
Abstract
Hydrodesulfurization reaction, as the last step of hydrothermal cracking reaction, is of great significance for the reduction of viscosity and desulfurization of heavy oil. Based on Density Functional Theory and using Dmol3 module of Materials Studio, this research simulated the adsorption and hydrodesulfurization of thiophene on Ni2P (001) surface, and discussed the hydrodesulfurization reaction mechanism of thiophene on Ni2P (001) surface. It was found that the direct hydrodesulfurization of thiophene had more advantages than the indirect hydrodesulfurization of thiophene. Finally, the optimal reaction path was determined: C4H4S+H2→C4H6.
Introduction
With the increasing demand for oil resources, thin oil field exploitation of China has entered the late stage of development, and the production of thin oil is becoming increasingly scarce. Therefore, the exploitation of heavy oil has become the main means to make up for the shortage of production. However, the viscosity of heavy oil is very high, and there are a large number of heteroatoms in its structure, which makes it difficult to exploit and transport heavy oil. The hydrothermal cracking reaction that occurs in the process of steam injection recovery of heavy oil can crack a part of the polymer components to produce light oil and improve the quality and reduce the viscosity, which has attracted the attention of researchers. The last and most critical step of the hydrothermal cracking reaction is hydrodesulfurization. The hydrodesulfurization reaction is to break the C–S bond and then form hydrogen sulfide with the hydrogen in the system, which reduces the viscosity and removes the sulfur element. With the development of technology, the industrial demand for heavy oil desulfurization is becoming more and more strict, so it is particularly important to further reduce the sulfur content by deep hydrodesulfurization (HDS) (Bataille et al., 2000) of heavy oil components. In order to achieve the effect of deep desulfurization of heavy oil (Angelici, 2006), it is necessary to reduce the steric hindrance effect of some sulfur-containing compounds by hydrogenation reaction, but the traditional catalyst has poor catalytic effect and weak hydrogenation ability, so it is difficult to achieve the effect of deep desulfurization (Sun and Prins, 2008). Therefore, researchers from various countries are committed to developing an HDS catalyst with excellent desulfurization effect (Oyama, 2003) to achieve the purpose of deep desulfurization of heavy oil components.
At present, transition metal elements and their sulfides have been widely used, such as Mo, Ni, Co, etc. (Chu et al., 2013; Li et al., 2015; Song et al., 2014), which are common desulfurization catalysts nowadays. Recently, carbides, nitrides, and phosphides have appeared in the literature because of their good physical properties and excellent hydrodesulfurization characteristics. Such catalysts have not been widely used in industry, but their application has broad prospect. Okamoto et al. (Okamoto et al., 2003) prepared precious metal catalysts such as Pd and Pt, and their experimental studies showed that such catalysts had better hydrodesulfurization effects than traditional transition metal catalysts. Vasudevan et al. (Jayamurthy and Vasudevan, 1994) found that the Pt catalyst had better hydrodesulfurization activity than the Co-Mo/Al2O3 catalyst system. Diana et al. (Stephanie et al., 2003) synthesized a supported catalyst and found that the hydrodesulfurization activity of a nickel phosphide catalyst supported on the surface of silica was better than that of a conventional sulfide catalyst. Maccra et al. (McCrea et al., 1997) synthesized a β-Mo2C catalyst supported on the surface of alumina (A12O3), and found that its effect on thiophene hydrodesulfurization was better than that of traditional transition metal catalysts. Puello-Polo et al. studied the catalytic effect of Ni-Mo carbide alloy catalyst in the hydrodesulfurization of thiophene and found that its catalytic performance was better. In the deep hydrodesulfurization reaction, the stability and activity of transition metal phosphides are exceptionally high (Clark et al., 2002; Oyama et al., 2004; Phillips et al., 2002). Moreover, compared with commercial catalysts, transition metal phosphides have lower oxygen consumption and better hydrogenation selectivity. Therefore, transition metal phosphide is one of the research hotspots of new catalytic materials.
There are a variety of organic sulfur compounds in heavy oil. Because of the typical structural characteristics of thiophene and the difficulty of desulfurization, thiophene is generally used as a model molecule for research (Zhu et al., 2006). The development of industrial hydrodesulfurization (HDS) catalysts has roughly experienced transition metal sulfides, noble metals, transition metal carbides and nitrides, and transition metal phosphides. Among them, the transition metal phosphides, especially nickel phosphide (Ni2P), have excellent hydrodesulfurization activity, sulfur poisoning resistance and stability. Besides, transition metal phosphides have better hydrogenation selectivity and lower oxygen consumption than commercial catalysts, which is an important direction of hydrodesulfurization research at home and abroad (Song et al., 2012). Because the nickel phosphide crystal does not have a layered structure like ordinary sulfides, the activation edge and activation angle on the surface of the nickel phosphide crystal have a larger pore space, which can provide enough space for the reaction to promote the reaction (Hua et al., 2013). In addition, nickel phosphide has the characteristics of an approximate ellipsoidal structure, which makes it easier for nickel phosphide to form isotropic crystals and expose more active centers in the reaction process to promote the forward reaction (Nag et al., 1979). However, few studies are focused on exploring the viscosity reduction reaction mechanism by transition metal at the atomic. Therefore, the phosphide Ni2P is chosen as the catalyst in this study.
In this paper, the elementary reaction of heavy oil model molecules on catalyst surface HDS was studied theoretically at the atomic by combining the quantum chemistry method in computer simulation technology. Firstly, the MS (Materials Studio) software was used to simulate the heavy oil model molecules and the Ni2P catalytic system, then the thiophene adsorption and HDS processes on the Ni2P (001) surface were obtained. Secondly, the most likely reaction path for thiophene hydrodesulfurization on the Ni2P (001) surface was determined by calculating the structural parameters, adsorption energy and activation energy of the adsorption system. This study will more accurately get the theoretical reaction mechanism of thiophene hydrodesulfurization on Ni2P (001) surface and contribute to the further development of deep HDS.
Calculation method
In this paper, combining with the quantum chemistry method in the computer simulation technology, the Dmol3 module in the Materials Studio software package was used for the calculation. Based on the density functional theory (DFT) numerical calculation method, the thiophene adsorption and hydrodesulfurization reactions were simulated. The Generalized Gradient Approximation (GGA) and PW91 density functional method (GGA-PW91) were used to calculate the exchange correlation energy. In the calculation, the double numerical basis set plus orbital polarization function (DNP) was used to process the valence electron wave function, and the electronic structure was analyzed by Density of States (DOS). And LST/QST tools were used to search for transition states. The Density Functional Semi-Core Pseudopotential (DSPP) was used for the electrons in the core of a metal atom. While for other atoms, such as hydrogen, carbon, nitrogen, and sulfur atoms, All Electrons were used. All calculations used spin polarization. Considering the calculation efficiency and calculation accuracy comprehensively, the system accuracy was set to Medium accuracy, the track thermal occupation generally adopted the default 0.005 Hartree, and the Density Mixing was set as 0.17. The thermal occupancy value was compatible with the selection of the Density Mixing damping coefficient and they were suitable for the value calculated this time. The iterative convergence criteria for energy, gradient, displacement, and Self-Consistent Field (SCF) under Medium accuracy were 22 × 10−5 Hartree, 4 × 10−3 Hartree/Å, 5 × 10−3Å and 1 × 10−5 Hartree respectively.
Surface energy was a concept to characterize the degree of chemical bond destruction between molecules when creating material surfaces. In the theory of solid physics, atoms on the surface of a substance had more energy than atoms inside it. According to the principle of lowest energy, atoms would spontaneously move from the surface of a substance to the interior of a substance. Therefore, another definition of surface energy was the extra energy on the surface of a material relative to the inside of the material. Its definition formula was equation (1)
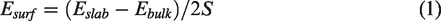
The smaller the surface energy calculated by the formula, the more stable the corresponding surface was.
Different adsorption methods corresponded to different adsorption energies. The definition of adsorption energy was shown in formula equation (2)
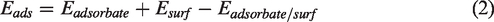
According to the above definition, a positive adsorption energy indicated stable adsorption, and the higher the adsorption energy value, the more stable the adsorption.
Simulation content
Construction and optimization of model molecules
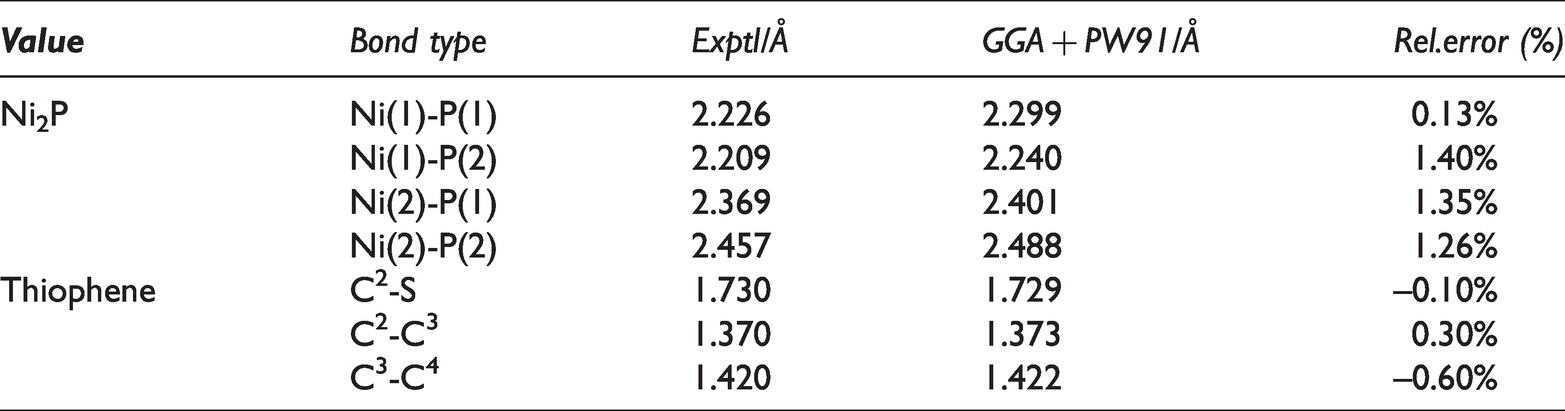
Ni2P was a metal-rich phosphide, and its crystal had a hexagonal structure with unit cell parameters were
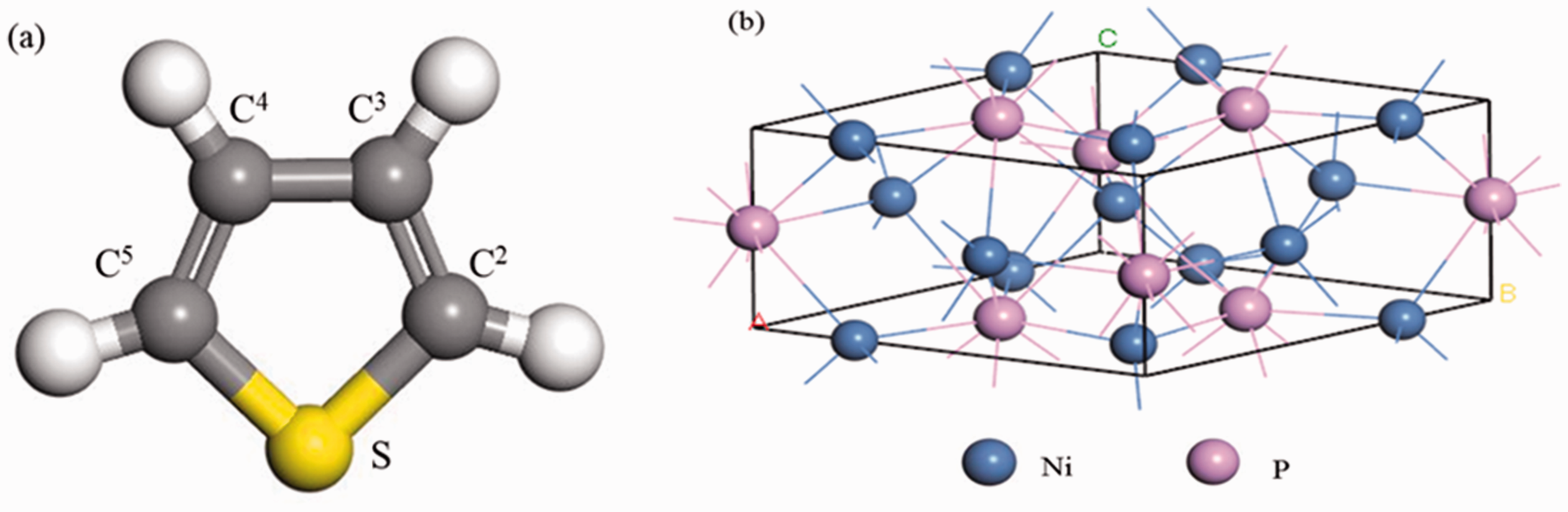
Schematic of thiophene and Ni2P.
The band lengths of Ni2P and thiophene model molecules before and after optimization.
Selection of Ni2P model adsorption surface
Ni2P crystal had a hexagonal crystal structure, its (001) crystal surface had the largest interplanar spacing and the largest surface density. The (001) crystal surface was more easily broken and exposed to the catalyst surface (Fuks et al., 2010; Nelson et al., 2006; Oyama et al., 2009), so the (001) surface was selected as the adsorption surface in this paper. Since Ni2P was a close-packed form of ABAB, there were two (001) crystal surfaces, as shown in Figure 2. The cut planes were shown in a 3 × 3 periodic model.
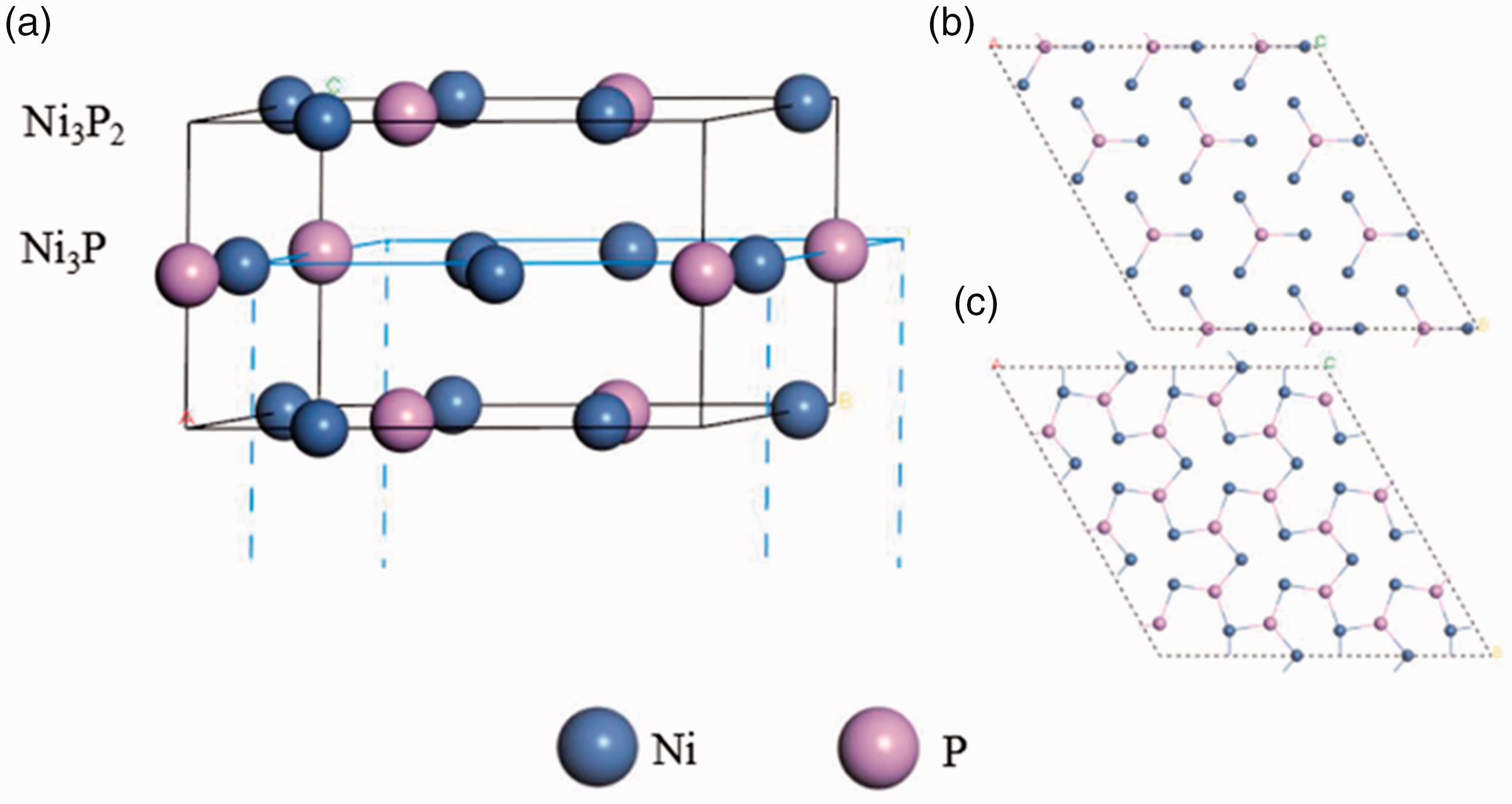
Tangential sketch of Ni2P (001) crystal surface (a), the structure is composed of Ni3P (b), and the structure is composed of Ni3P2 (b).
In this paper, a periodic 3 × 3 four-layer flat plate model was chosen to simulate two kinds of surfaces in the Ni2P (001) direction. The surface relaxation was considered in the calculation process, and the bottom atoms were fixed so that the upper three layers could move freely.
The surface relaxation optimization of two crystal surfaces Ni3P and Ni3P2 was carried out respectively to obtain the total energy Eslab of the substrate. Figures 3 and 4 were the optimized energy diagram and the convergence diagram, respectively.
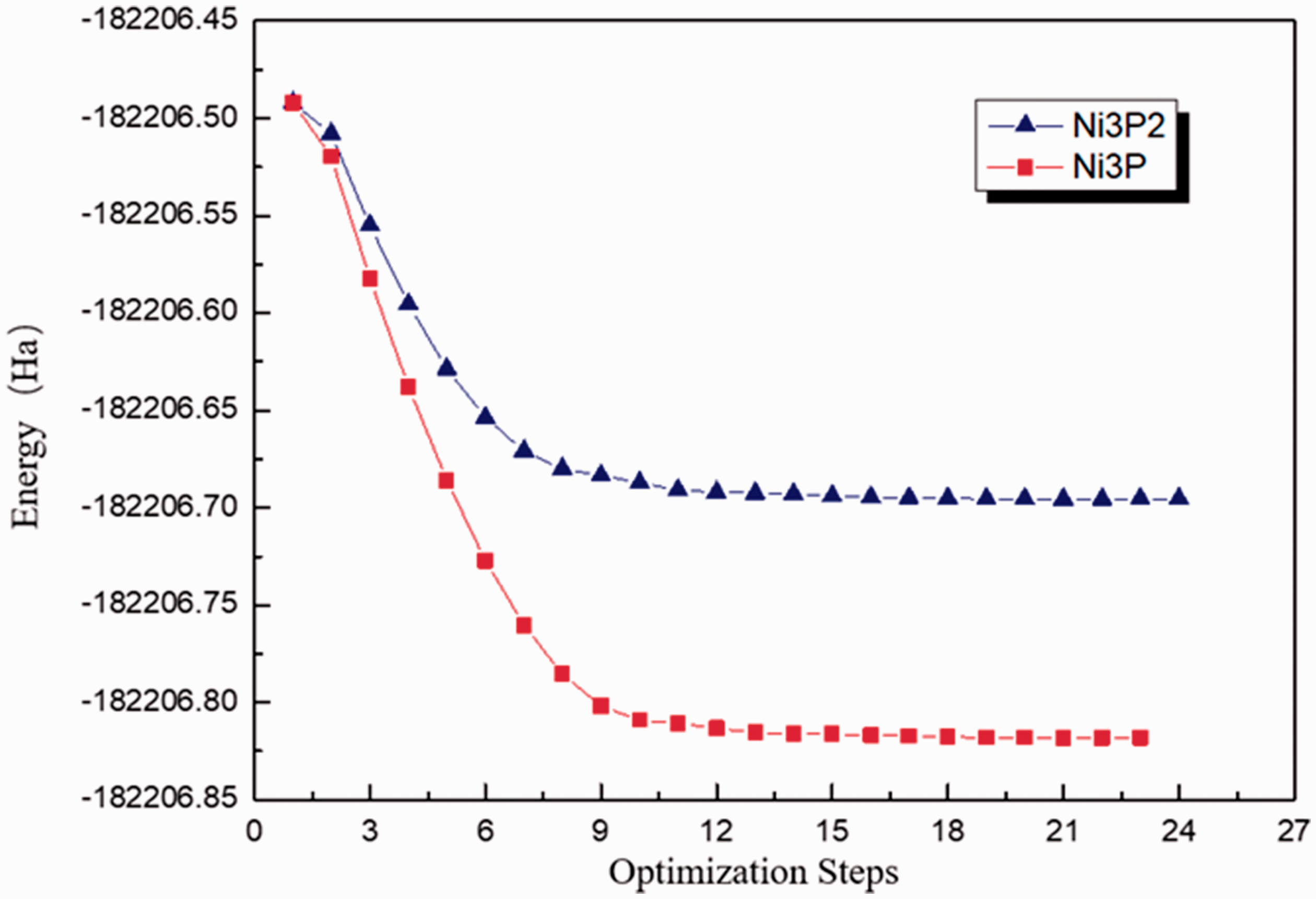
Energy diagram during optimization process of the section Ni3P2 and Ni3P.
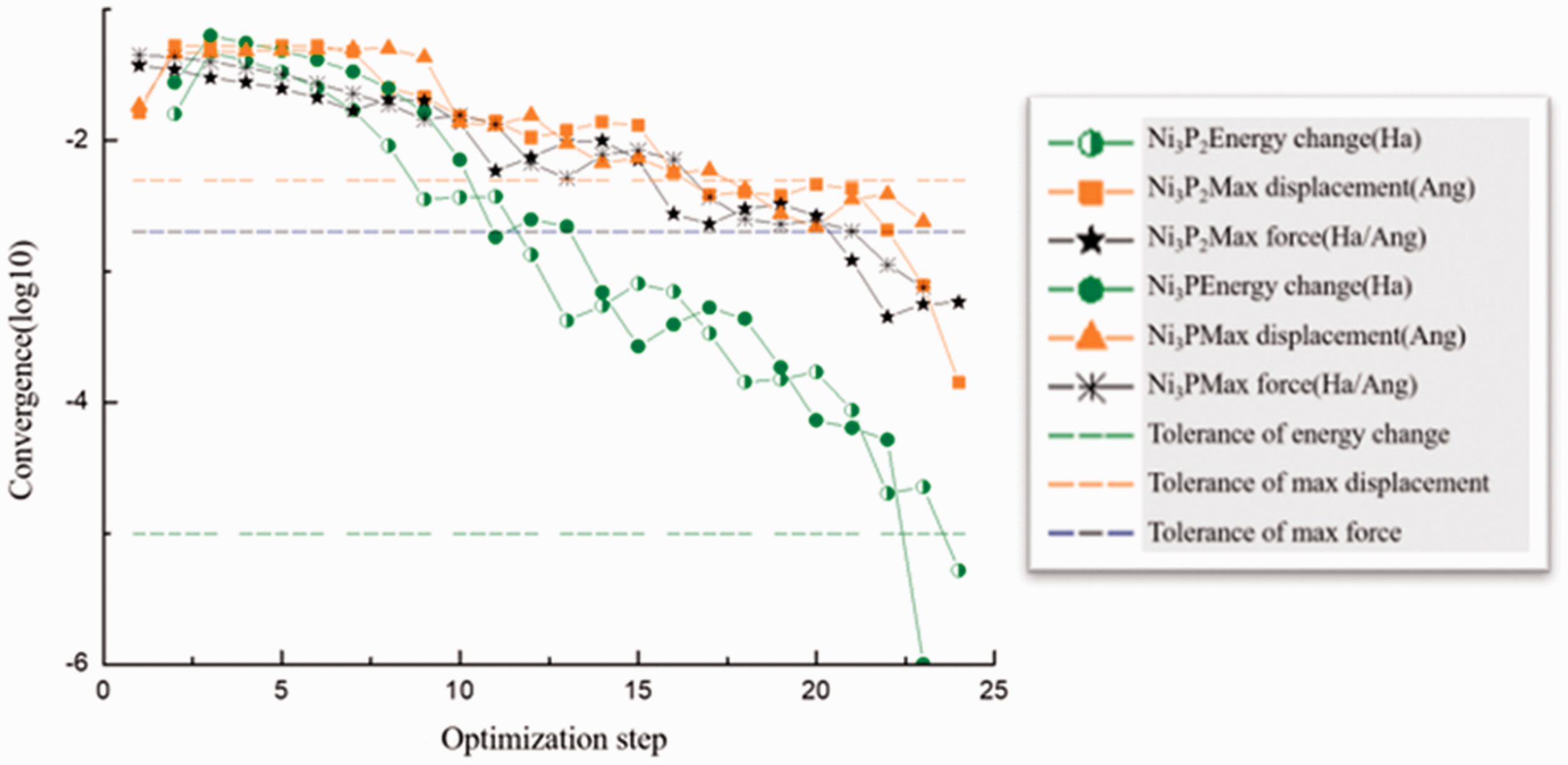
Convergence curve during optimization process of the section Ni3P2 and Ni3P.
Figure 3 showed that the energy value gradually decreased and tended to be stable during the optimization process. The horizontal line in Figure 4 represented the standard requirements of the iterative convergence. When the optimization was completed, the energy diagram and the convergence indicator diagram appeared when the convergence condition was satisfied. The surface energy of the two surfaces was calculated according to the calculation formula of the surface energy and the data obtained by optimization. The results were shown in Table 2.
Surface energy of two kinds of crystal surfaces on Ni2P(001).

As seen in Table 2, the surface energy of the Ni3P2 cut plane was about 0.0001924Ha/Å2 smaller than the surface energy of the Ni3P cut plane. The conversion factor 1Ha = 27.2114 eV was used to convert it into an electron volt eV of about 0.005 ev/A2, that is, the Ni3P2 cut plane was more stable than the Ni3P cut plane. Therefore, this paper adopted Ni3P2 to represent the crystal surface in the direction of Ni2P (001) as the research model.
Results and discussion
Adsorption of thiophene on Ni2P (001) surface
The thiophene molecule was an axisymmetric structure, and the C atoms in the thiophene ring were numbered, as shown in Figure 5. The atom labeling method of the thiophene ring in Figure 5 was also applicable to its hydrogenated derivative.
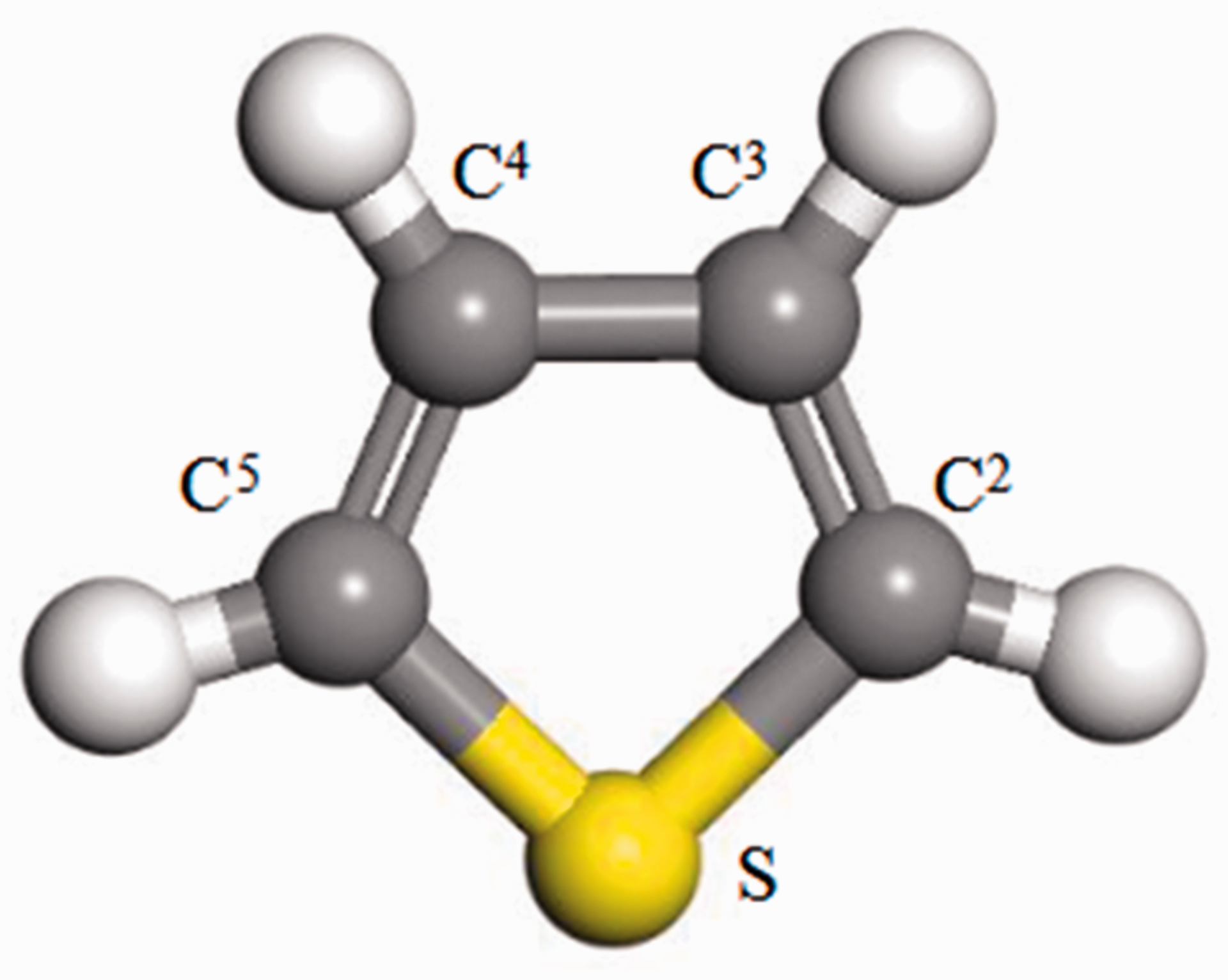
Schematic diagram of thiopene modecule. The letter X in CX represents the position of C atom in the thiopene ring.
For a certain surface, there were usually four adsorption sites for the adsorbed molecules, namely the top site, the bridge site, and the triple vacancy (hcp site and fcc site). Since Ni2P was a close-packed form of ABAB, its corresponding triple vacancy was only hcp site. There were two types of thiophene adsorption: parallel adsorption (flat mode) and vertical adsorption (end-connection mode).
An adsorption configuration was constructed on the Ni3P2 four-layer surface model. According to the relative position of the adsorption position, the adsorption of thiophene on the Ni2P (001) surface could be divided into 12 adsorption configurations as shown in Figure 6, among which (a)∼(f) belonged to flat adsorption. While (g)∼(l) molecules were perpendicular to the surface, and belonged to the end-connection adsorption. In the calculation process, each surface model was only allowed to adsorb one adsorptive thiophene molecule, so it corresponded to a lower 1/9ML surface coverage. Table 3 showed various adsorption structures, adsorption energy and some important binding structure parameters.
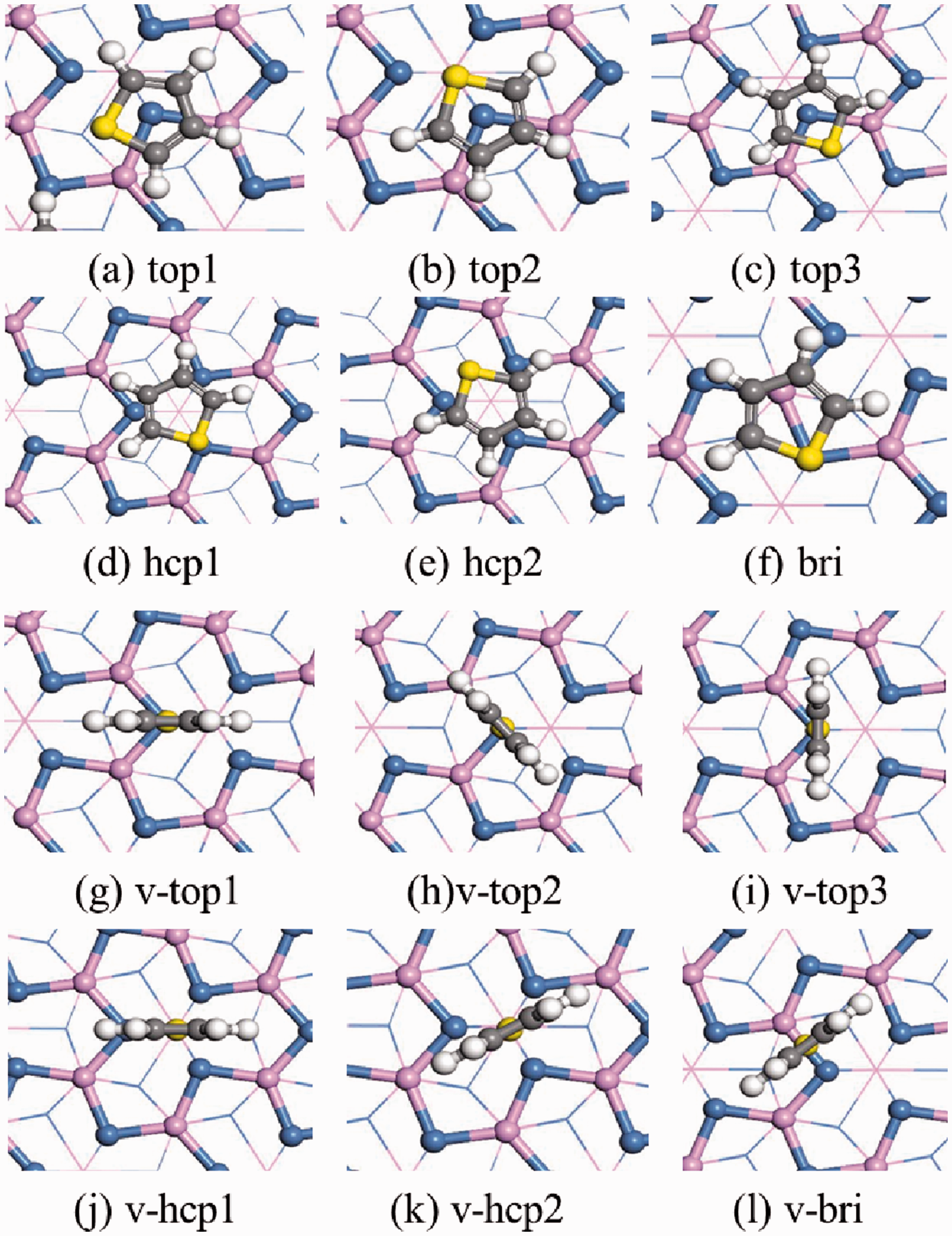
Adsorption structure of thiophene on the surface of Ni2P (001).
Adsorption sites, adsorption energies Eads (eV), and bond lengths (Å) for adsorbed thiophene derivatives on MoP(001).
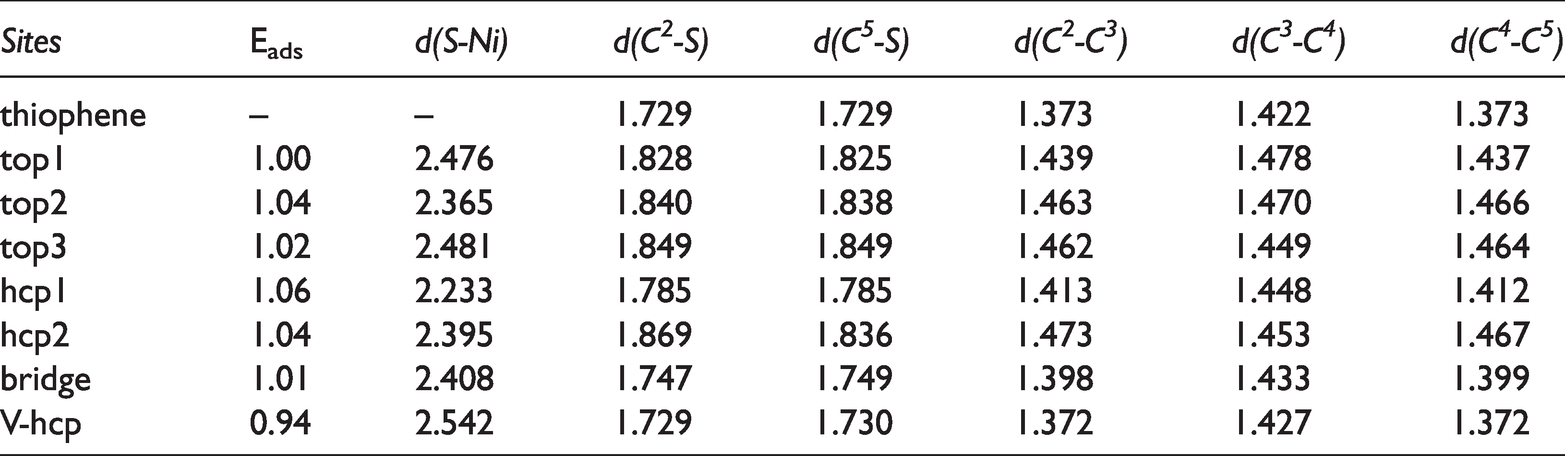
For (a)∼(f) adsorption configuration, the sulfur atoms in top1, top2, and top3 were located at the top position, the sulfur atoms in hcp1 and hcp2 were located at the triple vacancy, and the sulfur atoms in the bridge adsorption configuration were located at the bridge position. In (g)∼(l), the sulfur atoms were located at the triple vacancy in the V-hcp adsorption configuration. Compared with the end-connected mode adsorption configuration, the flat mode was more stable. Among the 12 adsorption configurations, the hcp1 configuration (Eads=1.06 eV) with the molecular ring located above the hcp position in the flat mode was the most stable, followed by the top2 and hcp2 configurations (Eads=1.04 eV). The adsorption energy of the other adsorption configuration was relatively low, and the adsorption energy of the V-hcp configuration was the lowest.
In the hydrogenation process, the most stable adsorption mode, that is, the hcp1 adsorption configuration was selected as the initial thiophene adsorption state. Hydrogen atoms were selected as the surface hydrogen source. An adsorption system was composed of thiophene adsorbed at the hcp site and H atoms adsorbed at adjacent vacancies, and used as the initial response state. The product structure formed by adding hydrogen atoms to different positions of thiophene was used as the final state of the reaction.
HDS reaction path of thiophene on Ni2P surface
Six possible thiophene hydrodesulfurization reaction pathways were summarized as shown in Figure 7 (Ni et al., 2013). Tables 4 and 5 detailed the specific reaction steps of the six thiophene hydrodesulfurization reaction paths (* indicated a certain amount of charge, and the product was named according to the position α or β where H was added).
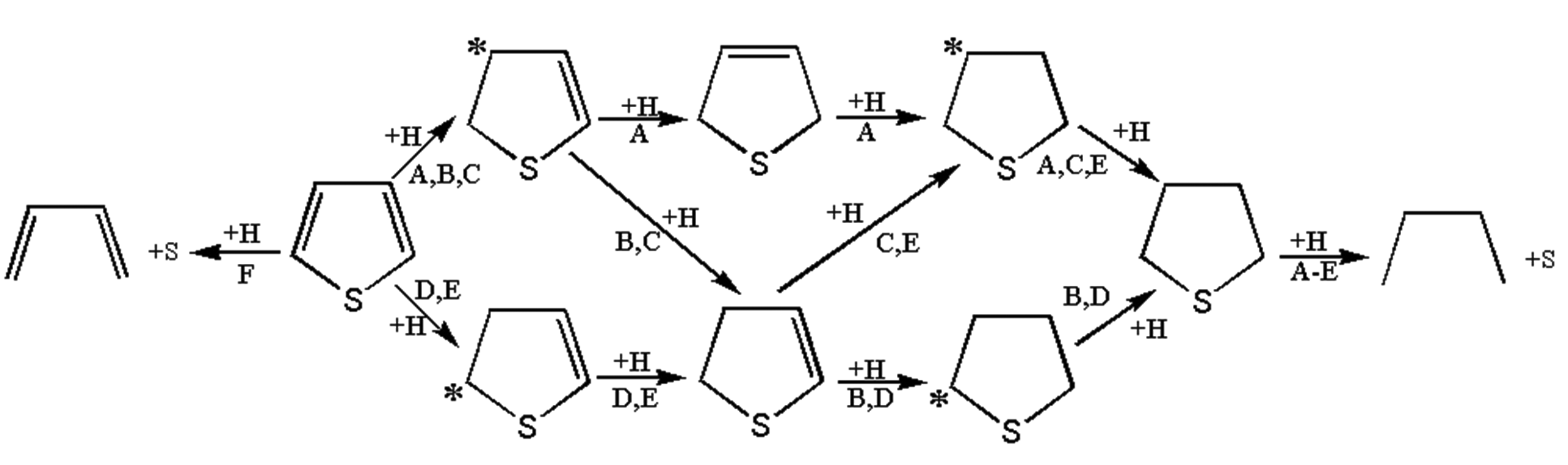
Different reaction pathways for the hydrogenation of thiophene.
Reaction mechanisms for the hydrogenation of thiophene on Ni2P (001) surface (A, B, C).
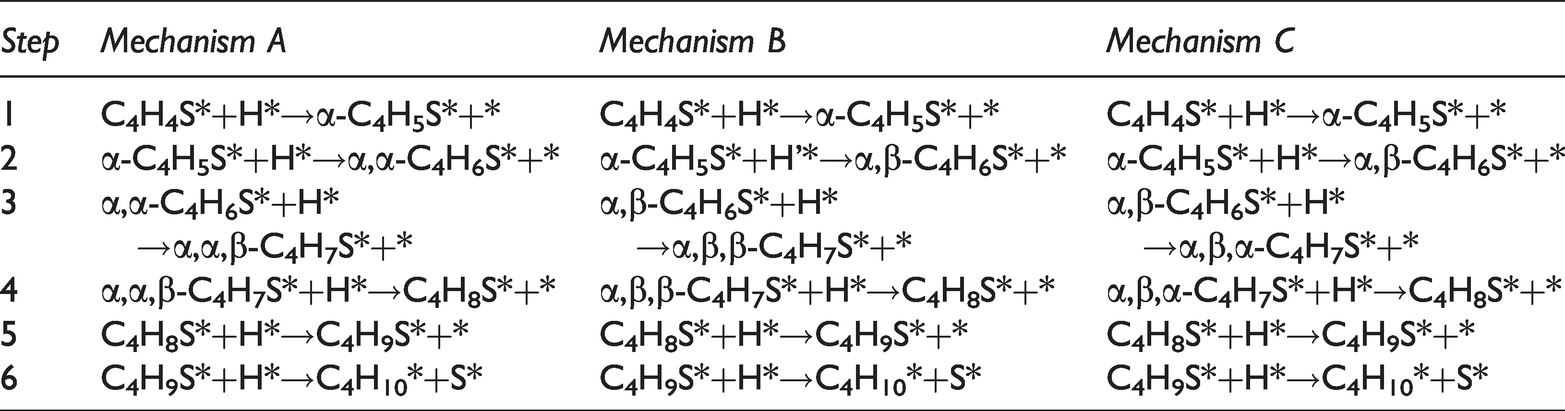
Reaction mechanisms for the hydrogenation of thiophene on Ni2P (001) surface (D, E, F).
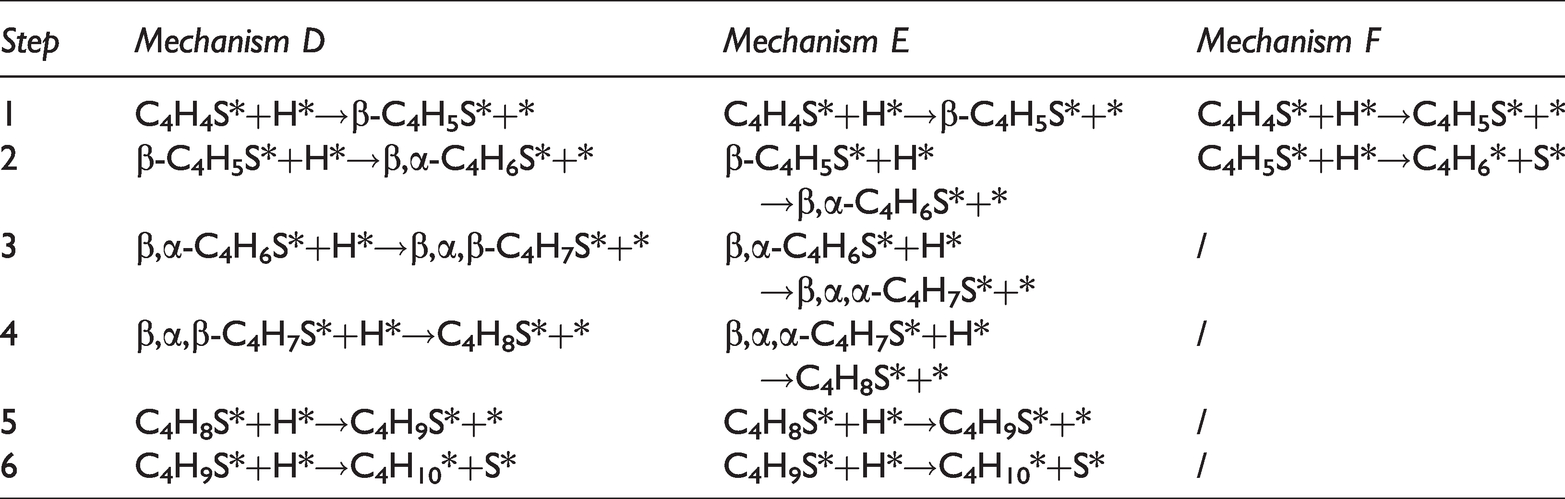
The reaction products were given different names to distinguish different reaction paths, such as α-C hydrogenation and β-C hydrogenation. According to whether the C-S bond in thiophene breaks directly, the hydrodesulfurization reaction of thiophene was divided into two types: direct hydrodesulfurization reaction and indirect hydrodesulfurization reaction. The indirect hydrodesulfurization reaction was A-E, and the direct hydrodesulfurization reaction was F.
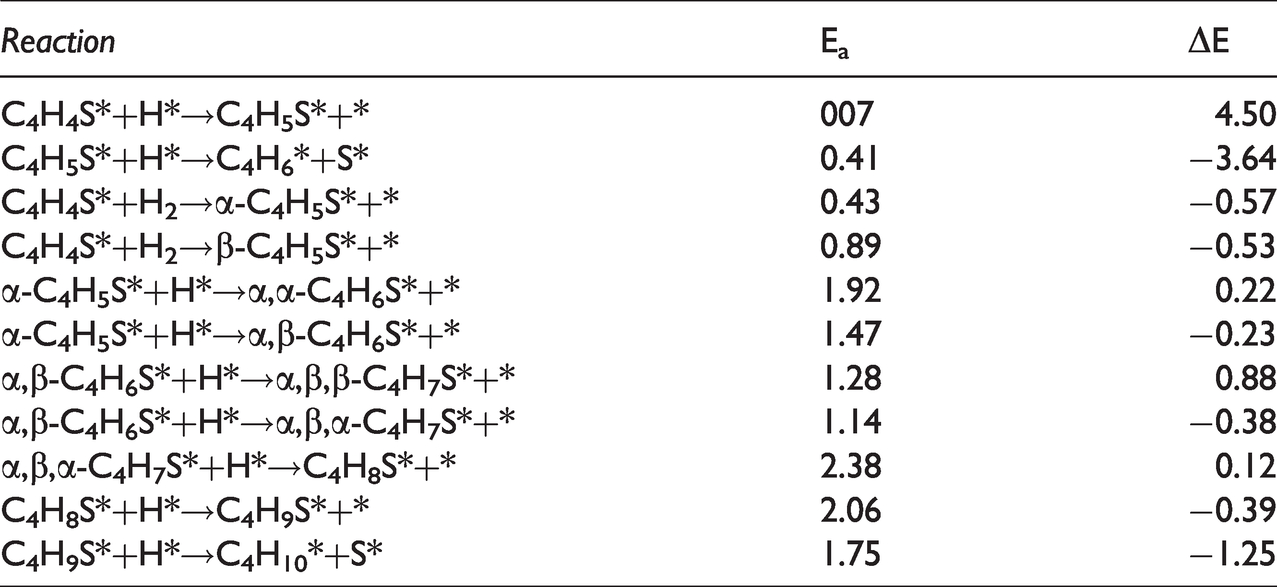
The adsorption systems consisting of catalysts, reactants and reaction products of the six reaction mechanisms in the reaction network diagram were optimized respectively. The optimized stable adsorption configurations were used as the initial state (IS) and final state (FS). Table 6 listed the activation energy Ea of each elementary reaction and the energy change ΔE before and after the reaction (Figure 8).
Activation energy (Ea, eV) and reaction energy (ΔE, eV) of each reaction on Ni2P (001) surface.
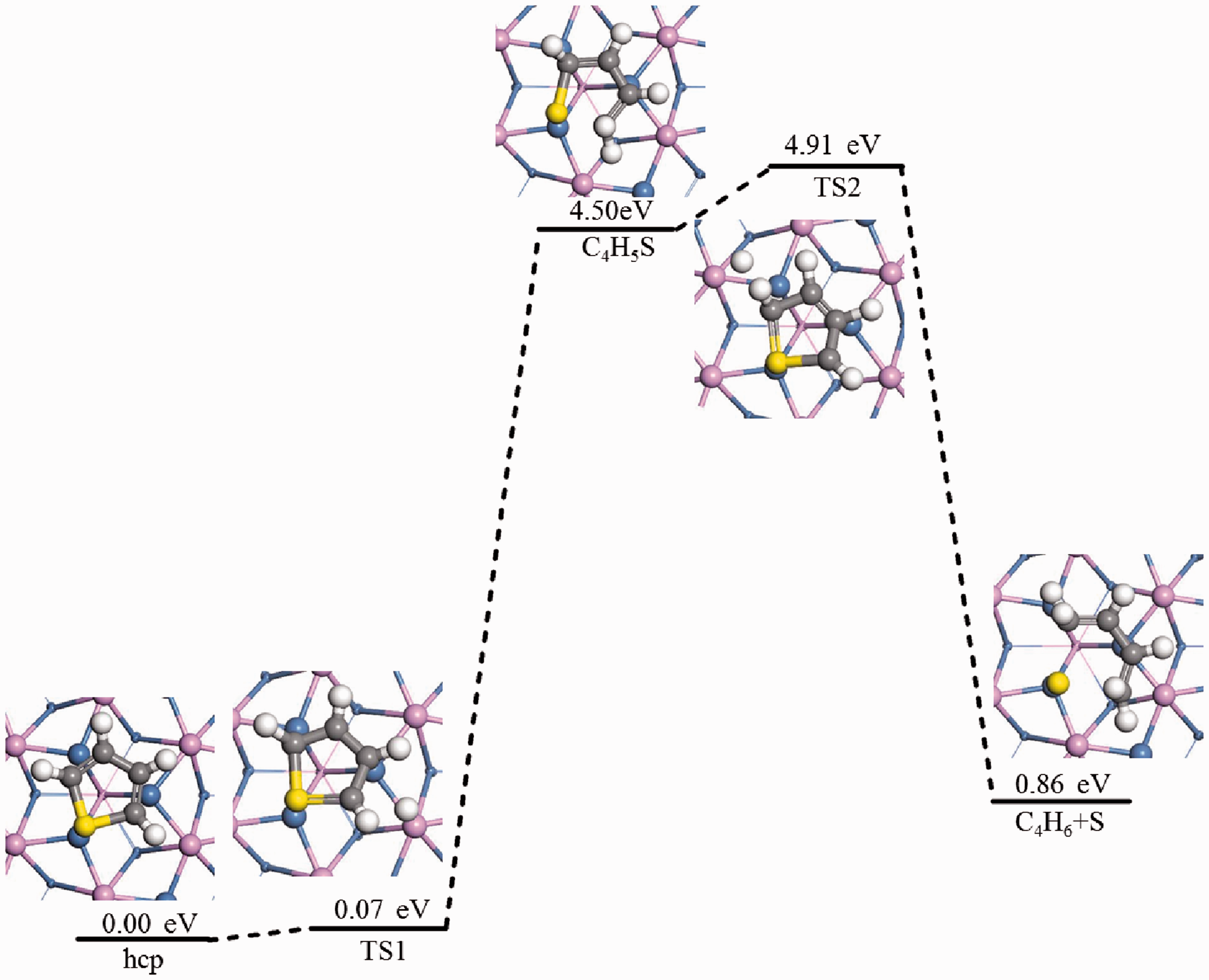
Direct hydrodesulfurization pathway of thiophene.
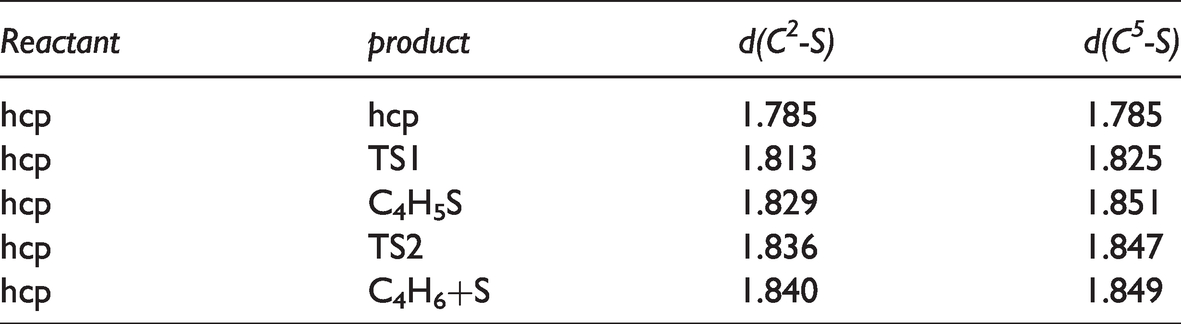
Reaction mechanism F was a direct hydrodesulfurization reaction. For the reaction C4H4S*+H2*→C4H6*+S*, two C-S bond breaks occurred directly after hydrogenation, and the products were butadiene and sulfur atoms. When the first C-S bond break occurred at C2-S, the S atom moved toward the adjacent Ni atom and formed a bond. In this transition state (TS1), the C2-S bond was broken. After TS1, the intermediate product C4H5S was formed as the final state of the reaction. In this step, as the hydrogen atom approached, the C2-S bond changed from 1.785 Å single bond to 1.813 Å longer double bond, and the C-C double bond became single bond. The final state of the reaction was further elongated relative to the initial C2-S bond. The energy barrier of the reaction in this step was 0.07 eV, and the endotherm was 4.50 eV. The second C-S bond break occurred at the C5-S bond, and the reaction product was a co-adsorption system of C4H6 intermediate and S atom. In this transition state (TS2), the activation energy barrier of the reaction was 0.41 eV and the reaction exotherm was 3.64 eV. The variation of C-S bond length in the direct hydrodesulfurization reaction was shown in Table 7.
C-S bond lengths (Å) in direct hydrodesulfurization reactions of adsorbed thiophene on Ni2P (001).
Reactions A∼E belonged to the indirect hydrodesulfurization process, in which the hydrogenation process and the broken process of the C-S bond competed with each other. The hcp configuration in the most stable flat mode was selected as the initial adsorption state of thiophene. The thiophene adsorbed at the hcp position and H adsorbed at adjacent vacancies were selected as the initial state of reaction. The product formed by adding hydrogen atoms to different positions of the thiophene was selected as the final state of the reaction. A series of hydrogenations that thiophene was hydrogenated to 2-MHT and 3-MHT at position 2 (same as position 5, collectively referred to as alpha position) and position 3 (same as position 4, collectively referred to as beta position) respectively. Then 2-MHT and 3-MHT were further hydrogenated to THT and then hydrogenated to n-butane and sulfur was studied.
For the reaction C4H4S*+H2→C4H6S*, it could be seen from Figure 7 that the main difference between the indirect hydrogenation reactions A-E was the difference of the initial hydrogenation position. Step 1 of the reaction mechanism A-C and step 1 of mechanism D and E were studied.
For the reaction C4H4S*+H2→α-C4H5S*+*, firstly, the stable adsorption configurations of C4H4S+H and α-C4H5S on the surface of Ni2P (001) were calculated, and used as the initial and final states of the reaction. For the reaction C4H4S*+H2→β-C4H5S*+*, the initial reaction model was the same as the step 1 of reaction mechanism A-C. Corresponding simulation calculation and transition state screening were performed on the initial reaction. In the transition state TS3, the H atom on the α-C in the C4H4S ring was tilted upward at a certain angle to shift the thiophene ring, and the free H atom gradually approached α-C and formed an α-C-H bond. The activation energy barrier of this reaction was 0.43 eV, and the reaction exotherm was 0.57 eV. In TS4, the thiophene ring did not shift significantly, and the H atom gradually approached β-C in C4H4S to form a β-C-H bond. The activation energy barrier of this reaction was 0.89 eV, and the reaction exotherm was 0.53 eV. Figure 9 showed the potential energy surface diagram (PES) of two reactions and the schematic diagram of the related intermediate structure.
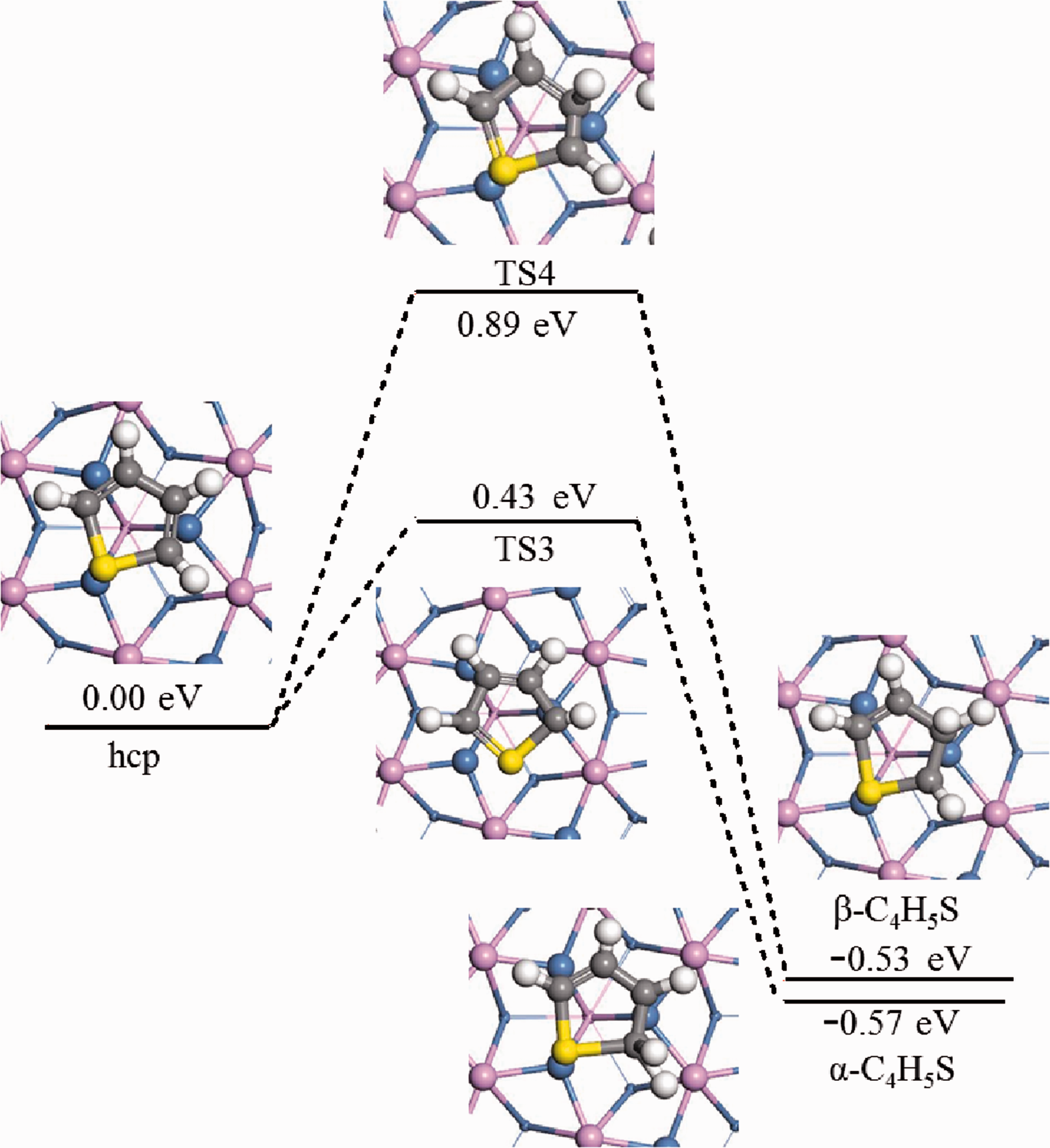
Hydrogenation pathways of thiophene on Ni2P (001) (Mechanisms A∼C(1),D,E(1)).
According to the data in Figure 9, it could be analyzed from the perspective of kinetics that the activation energy of step 1 of reaction A-C was 0.46 eV lower than that of step 1 of D and E, and H introduced in A-E step 1 was more inclined to combine with the α-C in C4H4S, which was consistent with the analysis of adsorption behavior. The C-S bond of the C4H4S ring in step 1 of reaction A-C was longer than that in step 1 of reaction D and E, which indicated that the C-S bond in step 1 of reaction A-C was more likely to break, and the reaction was more likely to progress to step 1 of reaction A-C. From the perspective of thermodynamics, the energy of the product in step 1 of A-C was 0.57 eV lower than that of the reactant, and the energy of the product in step 1 of D and E was 0.53 eV lower than the energy of the reactant. Both of these reactions were exothermic, and Lowering the temperature could promote the reaction.
α-C4H5S would have two different hydrogenation positions in the next step, resulting in two different reaction pathways. one was the hydroisomerization of α-C4H5S at α-C, and the other was hydrogenation of α-C4H5S at β-C. The two reactions formed dihydrothiophene (MHT).
For α-C4H5S*+H*→α,α-C4H6S*+* and α-C4H5S*+H*→α,β-C4H6S*+*, the dominant adsorption configurations of α-C4H5S+H and α,α-C4H6S, α-C4H5S+H and α,β-C4H6S on the Ni2P (001) surface were optimized. The configurations were used as the initial state and the final state to carry out the transition state search. The activation energy barrier of the reaction α-C4H5S*+H*→α,α-C4H6S* +* was 1.92 eV, and the heat absorption of the reaction was 0.22 eV. The activation energy barrier of the reaction α-C4H5S*+H*→α,β-C4H6S*+* was 1.47 eV, and the reaction exotherm was 0.23 eV. Figure 10 was a schematic diagram of a reaction potential energy surface map (PES) and related intermediate structures.
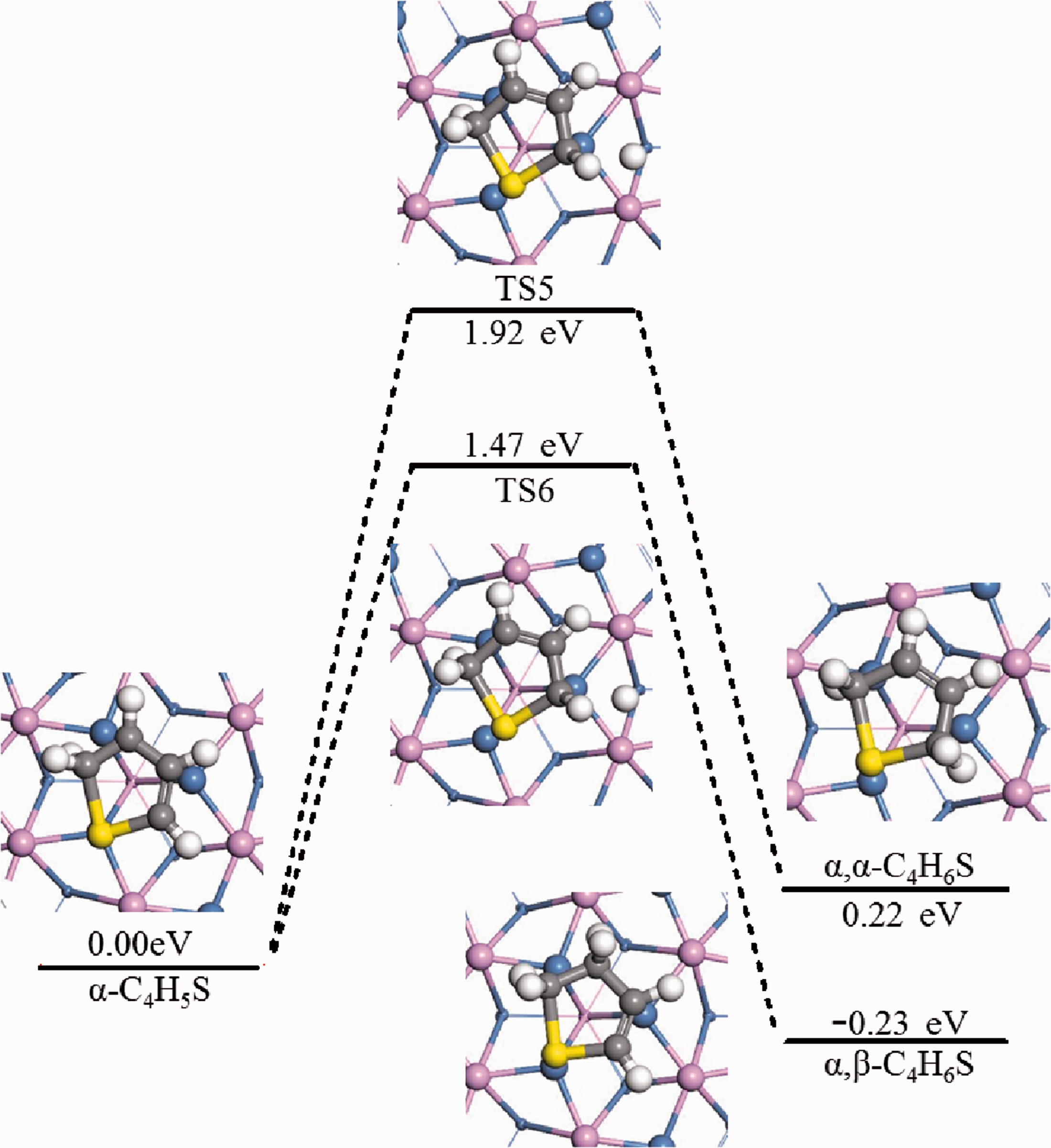
Hydrogenation pathways of thiophene on Ni2P (001)(Mechanisms A(2),B,C(2)).
As could be seen from the data, the activation energy of step 2 of reaction mechanism B and C was reduced by 0.45 eV compared with step 2 of reaction mechanism A, which indicated that the reaction was easier to proceed along reaction mechanism B and C. The energy difference before and after the step 2 of reaction A was 0.22 eV, which was an endothermic reaction; the energy difference between step 2 of reaction B and C was −0.23 eV, which was an exothermic reaction. Therefore, the second-step hydrogenation reaction of thiophene on the Ni2P (001) surface was easier to proceed along step 2 of mechanism B and C, and the product was α,β-C4H6S.
For the reaction α,β-C4H6S*+H2*→C4H7S* further hydrogenation of α,β-C4H6S might generate α,β,α-C4H7S or α,β,β-C4H7S. The IS, TS, FS and activation energy, etc. of reaction mechanism B, C step 3 were listed in Figure 11. For reactions α,β-C4H6S*+H*→α,β,β-C4H7S*+* and α,β-C4H6S*+H*→α,β,α-C4H7S*+*, the dominant adsorption configuration was calculated first, and the reaction energy barrier was shown in Figure 11. In TS7, the thiophene ring had a significant displacement, the hydrogen atoms on β-C were tilted away from the adsorption surface, and the free-state H atoms gradually approached β-C to form β-C-H bonds. After the β-C bond was hydrogenated, it further tilted up. The activation energy barrier of this reaction was 1.28 eV, and the heat absorption of the reaction was 0.88 eV. In TS8, the thiophene ring was deformed and the S-C2 bond had obvious elongation. The activation energy barrier of this reaction was 1.14 eV and the reaction exotherm was 0.37 eV.
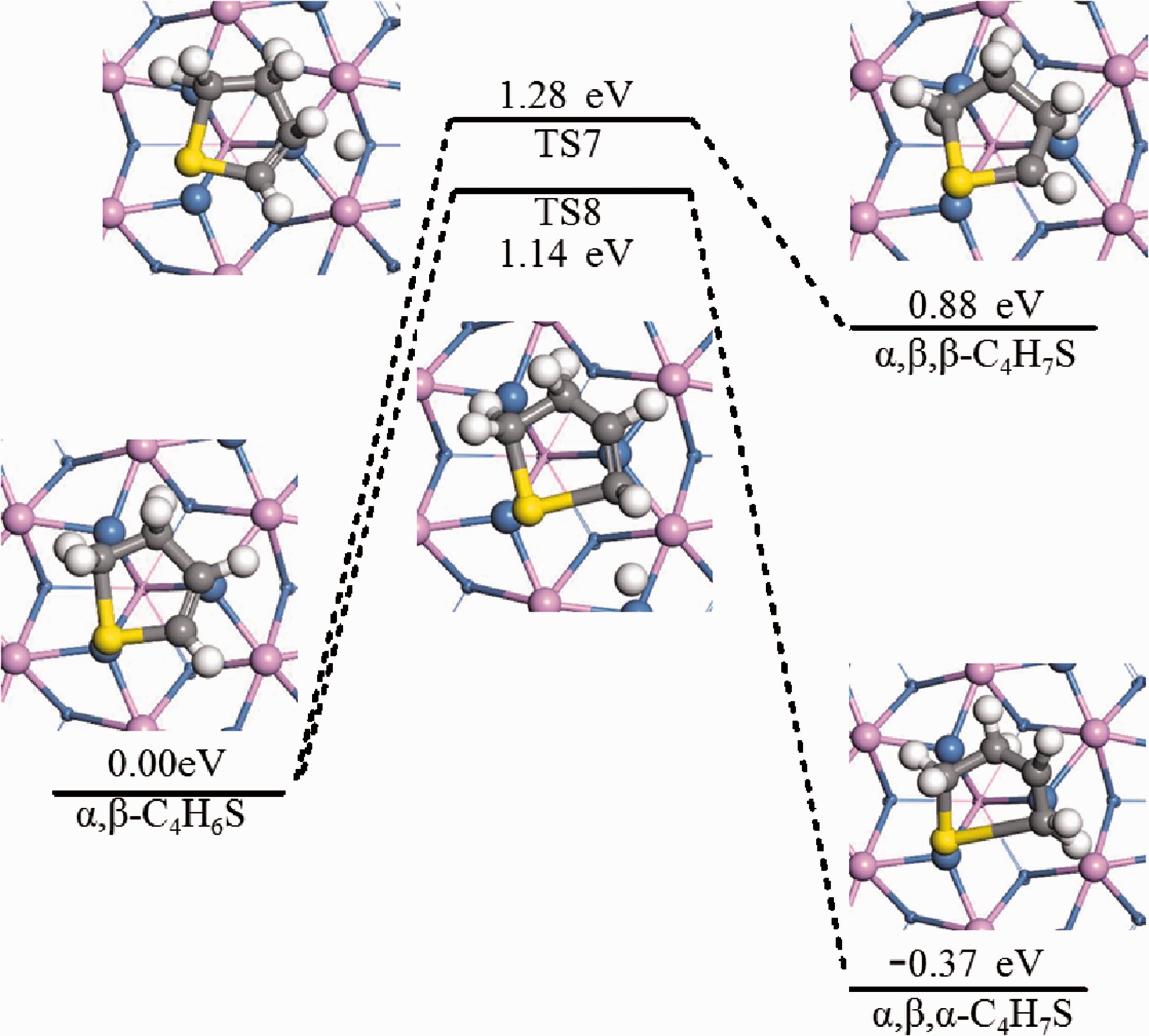
Hydrogenation pathways of thiophene on Ni2P (001) (Mechanisms B(3),C(3)).
It could be seen from Figure 11 that the activation energy of step 3 of reaction mechanism C was lower than that of step 3 of reaction mechanism B by 0.14 eV. Meanwhile, the energy difference before and after the step 3 of reaction C was −0.37 eV, and the reaction was exothermic. The energy difference before and after step 3 of reaction B was 0.88 eV, and the reaction is endothermic. Therefore, the hydrodesulfurization of thiophene on the Ni2P (001) surface was easier to perform according to step 3 of reaction mechanism C.
For the reaction α,β,α-C4H7S*+H*→C4H8S*+*, the dominant adsorption configurations of α,β,α-C4H7S+H and C4H8S on the Ni2P (001) surface were calculated. In TS9, the β-C slowly tilted upward, the steric hindrance effect of thiophene decreased, and at the same time, the free hydrogen atoms gradually approached the β-C to form the β-C-H bond. The product of this reaction was tetrahydrothiophene (THT). The activation energy barrier of the reaction was 2.38 eV, and the reaction endothermic was 0.12 eV (Figure 12).
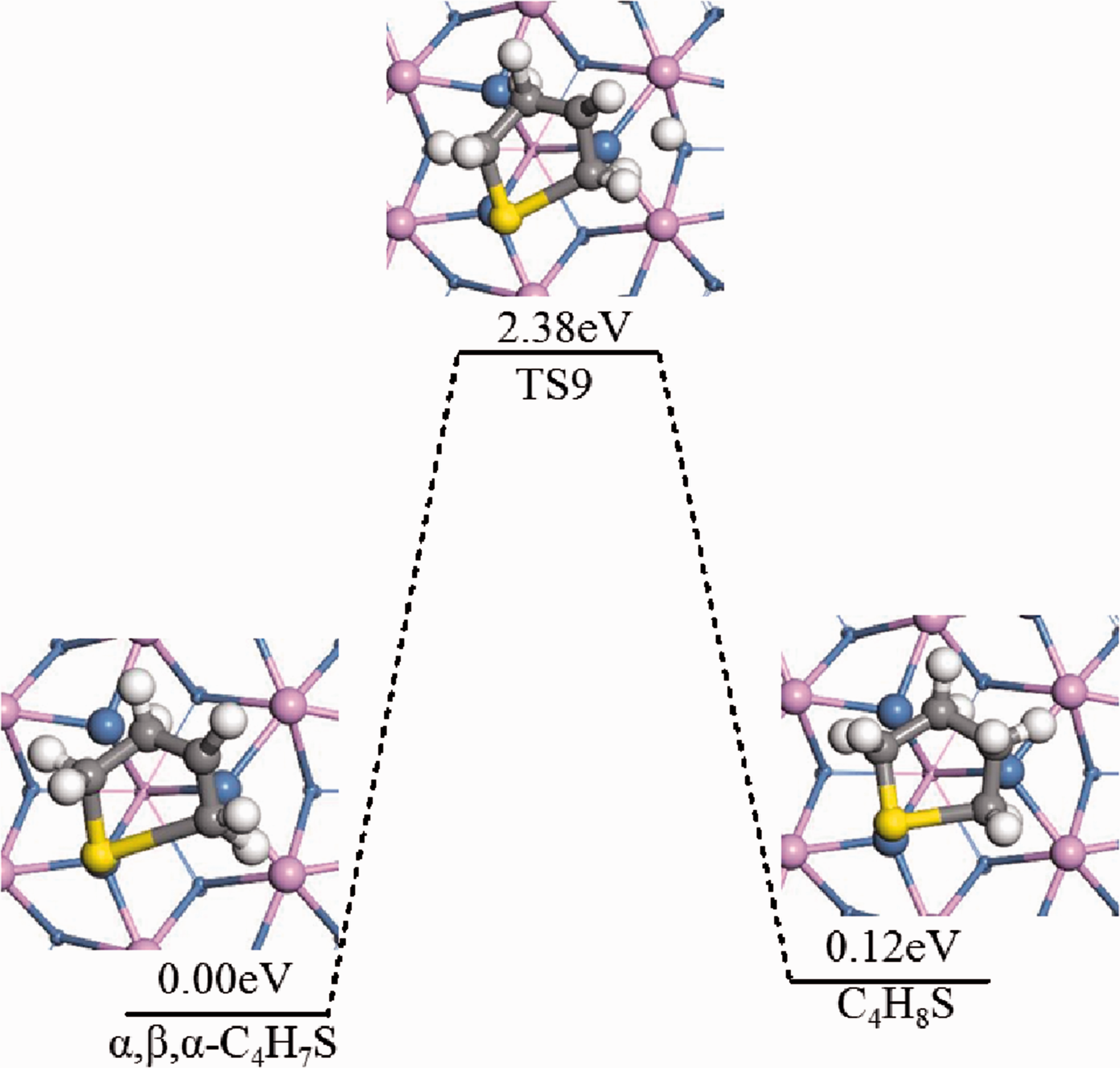
Hydrogenation pathway of thiophene on Ni2P (001) (Mechanism C(4)).
In the reaction C4H8S*+H2*→C4H10*+*, the hydrogenation reaction of tetrahydrothiophene caused the C-S bond to be broken. The initial state, final state, transition state, and reactive energy barrier of steps 5 and 6 of reaction mechanism were listed in Figure 13 respectively. For C4H8S*+H*→C4H9*+*, the structure optimization calculation of the dominant adsorption configuration was carried out, which was regarded as the initial state and the final state. In TS10, C4H8S had a slight displacement, the ring plane tilted upwards, and the free-state H atoms gradually approached α-C and formed the α-C-H bond. The activation energy barrier of this reaction was 2.06 eV and the reaction exotherm was 0.39 eV.
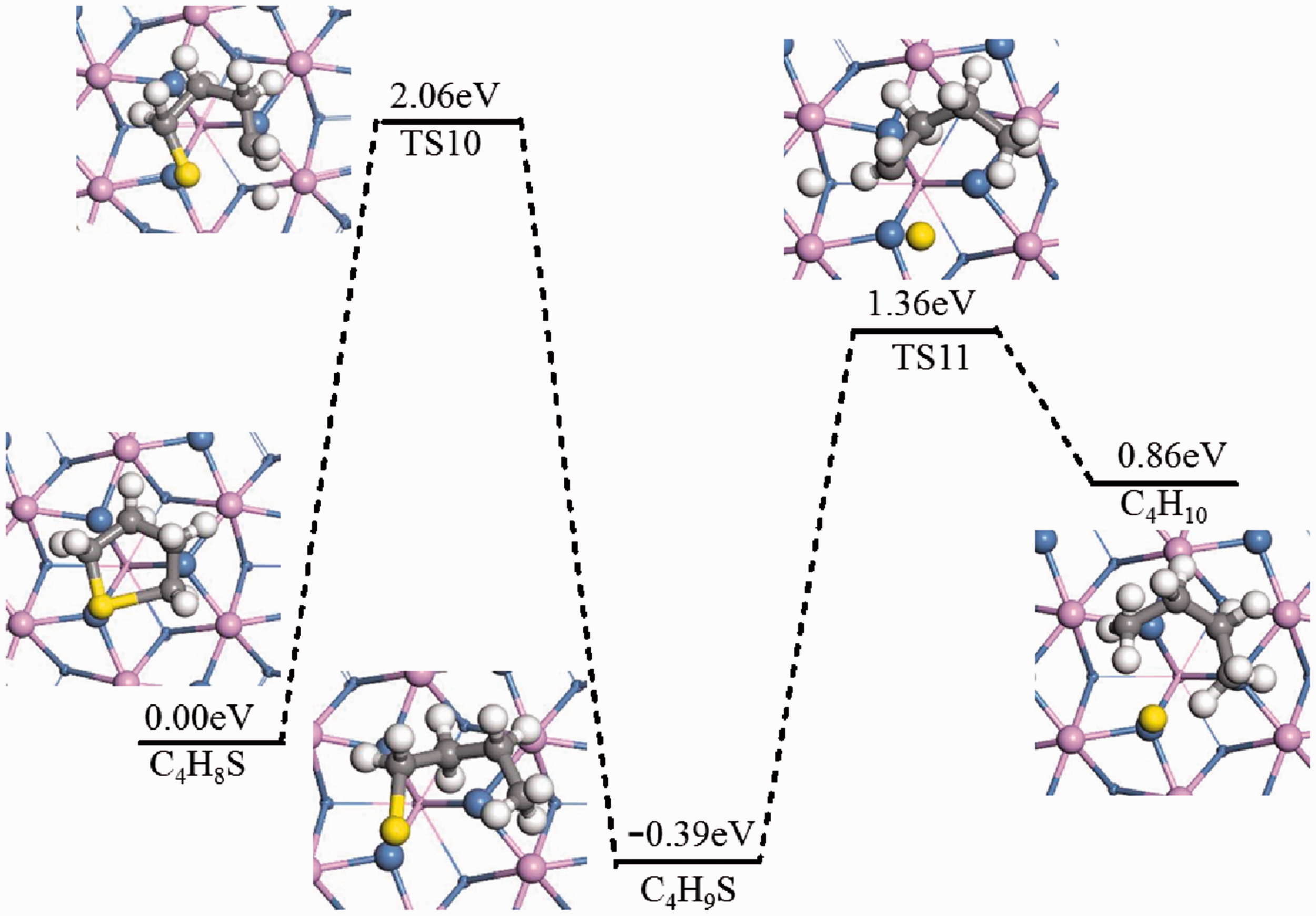
Hydrogenation pathway of thiophene on Ni2P (001) (Mechanism C(5),C(6)).
For C4H9*+H*→C4H10*+*, the structure optimization calculation of the dominant adsorption configuration was carried out, which was regarded as the initial state and the final state. In TS11, the α-C-S bond was broken to form C4H10 and S atoms, and the S atoms are adsorbed at the triple vacancy Ni atom. The activation energy barrier of this reaction was 1.75 eV, and the reaction exotherm was 0.5 eV.
In summary, the potential energy surface diagrams of the direct hydrodesulfurization and indirect hydrodesulfurization of thiophene on the Ni2P (001) surface were made as shown in Figure 14. As shown in the figure, the energy barrier of direct hydrodesulfurization of thiophene in the first step was significantly smaller than the energy barrier of indirect hydrodesulfurization of thiophene in the first step (Ea=0.07 eV<Ea=0.43 eV), therefore, the direct hydrodesulfurization of thiophene was more dominant on the surface of Ni2P (001).
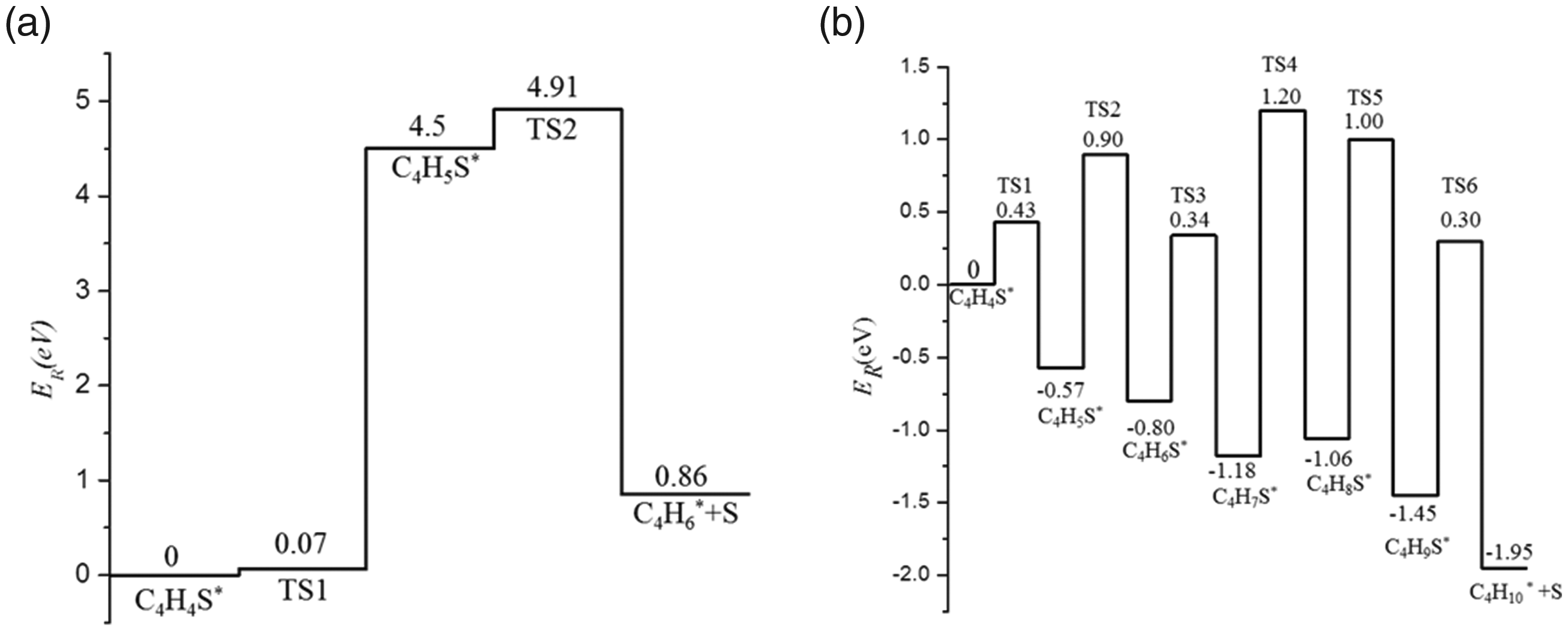
Sketch for potential relative energy of direct hydrodesulfurization and indirect hydrodesulfurization on Ni2P(001) surface.
Conclusions
When thiophene molecules were adsorbed in the triple vacancy of the Ni2P (001) surface by a flat mode, the adsorption energy was the largest and the adsorption configuration was the most stable. The direct hydrodesulfurization of thiophene had more advantages than the indirect hydrodesulfurization of thiophene. The direct hydrodesulfurization reaction was an endothermic reaction, and the increasing temperature could promote the reaction. The indirect hydrodesulfurization was an exothermic reaction, and cooling down could promote the reaction. In the indirect hydrodesulfurization reaction, the step 4 of reaction mechanism C (TS4) required the highest activation energy, which was the rate-limiting step of the entire reaction. Direct hydrodesulfurization reaction step was C4H4S+H2→C4H6.
Footnotes
Acknowledgements
The authors would like to acknowledge the financial support provided by the National Science and Technology Major Project of the Ministry of Science and Technology of China (2016ZX05012-002–005), National Natural Science Foundation of China (No. 51874333) and Natural Science Foundation of Shandong Province, China (No. ZR2017MEE030).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.