesis), stabilization, maturation and remodeling 30 and communicate with endothelial cells through peg-socket contacts as they – among other functions – jointly form and maintain the basement membrane. 27 Neurogenic locus notch homolog protein 3 (NOTCH3) signalling is crucial for the postnatal differentiation of vascular cells into their ‘correct’ arterial, capillary and venous phenotypes27,31 and their ability to adapt to changes in pressure and vascular strain.32,33 While pericytes are characterized by their relation to microvessels, they share cellular and functional characteristics with vascular smooth muscle cells (VSMCs) encircling arterioles and venules. The distinction between VSMC and pericytes recently became a matter of debate with regards to the attribution of CBF-regulation19,34,35 – see discussion in the literature. 36
Cerebral pericytes are contractile, and they have been shown to contract and relax in response to neuromediators, vasoactive drugs and, importantly, to sensory stimulation in brain slices as well as in vivo.18,19,35 Thus, cerebral pericytes have been shown to regulate capillary diameter during functional activation, 18 dilating about 1 s before arteriolar dilation and thereby possibly controlling both CBF and CTH. 19 Retinal pericytes have been characterized extensively in vitro: retinal pericytes constrict in response to high oxygen tension and relax in response to lactate and low pH, 37 possibly providing a mechanism by which capillary flows can redistribute to meet local cellular metabolic demands during activation. 37 Much like VSMCs, retinal pericytes react to a range of vasoactive substances, constricting when exposed to mechanical stretch, angiotensin II (AT2) 38 and endothelin-1 (ET1) 39 by a Ca++-dependent mechanism 40 and relaxing when exposed to adenosine, 41 ATP 42 and nitric oxide (NO)43,44 and in response to cholinergic 45 and adrenergic 40 stimulation. See the study by Attwell et al. 46 for a review of neurovascular coupling mechanisms and the control of VSMC and pericyte tone.
Pericyte loss and basement membrane thickening are observed in conditions that represent major risk factors for SVD, such as ageing, hypertension and diabetes – see Table 2. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL – Table 3) is associated with mutations in the NOTCH3 gene. 47 The receptor protein encoded by the NOTCH3 gene is expressed in both VSMCs and pericytes, 48 and recent evidence suggests that CADASIL is associated not only with degeneration and damage to VSMCs in small arteries and arterioles but also to microvascular pericytes. Dziewulska and Lewandowska 49 observed pericyte loss and pericyte fragments within thickened basement membranes in skin and muscle biopsies in CADASIL. They reported that pericyte-endothelial peg-socket contacts were disrupted, seemingly giving rise to basement membrane thickening, endothelial swelling and protrusions into the capillary lumen. 49 Recent animal models of CADASIL show evidence of reduced capillary density 50 and pericyte degeneration 51 and suggest that reduced pericyte coverage is related to impaired BBB function. 52
When exposed to viral and bacterial proteins, pericytes initiate inflammatory responses that facilitate the recruitment of immune cells from the blood stream – see the study by Hill et al. 53 for a recent review. As neutrophils pass through the surrounding basement membrane, they pass between embedded pericytes, which in turn remodel the laminin-rich basement membrane to permit extravasation. 54 Importantly, pericytes seemingly change their phenotype during inflammation to become migratory, 54 a phenomenon also observed in CNS injury. 55 The ability of pericytes to undertake BBB function 28 and capillary flow control 19 may therefore be compromised during infection and inflammation.
Pericyte exposure to parenchymal waste: Amyloid and hemoglobin
Molecules the size of haemoglobin and amyloid β (Aβ) are cleared from the subarachnoid space and brain parenchyma along the basement membranes of arteries and capillaries.56,57 Indeed, impaired glymphatic clearance has recently emerged as a potential therapeutic target. 58 Located in the layers of capillary basement membrane, pericytes are therefore exposed to amyloid during its perivascular removal and clearance across the BBB.59–61 Pericytes undergo degeneration when exposed to certain Aβ types in vitro,62,63 and although the role of pericytes in the pathogenesis of cerebral amyloid angiopathy (CAA) and Alzheimer’s disease (AD) remains poorly understood, evidence from Aβ transgenic mice and in vitro models suggest they may be involved in neurodegeneration.64,65 For recent reviews, see the study by Hamilton et al. 66 and Winkler et al. 67
Microbleeds are associated with SVD, and the vasoactive properties of hemoglobin breakdown-products are described in detail in the literature.68,69 Briefly, spontaneous autoxidation of oxyhemoglobin (HgbO) to methemoglobin and the iron released from hemoglobin cause the release of highly reactive superoxide radicals.68,69 Superoxides are thought to cause vasoconstriction by depleting vascular NO levels70,71 and to induce lipid peroxidation and peroxynitrite formation, which in turn cause vasoconstriction and structural damage to cerebral microvessels, including the endothelial cell layer. 72 The breakdown of heme into bilirubin under such oxidative conditions results in the formation of bilirubin oxidation products (BOXes) that change the contractility, signalling and metabolism in large vessels – see also the study by Pyne-Geithman et al. 73
Endothelial function
Endothelial cells are mechanically coupled by tight junctions, which ensure BBB integrity and prevent leakage of toxic molecules into the brain interstitium – see discussion in the literature.3,4 Endothelial cells are also electrically and metabolically coupled to each other as well as to nearby VSMCs via gap junctions composed of connexins. 74 This rapid, bidirectional signalling pathway seemingly provides efficient coordination of vessel function across the microvascular bed.75–77 Disruption of the signalling between endothelial cells has been shown to cause profound breakdowns in vascular control across the capillary bed, resulting in extreme degrees of capillary shunting through the shortest arteriolo-venular pathways. 78
In small-vessel vasculitides (Table 4) associated with antineutrophilic cytoplasmic antibodies (ANCAs), neutrophils adhere to capillary endothelial cells and cause the release of reactive oxygen species (ROS) and lysosomal enzymes. This abnormal inflammatory reaction causes endothelial cells to undergo necrosis and detach from the basement membrane, after which they can be found in peripheral blood. 79 While the activation of neutrophils and endothelial cells in the capillary lumen may disturb capillary flow patterns in itself, the disruption of endothelial cell-to-cell signalling described above is expected to cause severe capillary dysfunction.
Glycocalyx dysfunction
The luminal surface of the capillary endothelium is covered by a 0.5-µm thick glycocalyx.80,81 This carbohydrate-rich matrix affects the passage of blood cells through the capillary bed. 82 reducing capillary haematocrit to 20–50% of that found in the systemic circulation. Glycocalyx damage, in turn, disrupts capillary flow regulation, causing capillary hematocrit to approach that of the systemic circulation.83,84 The glycocalyx constitutes a fluid barrier in the vascular system and has been implicated in oedema formation.85,86 The glycocalyx is degraded by direct oxidative stress and exposure to oxidized lipoproteins,83,87,88 by hyperglycaemia 89 and by ischemia.88,90
Blood viscosity
The dimensions of white blood cells (WBC) and erythrocytes exceed the average capillary diameter. Experimental studies have shown that capillary flow patterns are sensitive to the size, deformability, viscosity, number and endothelial adhesion of blood cells. In infections and low-grade vascular inflammation, blood is hence increasingly redirected to thoroughfare channels which act as functional shunts. 91 In a classical study, hyperviscosity was demonstrated in diabetic patients (Table 2) compared to age-matched controls and the viscosity found to correlate with the extent of microvascular diabetic complications. Accordingly, erythrocyte deformability was lower in those diabetic patients who had the most extensive microangiopathy compared to either diabetics with no complications or to controls. 92
In cryoglobulinaemic vasculitis (Table 5), cryoglobulins precipitate and lead to hyperviscosity. 26 Hematocrit is lower in capillaries than in the systemic circulation, and since cryoglobulins precipitate more easily under conditions of serum excess, capillary flows may be particularly sensitive to this phenomenon.
Signs of capillary dysfunction
The previous section suggests that capillary morphology and function may be disturbed in conditions considered risk factors for SVD. Neuropathological data and direct in vivo observations of the microcirculation in these conditions are sparse, however, and it remains unclear whether capillary dysfunction might antedate changes in the morphology and function of upstream arteries and arterioles.
The neurovascular coupling mechanisms, 93 which control local CBF according to the metabolic needs of tissue, are expected to account for the oxygen extraction efficacy downstream, including changes related to capillary dysfunction. Below we discuss how CBF has to be adjusted in order to compensate for the reduced oxygen extraction efficacy that accompanies various degrees of capillary dysfunction. Some CBF changes are highly suggestive of capillary flow disturbances as opposed to a flow-limiting pathology, and neurovascular coupling studies may therefore serve as an indirect means of addressing the role of capillary dysfunction in the evolution of SVD from its risk factors. These changes are listed in the right-most column in Tables 2–7.
We used Web of Science™ and PubMed to search for occurrences of the terms listed in the leftmost column of Tables 2–7 in combination with ‘CBF’, ‘blood-oxygen-level-dependent (BOLD)’, ‘oxygen’, ‘metabolism’, ‘functional hyperemia’, ‘vasoreactivity’ and ‘stroke’. Literature searches for column 3 in Tables 2–7 were conducted concurrently with those for column 2 (previous section).
Mild capillary dysfunction: CBF increases in conditions that represent risk factors for SVD
For mild capillary disturbances, the reduction in oxygen extraction efficacy is so small that it can be compensated for by elevated CBF. Observations of increased resting CBF or increased CBF responses early in the course of SVD precursors therefore suggest that capillary dysfunction (elevated CTH), rather than primary changes in the morphology or function of upstream arterioles, is involved in the early etiology of the condition. Meanwhile, the BOLD signal is often used to localize brain activity through its sensitivity to tissue deoxyhemoglobin concentrations [dHgb]. Mild capillary dysfunction is characterized by reduced OEF and proportionately higher CBF responses during functional activations, both of which reduce [dHgb] and thereby increase BOLD signal amplitudes before resting CBF and OEF become affected. In asymptomatic subjects presented with identical tasks, BOLD responses are therefore expected to be higher in those with mild capillary dysfunction than in controls, despite identical changes in metabolic activity. CBF and BOLD responses are recorded in grey matter and are hence sensitive to changes in capillary function in the cortex and subcortical nuclei. Subcortical lesions may result in secondary changes in cortical function, and, in theory, even compensatory hyperactivity in some regions.
In streptozotocin (STZ)-induced diabetes (Table 1) in rats, both total and cortical CBF values are indeed elevated compared to control animals early after induction of the disease,94–96 and cortical oxygen tension elevated. 96 In early-stage hypertension (Table 2), elevated CBF and BOLD responses to hypercapnia have been reported in spontaneously hypertensive rats compared to age matched control animals. 97 The apolipoprotein (APOE) ɛ4 allele is associated with both CAA and the development of AD 98 (Table 2), and carriers of this allele have therefore been studied extensively. In asymptomatic APOE ɛ4 carriers aged 19–28, both resting- and activity-related CBF levels are elevated,99,100 and BOLD signal changes during memory encoding tasks are elevated in the asymptomatic APOE ɛ4 carriers,101–103 consistent with reduced OEF and compensatory hyperaemia. Similarly, BOLD responses are elevated in patients with systemic lupus erythematosus (SLE – Table 6) but little or no cognitive defects, compared to controls. 104 In asymptomatic human immunodeficiency virus 1 (HIV-1) infected patients (Table 5), elevated BOLD signals can be observed 105 in proportion to signs of glial activation. 106 The notion that viral replication in brain parenchyma is associated with mild capillary dysfunction is consistent with findings that BOLD signal amplitudes are elevated in seropositive patients with low BBB penetration of combination antiretroviral therapy, but comparable to those found in controls and in patients with high penetrance and thereby virological control. 107 It should be noted that relative BOLD signal changes depend on both resting and activation-related CBF and OEF levels. The interpretation of such changes in terms of the underlying microvascular pathology therefore requires detailed analysis of the underlying physiology and magnetic resonance signal mechanisms.17,108
Moderate capillary dysfunction: Transition from hyperperfusion to CBF suppression
If capillary flow disturbances become more severe, then hyperemia fails as a means to compensate for reduced oxygen extraction efficacy. Instead, CBF responses – and ultimately resting CBF – must be attenuated in order to limit functional shunting. The transition from mild to moderate capillary dysfunction is therefore predicted to require dramatic, yet characteristic changes in CBF in order to maintain flow-metabolism coupling.
Such a transition, from hyper- to hypoperfusion, was indeed observed in a follow-up study of asymptomatic APOE ɛ4 carriers and controls. 109 At the time of their initial examination, APOE ɛ4 carriers showed higher resting CBF values in vulnerable brain regions than did control subjects, while CBF reductions in these regions 8 years later were significantly larger in APOE ɛ4 carriers than in controls. 109
If flow responses are limited by a physical, flow-limiting condition, then CBF cannot increase beyond a certain upper limit, irrespective of the metabolic requirements of brain tissue. If CBF suppression instead serves to maintain flow-metabolism coupling, then CBF, tissue metabolism and the extent of capillary dysfunction determine whether flow suppression is necessary. We briefly discuss how this phenomenon might have revealed itself in studies of SVD and its precursor states.
In a mouse genetic model of CADASIL, Joutel et al. 50 examined CBF responses to functional activation, CO2 inhalation and reduced perfusion pressure in 5- to 6-month-old animals, at a time-point where no changes in arterial structure or signs of BBB breakdown could be observed. Animals with and without the CADASIL-causing NOTCH3 point-mutation had similar blood pressure, similar resting CBF and similar CBF responses to CO2 inhalation, a vasodilatory stimulus. CBF responses to functional activation, however, were attenuated in CADASIL mice. 50 These findings are consistent with capillary dysfunction that only limits tissue oxygenation during functional activation – either by deficient pericyte-mediated capillary dilation (and CTH reduction) during functional activation, 19 or because only the combination of elevated CBF and increased metabolic demands ‘unmasked’ capillary dysfunction at this early point in the development of characteristic disease signs.
In asymptomatic APOE ɛ4 carriers aged 50–65, elevated resting CBF values have been reported at a time where their CBF- and BOLD-responses to functional activation had been suppressed. 110 Suppression of CBF-responses to sensory stimulation at a time where resting CBF is elevated has also been observed in spontaneously hypertensive rats, prior to any changes in their microvasculature. 111 These findings are again consistent with the gradual transition from mild to moderate capillary dysfunction which first becomes apparent in states of high metabolic demand.
The unmasking of capillary dysfunction by combinations of high CBF and CTH that necessitate flow suppression was recently illustrated in a study by Suri et al. 112 who observed flow suppression during hypercapnia (a strong vasodilator) in young APOE ɛ4 carriers compared to noncarriers. These young APOE ɛ4 carriers showed increased hippocampal BOLD responses to memory tasks, and the attenuation of their CBF responses during hypercapnia accounted for 70% of this increase, 112 consistent with the prediction that mild (asymptomatic) capillary dysfunction links the two findings.
Attenuation of functional hyperemia can also be observed immediately after administration of AT2 and Aβ in animal models of hypertension and AD/amyloidosis, respectively (Table 2)113–115 and antedates any morphological changes in the vessel wall or brain parenchyma and even the development of high blood pressure in the model of hypertension. 116 Although the effects of AT2 and Aβ on pericyte tone in living brain remain to be studied, these findings are consistent with capillary dysfunction caused by pericyte constrictions and compensatory increase in VSMC tone to limit flow responses.
Flow suppression and small vessel changes
The flow suppression observed in animal models of hypertension and AD/amyloidosis is caused by vascular production of ROS,117,118 which reacts with NO to form peroxynitrite. 119 Both NO depletion and peroxynitrite cause vasoconstriction by impairing normal smooth muscle cell relaxation, 120 but also long-term remodelling and thickening of the vessel walls and VSMC damage. 121 By evoking flow suppression, capillary dysfunction may therefore contribute to – or even antedate – the wall damage observed in small arteries and arterioles before SVD becomes overt. Note that wall thickening narrows the vascular lumen and may reduce CBF and attenuate CBF responses. This ‘mechanical’ flow suppression would therefore be expected to make flow suppression by oxidative stress superfluous over time.
Oxygen diffuses through arteriolar walls to supply the capillary-free area around arterioles, and it can reach tissue with capillary supply as well. In fact, recent studies suggest that arterioles account for as much as 50% of the total oxygen extraction during rest, while capillaries serve as the primary site of oxygen extraction during functional hyperemia. 122 Arteriolar oxygen extraction is insensitive to CTH downstream, and the increased arteriolar tortuosity and length observed in some SVD precursor states 123 may therefore, paradoxically, increase arteriolar oxygen extraction capacity by increasing arteriolar surface area and transit time. The long, tortuous medullary arteries observed in SVD, however, are generally thought to cause hemodynamic insufficiency. Studies of retinal vessels in SVD patients suggest that venules are generally wide in SVD precursor states.124,125 While the restricted venular lumen observed in venous collagenosis 123 (Table 7) would be expected to facilitate tissue oxygen extraction by prolonging blood’s transit time through the capillary bed, complete venous occlusions, instead, cause venous infarcts to develop.
Moderate capillary dysfunction: Failure to suppress CBF
If the microcirculation cannot evoke intrinsic mechanisms to limit CBF and CBF responses, then functional shunting can become so severe that stroke-like symptoms and hypoxic tissue injury may develop at CBF values well above the classical ischemic threshold.
Fabry’s Disease (Table 3) is associated with cerebral hyperperfusion,126–128 which is reversed or attenuated by therapies that remove the capillary deposits shown in Figure 2.126,127 This hyperperfusion might, in principle, represent flow metabolism coupling, with hyperaemia compensating for mild capillary dysfunction. The relative hyperemia is, however, observed in patients with severe neurological symptoms
127
and seemingly antedates the development of white matter lesions that may occur in the disease,
129
suggesting that tissue damage is the result of insufficient suppression of excessive vasodilation and functional shunting in this disease.
Panel (a) illustrates the organization of endothelial cells, basement membrane and pericytes in the vessel wall. Capillaries are ensheathed by astrocytic endfeet, and neuronal terminals are closely apposed to capillaries and pericytes.
66
Source: Reproduced from Hamilton et al.
66
according to the Creative Commons terms. Panel (b) shows a cross section of normal capillary with a thin basement membrane (arrow) and normal appearing endothelial cell (e). In ageing (Panel (c)), thickened basement membranes (arrows), pericapillary fibrosis and pericyte loss are often found. Source: Panels (b) and (c) are reproduced from Farkas et al.
188
Panel (d) shows a capillary cross section from the skin of a patient with Fabry’s disease. Note the lamellar sphingolipid inclusions in the capillary endothelium (arrow). These inclusions disappear upon enzyme replacement therapy. Source: Reproduced from Eng et al.
189
Panel (e) shows typical cerebral capillary wall pathology in human AD. The arrow indicates pericyte degeneration. The symbols denote lumen (l), endothelial cell (e), basement membrane (*) and pericyte (p). Source: Reproduced from Farkas et al.
190
Panel (f) shows a cross section of a muscle capillary from a patient with MELAS. Note the thickened basement membrane and increased number and size of mitochondria in the pericyte. Source: Reproduced from Sakuta and Nonaka.
191
Panel (g) shows a cross section of a cerebral capillary from the motor cortex of a MELAS patient with accumulation of mitochondria in the endothelial cell. Source: Reproduced from Ohama et al.
192
with permission from the publisher. AD: Alzheimer’s disease; MELAS: mitochondrial encephalopathy with lactic acidosis and stroke-like episodes.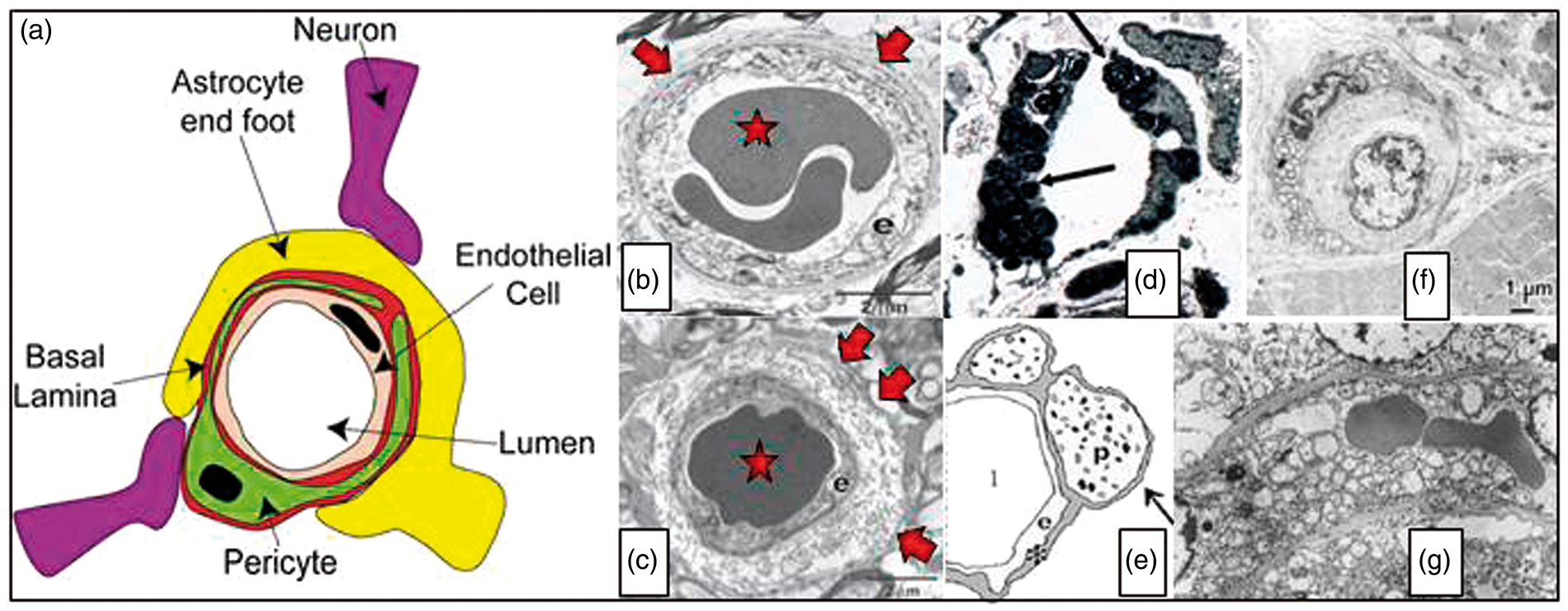
Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS – Table 3) is also associated with severe hyperperfusion, which seemingly contributes to tissue infarction. Studies have revealed globally elevated CBF, low OEF and reduced cerebral metabolic rate of oxygen (CMRO2) in MELAS patients both before130–133 and after131,132,134 stroke. In these patients, the mitochondrial enzymes needed for oxidative phosphorylation are deficient, and it is therefore unclear how tissue levels of oxygen, ATP and lactate affect neurovascular coupling. The lack of metabolic feedback mechanisms to limit excessive vasodilation in this condition is underscored by the finding that normal vasodilation is preserved, even in hyperperfused stroke lesions. 135
Vascular endothelium is extremely vulnerable to radiation and post-radiation angiopathy (Table 7) is associated with capillary rarefaction and tissue hypoxia.136,137 Mineura et al. 138 found hyperperfusion and low OEF early after radiation therapy in humans, and Hahn et al. 139 demonstrated a dose-dependent CBF increase three months after irradiation. If this hyperaemia was a compensatory response to mild capillary dysfunction, then tissue oxygen tension would be elevated rather than reduced and hyperaemia limited by flow-metabolism coupling rather than radiation dose. Instead, these observations suggest that microvascular signalling is severely disturbed, possibly due to a loss of endothelial regulatory signalling. After radiation exposure, initial hyperperfusion is often followed by normalization of CBF, 138 but OEF remains low, 138 indicating reduced tissue metabolism as a result of tissue damage. Hahn et al. 139 observed that neuropsychological performance deteriorated when the dose-dependent CBF increase three months after irradiation was followed by a reduction in CBF by six months. These findings suggest that uncontrolled hyperemia contributes to on-going tissue damage at least three months after radiation, but also lends hope to the notion that dose fractionation might ameliorate tissue damage and cognitive decline.
Severe capillary dysfunction
In severe capillary dysfunction, oxygen availability gradually approaches the metabolic needs of brain tissue. The parallel reduction in tissue oxygen tension, in turn, is highly conducive to BBB breakdown, Aβ deposition and loss of trophic support. See literature7,140 for a discussion of capillary dysfunction in dementia and preliminary data.
If capillary changes become so severe that CTH approaches CTHmax, then even increases in viscosity might reduce tissue oxygenation enough to trigger neurological symptoms or tissue injury. This suggests a mechanism by which dehydration or bacterial infections (during which WBC count is elevated) can cause neurological deterioration or stroke-like symptoms in patients with preexisting, capillary dysfunction. 6 We discuss therapeutic implications of this pathophysiological process below.
In severe capillary dysfunction, resting CBF is predicted to approach the classical ischemic threshold at which oxygen extraction is optimal, even for high CTH values. This biophysical feature implies that, paradoxically, tissue oxygenation may be improved to meet the metabolic demands of functional activation by reducing CBF to reduce physiological shunting. Indeed, Ferraccioli et al. 141 observed inverted CBF responses to painful stimuli (Raynaud’s phenomenon) in the sensorimotor cortex of patients with scleroderma or SLE vasculitis (Table 6).
What determines the course of tissue damage in SVD?
The analysis presented here suggests that progressive capillary dysfunction inevitably leads to tissue damage – either gradually, by loss of trophic support, amyloid pathology, hypoxic injury or inflow of toxic substances through a failing BBB – or suddenly, in relation to stroke-like episodes. The precise cause of the small subcortical infarcts, lacunes, white matter hyperintensities, microbleeds and cortical atrophy that accompany SVD has been the subject of intense scrutiny – see recent work by Wardlaw et al.3,4,142
Hassan et al. 143 showed that the intron 4ab polymorphism of the endothelial NO synthase (eNOS) gene protects against the development of SVD in the form of symptomatic, small subcortical infarctions, but not of SVD with white matter hyperintensities, suggesting that insufficiency of NO may be associated with the development of small subcortical infarctions. Similarly, the intron 4aa genotype appears to be protective for lacunar infarctions. 144 In mammals, dietary nitrite is a major source of NO, 145 and eNOS may be involved in the conversion of nitrate to NO in tissue. 146 The finding that blood nitrite levels depend on endothelial nitric oxide gene haplotype 143 may indicate that interactions between genotype and dietary habits (see below) is one of many factors which determine whether SVD progresses along a ‘stroke-like’ clinical presentation or along one of white matter hyperintensities and cognitive decline.
Cognitive decline or stroke-like phenotype?
Both capillary dysfunction and arteriolar narrowing can lead to hypoxic tissue injury – but what determines whether a SVD precursor state develops along a ‘dementia-like’ 7 or a ‘stroke-like’ 6 pathway? Although ‘mixed’ cases exist, chronic spirochete infections by Treponema pallidum and Borrelia burgdorferi both come in a meningovascular form, which is dominated by lymphoplasmacytic infiltrates and intima-proliferation in the walls of leptomeningeal arteries, veins and vasa vasorum and associated with the development of multiple cerebral infarcts, that is, a stroke-like phenotype. In their parenchymal form, spirochetes invade brain tissue to aggregate around microvessels, and particularly capillaries. 147 In this form, the infection is instead associated with cortical atrophy and dementia – and often amyloid deposits and neurofibrillary tangles. In severe neurosyphillis, antibacterial treatment in some cases leads to significant increases in CMRO2 without increases in CBF from its near-ischemic levels. 147 Such an ‘isolated’ improvement of OEF and cerebral metabolism is difficult to reconcile with flow-limiting SVD, or the reversal of infection-specific suppression of neuronal function. 147 We speculate that the pericapillary invasion of spirochetes may be associated with severe capillary dysfunction, causing CTH to exceed CTHmax in general paresis. Reduction of capillary flow disturbances by antibiotic therapy would then be expected to improve CMRO2 and neurological function, but only to normalize CBF if CTH was reduced considerably. In patients with neuroborreliosis and less severe neurological symptoms, cerebral hypoperfusion is partially reversed by antibiotic therapy. 148
Similar correlates between microvascular histopathology and the progression along pathways dominated by stroke-like symptoms or cognitive decline may exist for other SVD risk factors and help elucidate what separates these etiologically related, but clinically separate, presentations.
SVD risk factors versus comorbidities
Many regard hypertension as the single strongest risk factor for SVD, but not all patients with SVD have hypertension or diabetes, and it has been debated whether hypertension might be secondary to impaired cerebral perfusion. Here we briefly discuss whether hypertension and type-2 diabetes are risk factors for SVD – or whether they themselves reflect evolving capillary dysfunction.
We argued above that the attenuation of functional hyperemia observed after administration of AT2 – a likely source of capillary dysfunction 38 – may represent flow suppression to maintain flow-metabolism coupling in brain tissue. This phenomenon was shown to antedate the development of high blood pressure in AT2 animal models of hypertension. 116 Is it conceivable that hypertension itself represents a systemic response to maintain tissue oxygenation in certain organs as their level of capillary dysfunction reaches the thresholds at which flow-suppression becomes necessary? If so, which organs? Dickinson proposed that essential hypertension serves to overcome increased vascular resistance in the brain’s large, feeding vessels. 149 While studying large vessel resistance in hypertension, however, he points out that histopathological studies show early changes in capillaries and veins, rather than small arteries and arterioles, in brain tissue from hypertensive patients.149,150 These observations are consistent with capillary changes as a primary event in hypertension and with the notion that capillary dysfunction require proximal adjustments of vascular resistance to preserve brain oxygenation.
If hypertension represents an early sign of capillary dysfunction, either in brain or other in other organs, then antihypertensive therapy may be warranted at an early stage in patients at risk of SVD. 151 Further studies should address whether the antihypertensive effects of AT2 inhibitors, angiotensin converting enzyme (ACE) inhibitors and Ca++ channel blockers are linked to inhibition of AT2 and Ca++-dependent pericyte constriction. Specific antihypertensive agents may also prove useful in targeting organ- and cell-specific aspects of capillary dysfunction.
Measurements of glucose-analogue extraction in the human brain suggest that capillary transit time heterogeneity limits glucose extraction. 152 Although glucose extraction differs somewhat from that of oxygen, 153 our analysis suggests that capillary dysfunction also limits the clearance of glucose from blood.5,154 In prediabetic rats, impaired glucose tolerance is indeed accompanied by reduced oxygen extraction efficacy, 155 and the beneficial effects of Mediterranian Diet (MD) (see discussion below) extend to both ambulatory blood pressure and blood glucose levels. 156 Our preliminary analysis indicates that capillary dysfunction favours the extraction of glucose over that of oxygen. 157 As capillary dysfunction becomes severe, tissue is therefore predicted to reveal some degree of aerobic glycolysis. 157 This property may contribute to findings of reduced respiratory coefficient in patients with severe hypertension. 149
Therapeutic implications
Despite the heterogeneity and complexity of the conditions known to contribute to the development of SVD and to subsequent stroke or cognitive decline, we speculate that capillary flow disturbances may be a shared feature of some if not most of these conditions. Below, we therefore discuss aspects of capillary dysfunction that can be prevented or alleviated and therefore might be of general benefit to patients at risk of SVD, stroke or cognitive decline.
Blood viscosity
Dehydration and infection-induced leukocytosis increase blood viscosity. These common occurrences can therefore reduce the brain’s oxygen supply by increasing CTH. Influenza vaccinations are offered to the elderly in order to reduce the incidence of influenza and secondary infections, and this is known to reduce the number of stroke deaths.158–160 Infections may trigger thromboembolism by destabilizing atheromatous plaques in the walls of cerebral vessels, but we speculate that increases in blood viscosity may elicit stroke-like symptoms in patients with preexisting, severe capillary dysfunction as well. Therefore, influenza vaccinations may be beneficial for SVD patients, irrespective of age. The impact of bacterial infections on cerebral oxygenation and thereby cognition may also be reflected in the observation that eradication of chronic helicobacter pylori infections in AD patients dramatically improves their cognitive scores and overall survival.161,162
CTH may be reduced by lowering blood viscosity, and aggressive management of hyperlipidaemia may therefore be warranted in SVD patients. Similarly, high homocysteine levels increase blood viscosity, increase the adhesion of monocytes to the capillary wall and cause oxidation of low-density lipoproteins 163 and could therefore represent a source of capillary dysfunction. Indeed, Hassan et al. 164 showed that elevated homocysteine levels represent an independent risk factor of SVD. A meta-analysis has suggested that homocysteine-lowering therapy may reduce stroke risk in regions where folate levels are low. 165
Phosphodiesterase (PDE) inhibitors reduce platelet aggregation, 166 decrease blood viscosity 167 and increase the flexibility of erythrocytes. 167 While reduced platelet aggregation might increase bleeding risk, the haemorheologic PDE inhibitor effects would be expected to reduce CTH and thereby improve tissue oxygenation. For a recent review of pharmacological approaches to SVD management, see literature. 168
Nicotine up-regulates the expression of adhesion molecules in the capillary endothelium 169 and increases leukocyte rolling, 170 keeping in mind that the latter is observed mainly in post-capillary venules where selectin density and glycocalyx properties provide optimal adhesion for leukocyte recruitment. 171 In additions to nicotine’s effects on larger vessels and large vessel atheromatous plaques, smoking would therefore be expected to worsen capillary flow disturbances, to accelerate the development of SVD from its risk factors and to increase the risk of an ischemia-like event. Indeed, pack years of smoking are associated with an increased risk of stroke in CADASIL 172 and with a higher burden of SVD lesions in patients with sporadic lacunar stroke. 173 Patients with SVD precursor states should therefore receive help to reduce not only smoking, but also nicotine consumption in general, in order to reduce their risk of later cognitive decline or stroke.
Hyperaemia, anaemia and poor oxygen saturation
Hyperaemia is predicted to be harmful in patients with capillary dysfunction.
Obstructive sleep apnea (OSA) is associated with periods of severe nocturnal hypercapnia and hypoxemia, both of which cause dramatic increases in CBF in the normal brain. In addition, reductions in oxygen saturation cause a proportionate reduction in the net oxygen metabolism that can be supported for a given level of capillary dysfunction – increasing the risk of neurological symptoms. The risk that hyperaemia poses to patients who suffer from capillary dysfunction may in part be reflected in the observation that continuous positive airway pressure (CPAP) treatment reduces the incidence of strokes. 174
The vulnerability of patients with capillary dysfunction to increased blood viscosity and reduced arterial oxygen content may contribute to the strong association between delirium and dementia: 175 declining haemoglobin levels, dehydration and minor infections are known to herald delirium in the elderly. In patients with severe capillary dysfunction, small reductions in haemoglobin concentration or blood saturation, as well as increases in blood viscosity, may trigger regional cerebral hypoxia and thus, in principle, contribute to delirium symptoms. For patients with SVD precursor states, stricter definitions of anaemia and special attention to pulmonary function may therefore be warranted to reduce the risk of delirium and to alleviate the long-term consequences of chronic brain tissue hypoxia.
NO depletion
Capillary NO depletion due to oxidative stress and tissue hypoxia may represent a modifiable aspect of capillary dysfunction. Mediterranean diet is rich in green-leafed vegetables, a major source of dietary nitrate (see above). One might expect this diet to offer some protection towards NO depletion in patients with capillary dysfunction, and hence the development of SVD pathology. Preference for MD is indeed associated with a lower burden of white matter hyperintensities 176 and, more generally, with a lower risk of ischemic stroke.177,178 Keeping in mind that this protective effect may be attributable to other MD constituents, these observations may warrant further studies of dietary interventions in SVD and its risk factors. See also the study by Bath and Wardlaw. 168
Stroke management in SVD patients
Given the deleterious effects of dehydration and poor saturation on tissue oxygenation, pre-hospital rehydration and efforts to reach full blood saturation may be warranted in patients with capillary dysfunction. While recanalization therapy clearly limits infarct size if large vessel occlusion is the cause of tissue hypoxia, it is important to keep in mind that means of restoring capillary flow patterns should also be explored.19,24,179 Since SVD risk factors are generally prevalent in the stroke population irrespective of mechanisms of individual events, these interventions may be of general benefit.
Diagnostic considerations
Flow-limiting conditions and moderate capillary dysfunction are both predicted to reduce cerebrovascular reserve capacity and increase OEF. We showed recently that elevated CTH can contribute significantly to the elevated OEF observed in patients with carotid stenosis 140 and proposed that concurrent capillary dysfunction may contribute to the non-superiority of bypass surgery over aggressive cardiovascular risk factor management for stroke prevention in patients with severe carotid disease. 180 Assessment of CTH and thus the ‘capillary contribution’ to elevated OEF may therefore be warranted when revascularization therapy is considered in patients with carotid stenosis and SVD progenitor states such as diabetes and hypertension.
The detection of capillary dysfunction may also prove important in the management of acute stroke in SVD patients. In acute ischemic stroke, perfusion weighted imaging (PWI) is widely used in the identification of salvageable tissue prior to recanalization therapy. Based on dynamic computerized tomography (CT) or magnetic resonance imaging (MRI) during bolus injection of an intravascular contrast agent, concentration–time curves (CTC) can be estimated in each image voxel and corrected for arterial contrast profile in each patient. The resulting transit time metrics, such as the mean transit time (MTT) and time-to-maximum (Tmax), can then be compared to thresholds above or below which literature studies have shown high likelihood of infarction in the absence of recanalization. Such studies of the relation between perfusion metrics and tissue outcome have found inconsistent transit time thresholds. 181 We speculate that this finding relates to the fact that tissue outcome depends on the extent of capillary dysfunction in ‘hypoperfused’ tissue: first, the tissue CTC recorded during PWI depends on both CBF and CTH, but the PWI algorithms used in studies so far cannot disentangle the two. 182 The success of current recanalization therapies, however, clearly hinges on whether hypoperfusion is the result of a vascular occlusion, exacerbated capillary dysfunction, or both. Differences in the proportion of stroke subtypes and the incidental dependence of preferred transit time metric on oxygen extraction efficacy 6 may therefore explain the difficulty in establishing ‘universal’ transit time thresholds to define tissue-at-risk in acute stroke patients from cohort studies.
Figure 3 shows acute PWI maps from an 84-year-old man with a history of hypertension and smoking who presented with mild (NIHSS 4) stroke symptoms. We note that MTT was prolonged and OEF predicted to be high in relation to tissue that subsequently went on to infarction (red circle). Prolonged MTT and high OEF are indeed central characteristic of penumbral tissue,
22
yet areas with white matter hyperintensities showed similar changes in this patient. Our preliminary data suggest that knowledge of CTH is necessary to predict OEF as obtained by positron emission tomography (PET).
140
Needless to say, further studies are needed to disentangle the effects of limited flow (low CBF) and capillary dysfunction (high CTH relative to MTT) on oxygen extraction in SVD and SVD-related strokes.
The two leftmost columns in Figure 3 show acute FLAIR and ADC at four identical slice-positions in an 84-year-old patient who presented with acute stroke symptoms three hours earlier. Acute ischemic changes are visible as areas of low ADC (red circles), consistent with reduced extracellular water diffusion and often ascribed to anoxic depolarizations. The FLAIR images show discrete (purple ellipses) and confluent (brown circles) white matter hyperintensities. The rightmost column show FLAIR images in the same slice positions 30 days later. The green overlays on bright tissue lesions (within the red circles) indicate tissue that infarcted in relation to the stroke episode. Note that areas of elevated CTH and MTT are observed in relation to the area of low ADC.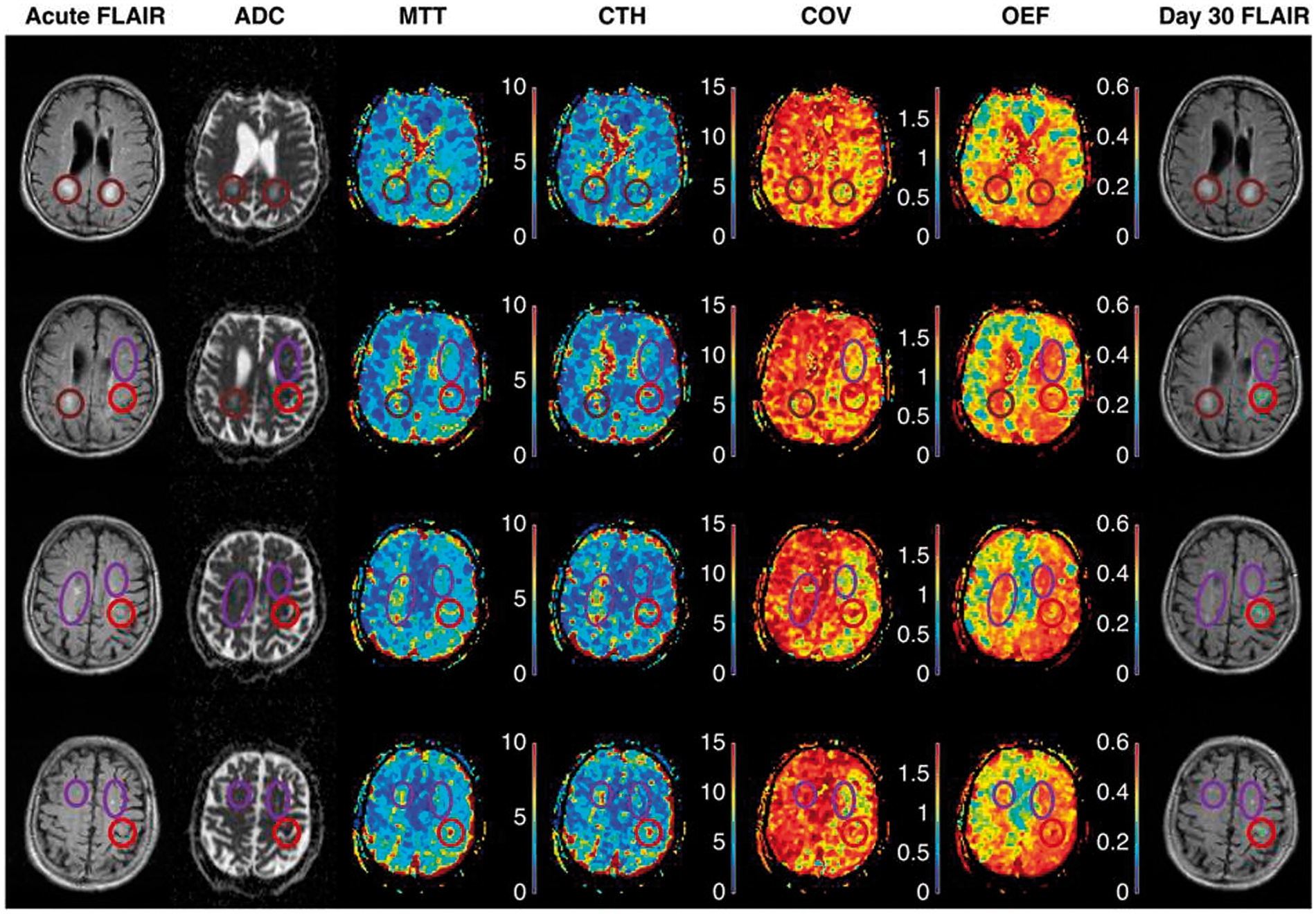
Conclusion
The morphology and function of cerebral capillaries in conditions considered risk factors for SVD remains relatively understudied. This review suggests that capillary dysfunction may be an early and shared feature of SVD risk factors, and a source of neurodegeneration, stroke and cognitive decline, despite considerable differences in the aetiologies and clinical presentations of these syndromes. We propose that the study of parallel changes in capillary and arteriolar morphology and function may represent an important area of preclinical SVD research. Except for studies in animal models of hypertension, diabetes and CADASIL and in human APOE-ɛ4 carriers, we identified few reports of neurovascular coupling in the early or presymptomatic phase of SVD risk factors. Given that altered neurovascular coupling may reveal capillary dysfunction before symptoms are predicted to arise, studies using BOLD contrast or CBF-sensitive methods might provide new insights into the aetiopathogenesis of SVD. Similarly, direct measurements of CTH as an index of capillary dysfunction should be applied to SVD risk factors to test the sensitivity of PWI as an investigative or diagnostic tool.
In this review, the pericyte emerged as a critical determinant of several aspects of capillary function. Recent breakthroughs in the understanding of this cell19,24,64,67,183–185 have already contributed to our understanding of neurodegeneration and stroke. Studies in stroke 179 and diabetic retinopathy 186 lend hope to the notion that pericytes and other components of the neurovascular unit that are affected in SVD may represent targets in future efforts to prevent capillary dysfunction.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: LØ, TSE and MBH were supported by the Danish National Research Foundation (CFIN), The Danish Ministry of Science, Innovation, and Education (MINDLab) and the VELUX Foundation (ARCADIA). HSM is supported by an NIHR Senior Investigator award and by the Cambridge University Hospitals NIHR Comprehensive Biomedical Research Centre. FM was supported by a Chief Scientist Office of Scotland project grant. JW was supported by the Scottish Funding Council and Chief Scientist Office through the Scottish Imaging Network A Platform for Scientific Excellence (SINAPSE), the Wellcome Trust and the Medical Research Council through the Centre for Cognitive Ageing and Cognitive Epidemiology (CCACE).
Acknowledgments
The authors wish to thank Marie-Germaine Bousser, Anne Joutel, Richard Buxton, David Brooks, Simon Fristed Eskildsen, Rikke Beese Dalby, Kim Mouridsen, and Sune Nørhøj Jespersen for inspiration and helpful comments to our manuscript.
Declaration of conflicting interests
MBH owns stock in COMBAT Stroke.