
Abstract
The interaction mechanisms of undoped, silicon- and boron-doped C20 fullerenes and 1-acetylpiperazine (1-ap) were investigated. Stability, electronic properties, influence of water on the solubility and stability, molecular parameters, descriptive vibrational bands and nuclear magnetic resonance shielding values are reported. The quantum mechanical calculations were carried out using the M06-2X functional and the 6-31G(d) basis set. It is observed that all the complexes are more stabilized in water compared to the gas phase. The most stable complex was found as silicon-doped fullerene interacting with the carbonyl edge of 1-ap releasing energy of 64.13 kcal/mol in water.
Introduction
C20 fullerenes, comprising only pentagons and having the largest mass density among the fullerene family, are the smallest members of fullerenes (Han et al., 2000; Li and Zhang, 2013). They were first experimentally synthesized by Prinzbach et al. (2000). Due to their useful and versatile properties, fullerenes have found a wide range of applications expanding from medical, bio-nano technological research to the drug industry (Beyeh et al., 2010; Hatnapure et al., 2012; Kharb et al., 2012; Kavitha et al., 2013; Zhou et al., 2010).
The development of different types of materials with enhanced physical, chemical and biological properties is of current research interest. Theoretical approaches provide useful insights for studying molecular systems before going into experimental investigations. Density functional theory (DFT) has been widely used as a theoretical approach (Anafcheh et al., 2016; Anafcheh and Ghafouri, 2014; Dheivamalar and Sugi, 2015; Handy et al., 1993; Handy et al., 1996; Hassani and Tavakol, 2014; Helgaker et al., 1999; Lee and Boo, 1996; Li et al., 2015; Stephens et al., 1994). Piperazine and its derivatives have been the target of research in the production of anti-microbial and anti-carcinogenic pharmacological agents, in a series of therapeutic processes and in the synthesis of complexes and clathrates (Beyeh et al., 2010; Hatnapure et al., 2012; Long et al., 2010; Parlak et al., 2009a, 2009b).
The scope of the present research involves the study of the interaction mechanisms of 1-ap and undoped C20 and B-, Si-doped C20 fullerene complexes based on computational methods with the M06-2X functional and 6-31G(d) basis set (Zhao and Truhlar, 2008). We, hereby, report the results obtained, discuss these results and hope that this study catalyses further investigations.
Computational details
Molecular structures of the complexes were examined using the M06-2X/6-31G(d) method in the gas phase and in the water as the solvent. Vibrational frequency calculations and computations of the chemical shift values for 11B and 29Si nuclei were also performed at the same level of theory. The optimization processes were repeated with no geometric constraints until imaginary frequencies were not observed. The optimized structures of the complexes obtained are given in Figure 1. In order to evaluate the stability of the complexes, binding energies (Eb) were calculated using equation (1) (Hazrati and Hadipour, 2016):
Optimized molecular structures for the investigated systems.
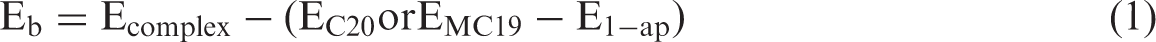
In equation (1), Ecomplex, EC20 or EMC19 and E1-ap represent the total electronic energies of the ligand interacted complex system, undoped and B-, Si-doped fullerene complex and 1-ap molecule, respectively.
When dealing with the minimization of energy of the systems with multiple components, basis set superposition error (BSSE) appears as an important factor affecting the results of Eb values (Liu and McLean, 1973). In this work, BSSE was taken into account using the counterpoise correction method (Boys and Bernardi, 1970; Gutowski and Chałasinski, 1993; Simon et al., 1996). Charge transfer direction and electrophilic character assessments of the complexes were examined by calculating electrophilicity indexes (ω) and chemical hardness (η) as given by equations (2) and (3), respectively (Parr et al., 1991; Pearson, 1986):

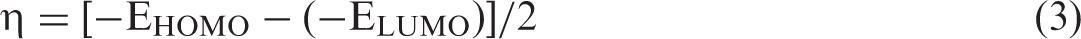
In order to have insights of the solvent effect, all the calculations were conducted both in the gas phase and water as the solvent. Gaussian 09 was used for all calculations (Frisch et al., 2009). The optimized molecular structures were viewed with the GaussView program (Dennington et al., 2008).
Results and discussion
Firstly, electrostatic potential surfaces (EPSs) were obtained for 1-ap ligand molecule and they are illustrated in Figure 2. Two possible interaction edges, C=O and NH, were located based on the EPS findings. As for undoped C20+1-ap interaction, the complex system was analysed as different structures and the lowest energy configurations for the complex systems are shown in Figure 1. Interaction sites for doped fullerene systems were chosen as the boron and silicon atoms similar to the earlier reports (Hazrati and Hadipour, 2016; Mitschker and Kluner, 2012). Binding energy can be described as the difference between total energy of the optimized complex and the total energy of the each fragment, which are free 1-ap and undoped C20 or B-, Si-doped C20 (Ivanov et al., 2011).
Electrostatic potentials on 1-ap. Surfaces are defined by the 0.0004 e/Å3 contour of the electronic density. Color ranges (a.u): blue, more positive than 0.03 and red, more negative than −0.03.
BSSE-corrected binding and solvent energies (kcal/mol) of the investigated structures.

Eb: binding energy; Es: solvent energy.
Some energetic parameters (eV) of the investigated structures.

C=O and NH stretching vibrations (cm-1) for the investigated structures.

The nuclear magnetic resonance chemical shifts of boron and silicon atoms were also identified to support the results of structural findings. It was observed that the resonance frequencies of 11B and 29Si nuclei in water changed around 71/68 ppm from BC19 to BC19+1-ap (NH)/BC19+1-ap (C=O) and 301/322 ppm from SiC19 to SiC19+1-ap (NH)/SiC19+1-ap (C=O) upon adsorption and these reflect the possible interaction.
Conclusions
In the current theoretical search, based on the DFT calculation using the M06-2X/6-31G(d) method, the interaction mechanisms of undoped, B- and Si-doped C20 fullerenes and 1-ap were investigated both in the gas phase and water as the solvent. For the calculations conducted in the gas phase, within fullerene system, the complex SiC19+1-ap (NH) with a binding energy of −54.42 kcal/mol was found the most stable, while C20+1-ap (C=O) was found as the least stable with a binding energy of −1.77 kcal/mol. In water, SiC19+1-ap (C=O) complex appears as the most stable with a binding energy of −64.13 kcal/mol and C20+1-ap (C=O) is the least stable with a releasing energy of 10.49 kcal/mol. In the gas phase, the interaction of C20 with C=O edge of 1-ap released was around 1.8 kcal/mol energy, the value of which lies in the range of physisorption. On the other hand, if the interaction occurs at NH edge, approximately 20 kcal/mol is released and this indicates the possibility of a chemisorption.
Footnotes
Acknowledgements
We acknowledge the computing resources provided by Fencluster system in the Science Faculty of Ege University. The authors acknowledge the useful comments from the reviewers to improve the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.