
Abstract
Fullerenes and piperazines have been investigated, particularly, in the field of nanoscience and medicinal chemistry. In the present research, besides discussing structural and electronic properties, the most probable interaction mechanisms between C20, B-, Si-, Al-, Ga-doped C20 and 1,4-diformylpiperazine (1,4-dfp) were studied by employing density functional theory (DFT) in both the gas phase and water as the solvent. Stabilities of the investigated complexes were discussed based on the binding energy and electronic properties such as band gap energy, chemical hardness and electrophilicity index. It is found that doped complexes are more stabilized in water compared to the gas phase. However, the interaction between C20 and 1,4-dfp weakens upon the introduction of water as the solvent.
Introduction
The development of new drugs as a result of growing microbial resistance is a current issue among researchers. It is an urgent and very crucial subject to produce new generation of anti-microbial drugs. Piperazine and its derivatives have found potential applications for the production of anti-microbial agents and in a series of therapeutic processes because of their anti-bacterial, -fungal and -carcinogenic pharmacological activities (Beyeh et al., 2010; Bogatcheva et al., 2006; Hatnapure et al., 2012; Kavitha et al., 2013; Kharb et al., 2012).
C20 molecule having a dodecahedral cage structure is the smallest member of the fullerenes. The production of the smallest fullerene is relatively much more difficult than that of C60. Prinzbach et al. (2000) synthesized C20, starting from dodecahedrane C20H20 (Paquette et al., 1983; Ternansky et al., 1982), by replacing the hydrogen atoms with bromine and debrominating the compound to obtain gas phase the C20 cage.
The 20 carbon atoms in the cage form 12 pentagons and 30 bonds. The molecular diameter is about 3.1 Å. (Sattler, 2011). It has the three major isomers: cage, bowl and ring. There are several calculations on the ground state energy and relative stability of C20 isomers. However, different ground state energy orderings were predicted by calculations and these depend on the method used such as Hartree-Fock (HF) (Ering < Ebowl < Ecage) (Feyereisen et al., 1992; Parasuk and Almlöf, 1991), local density approximation (LDA) (Ecage < Ebowl < Ering) (Brabec et al., 1992; Wang et al, 1996), generalized gradient approximation (GGA) (Ering < Ebowl < Ecage) (Raghavachari et al., 1993), Hybrid HF/DFT (Ering < Ebowl < Ecage) (Allison and Beran, 2004), DFT/B3LYP (Ering < Ebowl < Ecage) (Xu et al., 2006), coupled cluster singles and doubles (CCSD) (Ecage ≈ Ebowl < Ering) (Taylor et al., 1995), CCSD (Ebowl < Ecage< Ering) (An et al., 2005), tight binding (Ecage < Ebowl < Ering) (Cao, 2001), quantum Monte Carlo (QMC)-pseudopotential (Ebowl < Ering < Ecage) (Grossman et al., 1995) and QMC-all electron (Ebowl < Ering < Ecage) (Sokolova et al., 2000). A DFT study on the possible forms of solid C20 phase has predicted the simple cubic lattice to be the most stable one. It has also been suggested that a one-dimensional chain of C20 cages can have a higher transition temperature if it is doped with either electrons or holes to shift the Fermi level to the peak of the density of states (DOS) (Miyamoto and Saito, 2001). Further, DFT calculations showed that the orthorhombic and tetragonal lattices with the polymerized C20 cage as a building block have lower energies than the simple cubic lattice. Calculations of DOS reported that these lattices are semiconductors with energy gaps of 1.4 and 1.7 eV correspondingly (Okada et al., 2001).
In order to make a more targeted delivery in medicine and nanoscience, different sizes of fullerene as the vehicle interacting with drugs and different types of molecules were reported (Baei et al., 2013a, 2013b; Bakry et al., 2007; Hadipour et al., 2015; Kroto et al., 1985; Peyghan and Noei, 2014; Peyghan et al., 2013; Renz et al., 2008; Singh and Lillard, 2009). In the current research, the interaction mechanism of 1,4-dfp molecule and a series of aluminium, boron, silicon, gallium doped and undoped fullerene systems for C20 cage were examined based on the quantum mechanical calculations. The primary intension is to provide insights before further experimental or theoretical investigations. We hereby report the findings of this theoretical research.
Computational details
In order to obtain the most stable configurations of the investigated systems, the structures were built and optimized at the level of M062X/6-31 G(d) in the gas phase and water. The polarizable continuum model was used to observe the solvation effects (Tomasi et al., 2005). Geometrical constraints were not imposed during the optimization process. Vibrational frequencies were calculated at the same level of theory to confirm the ground state structures without any imaginary frequency. Cartesian coordinates and vibrational wavenumbers of the optimized structures are provided as supplementary materials. In order to assess the stability, the electronic binding energies (Eb) of the systems were calculated as given by equation (1) (Parlak et al., 2017).
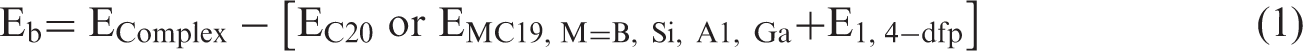
In equation (1), EComplex, EC20 or EMC19, M = B, Si, Al, Ga and E1,4-dfp stand for total electronic energies of the ligand-interacted complex system, undoped and Al-, B-, Si-, Ga-doped fullerene systems and 1,4-dfp, respectively. To make an assessment about the charge transfer, electrophilic character and behaviour of the investigated molecular systems, electrophilicity index (ω) and chemical hardness (η) were calculated as given by equations (2) and (3) (Parr et al., 1999; Pearson et al., 1986).

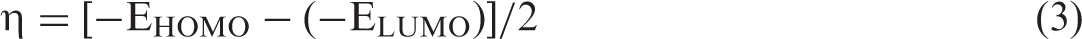
All computations were carried out using Gaussian 09 (Frisch et al, 2009). Optimized complex systems were visualized with GaussSum and GaussView programs (O’Boyle et al., 2008; Dennington et al., 2008).
Results and discussion
The optimized structures of the systems obtained are shown in Figure 1 and the electrostatic potential surface of 1,4-dfp is illustrated in Figure 2. An analysis of the electron density in Figure 2 allows the oxygen atom to be chosen as the active site of 1,4-dfp. The active sites for doped fullerene cages were chosen as B, Si, Al and Ga atoms in compliance with our previous study (Parlak et al., 2017). The calculated binding and solvent energies (Eb and Es) with M062X/6-31 G(d) level of theory in both the gas phase and water are collected in Table 1. In the implicit solvent model, the solvent used in the calculations, in this work it is water, is considered as a continuous medium instead of individual explicit solvent molecules (Tomasi et al., 2005). It is possible to observe the overall effect of the solvent in this model. Therefore, it is expected to observe the differences in the electronic properties and Eb energies of the investigated systems compared to gas phase calculations.
Optimized structures for the investigated molecular systems in the gas phase. Electrostatic potentials on 1,4-dfp. Color ranges, in a.u.: blue, more positive than 0.03 and red, more negative than −0.03. 1,4-dfp: 1,4-diformylpiperazine. Binding and solvent energies (kcal/mol) of the investigated molecular systems. Eb: binding energies; Es: solvent energy.

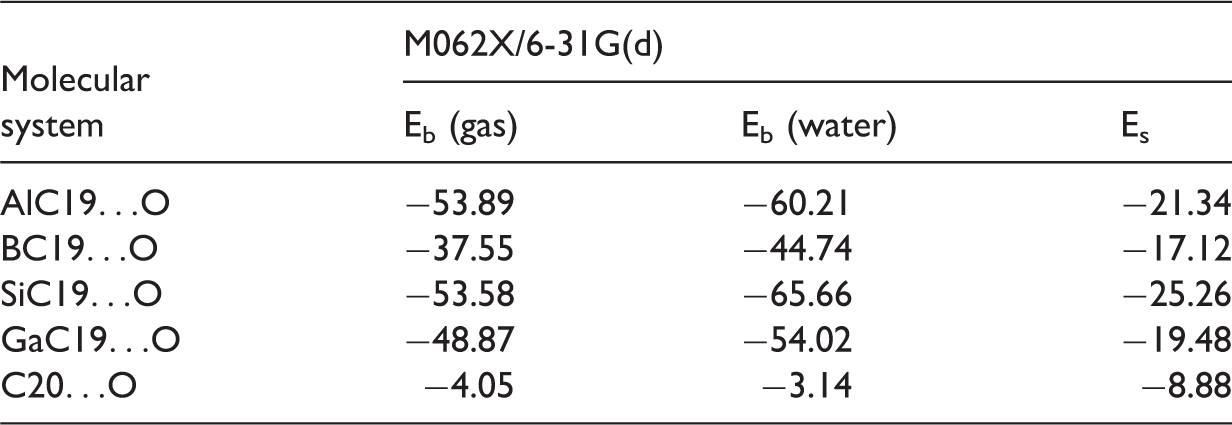
Except for C20…O complex, all the examined structures are more stable in water compared to the gas phase. When water is considered as the solvent, the binding energy of C20…O system is reduced in magnitude from 4.05 to 3.14 kcal/mol. Therefore, C20…O is more stable in the gas phase. The reported physisorption and chemisorption binding energy ranges are 1–2 kcal/mol and 10–100 kcal/mol correspondingly (Bhushan, 1999). The Eb energies for AlC19…O, BC19…O, SiC19…O and GaC19…O increase in magnitude by 6.32, 7.19, 12.08 and 5.15 kcal/mol, respectively. The interaction mechanism of C20…O in gas phase and water is closer to physisorption range. The dominant interaction mechanism for MC19…O in both environments can be considered as chemisorption. The Es energies given in Table 1 suggest that a higher solubility is possible for SiC19…O complex with −25.26 kcal/mol solvent energy compared to other systems.
Some energetic parameters (eV) of the investigated molecular systems.
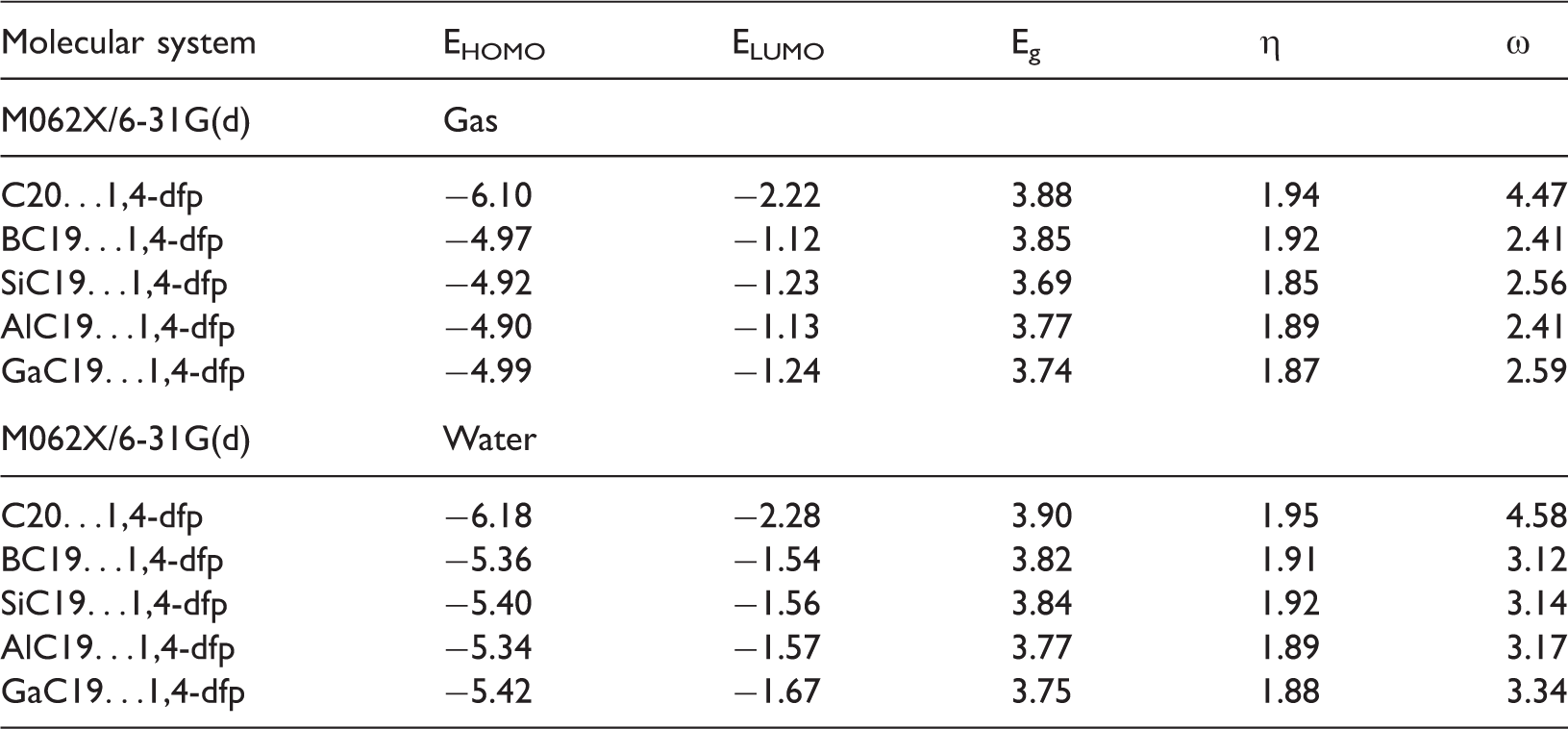
1,4-dfp: 1,4-diformylpiperazine.
The interatomic distances of Al…O, B…O, Si…O and Ga…O were calculated as 1.87, 1.54, 1.80 and 1.96 in gas phase and 1.81, 1.50, 1.73 and 1.91 Å in water. The consideration of water as the solvent reduces inter atomic distances, Eb increases in magnitude (Table 2) and complexes become more stable compared to the gas phase. In order to quantify the influence of doping to electronic properties of the C20 fullerene, the DOS graphs for the investigated structures were calculated, which are shown in Figure 3. Analysing Figure 3, it can be deduced that the electronic structure of the doped systems changes variably when water is considered as the solvent.
DOS spectra of the investigated molecular systems.
Conclusions
The nature of the interaction mechanisms of undoped, Al-, B-, Si- and Ga-doped C20 fullerenes and 1,4-dfp molecule was studied based on DFT with the M062X/6-31 G(d) method both in the gas phase and water as the solvent. In the gas phase, among the doped fullerene system, the Al-complex with binding energy of −53.89 kcal/mol is found to be the most stable one while the B-complex is the least stable one with a binding energy of −37.55 kcal/mol. In water, all the investigated samples get more stabilized except for C20+1,4-dfp complex. Water possibly hinders the effective interaction between C20 and 1,4-dfp which already has a weak interaction with a binding energy of −4.05 kcal/mol in the gas phase.
Footnotes
Acknowledgements
The authors acknowledge the computing resources provided by Fencluster system in the Science Faculty of Ege University.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.