
Abstract
Recently, we reported a new chiral stationary phase prepared using β-cyclodextrin functionalized with aromatic ionic liquid which is aimed to enhance the performance of enantioseparation of flavonoids and β-blockers. In this paper, the characteristics and performance of previously prepared chiral stationary phase denoted as β-CD-BIMOTs were compared with the newly synthesized chiral stationary phase denoted as β-CD-DIMOTs. β-CD-DIMOTs were prepared by functionalization of β-cyclodextrin with aliphatic ionic liquid. The obtained β-CD-BIMOTs and β-CD-DIMOTs stationary phases were compared with native β-CD stationary phase for the enantioseparation of non-steroidal anti-inflammatory drugs (NSAIDs) (ibuprofen, indoprofen, ketoprofen and fenoprofen). The β-CD-BIMOTs stationary phase showed greater chiral resolution capabilities rather than β-CD-DIMOTs and native β-CD stationary phases. Further, in order to understand the interaction of enantioseparation, the inclusion complex formation between NSAIDs and β-CD-BIMOTs was studied using 1H NMR, NOESY and UV/Vis. The enantioseparated NSAIDs were found to form multiple interactions with β-CD-BIMOTs-CSP.
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are drugs that have been used to provide analgesic, antipyretic and anti-inflammatory effects (Ye et al., 2010). Profen (2-arylpropionic acids) is an important group of NSAIDs, characterized by a chiral carbon atom next to the carboxylic acid group (Ye et al., 2010). This chiral structure of NSAIDs exhibits optical activity and causes the different biological properties of enantiomers (Ye et al., 2010). For example, for ibuprofen, the pharmacological activity resides in the S-enantiomer only (Núñez-Agüero et al., 2006). Consequently, the enantioseparation of NSAIDs is an important concern for pharmaceutical use.
High-performance liquid chromatography (HPLC) has been proven to be one of the most widespread techniques for the enantiomeric separation and analysis (Muderawan et al., 2006; Zhang et al., 2008). In this study, the enantioseparation of selected NSAIDs was performed using HPLC with β-cyclodextrin (β-CD)-based chiral stationary phase (CSP). β-CD is a doughnut-shaped cyclic oligosaccharides containing seven α-(1,4)-glycosidic linkages. β-CD has been used extensively as CSPs in HPLC because of its ability to recognize enantiomeric molecules through the formation of inclusion complexes (Xiao et al., 2012; Zhong et al., 2006) and its C7 symmetry axis. Fourteen hydroxyl groups located at the mouth of the cavity provide a number of potential interactions with enantiomers during the enantioseparation. Until 1990, most of the enantioseparation studies focused on the preparation of native β-CD-based CSPs modified by different linkage groups (Zhou et al., 2010). However, the application of native β-CD-CSPs was not always satisfactory (Zhou et al., 2010). Therefore, recently, researches have been focused on the preparation of modified β-CD to be used as CSPs (Xiao et al., 2012).
The addition of different substituent groups onto the rim of β-CD provides multiple interactions such as π–π, dipole–dipole interaction, electrostatic interaction, and hydrogen bonding which contributes significantly to effective enantioseparation. Ionic Liquids (ILs) are examples of new substituent groups that are been used to modify β-CD (Li and Zhou, 2014; Li et al., 2011). ILs is composed of organic cation and inorganic or organic anion (Wasserscheid and Keim, 2000). It is widely used in environmentally benign chemical processing and analysis (Pandey, 2006). ILs molecules consist of high charge region and low charge region (Canongia Lopes and Pádua, 2006) which contributes to enantioseparation through electrostatic and dispersive interaction (Anderson and Armstrong, 2003).
In our previous work, we introduced the preparation, characterization and chromatographic performance of β-CD-BIMOTs-CSP (Figure 1) (Rahim et al., 2016a, 2016b). It was observed that compared with native β-CD-CSP, β-CD-BIMOTs-CSP possessed excellent chiral recognition abilities towards the selected β-blockers and flavonoids. Herein, we demonstrate another β-CD functionalized IL denoted as β-CD-DIMOTs-CSP (Figure 1) to investigate the effect of the alkyl chain of imidazolium cation of IL on enantioseparation abilities. The characterization of β-CD-BIMOTs-CSP and β-CD-DIMOTs-CSP was compared with native β-CD-CSP using FTIR and thermal analysis. Additionally, the chromatographic performance of β-CD-BIMOTs-CSP, β-CD-DIMOTs-CSP and native β-CD-CSP was compared for the enantioseparation of NSAIDs (Figure 2). To the best of our knowledge, most studies on the interactions of enantioseparation using β-CD functionalized IL-based CSP are often elaborated through computational study (Li and Zhou, 2014; Li et al., 2011). However, none of those studies provided data via experimental data. As a solution to this problem, this article evaluates the inclusion complex formation between NSAIDs and β-CD functionalized IL CSP in enantioseparation using spectroscopic techniques (1H NMR, NOESY and UV/Vis).
The structure of β-CD-DIMOTs-CSP, β-CD-BIMOTs-CSP and native β-CD-CSP. The structure of selected NSAIDs.

Experimental
Materials
β-CD was purchased from Acros (Belgium) (99%). 1-benzylimidazole (1-BzlIm), 1-decyl-2-methylimidazole (C10MIm), 2,4-toluene diisocyanate (TDI) and NSAIDs were purchased from Aldrich (USA). Solvent used for HPLC and synthesis are LC and anhydrous grade solvents, respectively, purchased from Merck (Germany). Kromasil spherical silica gel with a mean pore size 100 Å and particle size of 5 µm was purchased from Merck (Germany). The stainless steel HPLC empty columns (250 mm × 4.6 mm) were purchased from Grace (USA).
Instruments
FT-IR spectra were performed on a Perkin–Elmer RX1 FT-IR (Perkin Elmer, Waltham, MA, USA) using KBr pellets. Thermogravimetric (TGA) analyses curves were examined using a TA Instrument Q500 (Perkin Elmer, Waltham, MA, USA). An elemental analysis of the sample was determined with a Leco Truspec CHN Analyzer (Saint Joseph, MI). 1H NMR, 13C NMR and NOESY spectra were recorded using an Avance spectrometer at 600 MHz (Bruker, Fällanden, Switzerland). Absorption spectra measurements were carried out with a Shimadzu UV 1800 (Shimadzu, Japan) spectrophotometer in the range of 190 to 800 nm. The employed HPLC system comprised an LC-20AT pump, an SPD-M20 detector, an SIL-20AHT auto sampler, a CTO-20AC column oven and CBM-20A communication bus module (Shimadzu, Japan).
Synthesis of CSPs
The β-CD-BIMOTs-CSP was synthesized according to the procedure reported previously (Rahim et al., 2016a, 2016b). Meanwhile, the preparation of β-CD-DIMOTs-CSP involved the following four steps. (i) preparation of p-toluene sulfonic anhydride (Ts2O), (ii) preparation of 6-O-Monotosyl-6-deoxy-β-CD (β-CDOTs), (iii) synthesis of Mono-6-deoxy-6-(3-decyl-2-methylimidazolium tosylate)-β-CD (β-CD-DIMOTs) and (iv) immobilization of β-CD-DIMOTs onto modified silica. The synthesis pathway of β-CD-DIMOTs-CSP is illustrated in Figure 3.
Synthesis pathway of β-CD-DIMOTs-CSP.
Ts2O was prepared according to our previous publications (Rahim et al., 2016a, 2016b), primarily by dissolving p-toluene sulfonyl chloride (2.00 g, 10.4 mmol) in dichloromethane (DCM) (12.5 mL). Then, p-toluene sulfonic acid (0.52 g, 2.63 mmol) was added gradually with vigorous stirring under nitrogen atmosphere. The resulting mixture was stirred overnight at room temperature. The mixture was then filtered to remove the unreacted p-toluene sulfonic acid. Hexane (50 ml) was added to the filtrate and a precipitate was obtained after drying overnight under reduced pressure. β-CDOTs was also prepared according to our previously reported method (Rahim et al., 2016a, 2016b). C10MIm (10 mol equivalent) was then added dropwise to a stirred solution of dry β-CDOTs (1.00 g, 0.78 mmol) in anhydrous DMF (40 ml) to prepare β-CD-DIMOTs. Stirring was continued at 90℃ under nitrogen atmosphere for a further 48 h. After cooling to room temperature, acetone was added to precipitate the product. Thereafter, the mixture was then stirred for 30 min and the product was filtered and washed with excess amount of acetone. A white yellow precipitate was obtained as the final product. The structure of β-CD-DIMOTs is shown in Figure 4.
The structure of β-CD-DIMOTs.
The immobilization of β-CD-DIMOTs onto silica was first prepared by modifying silica gel as reported by Yatabe and Kageyama (1994). The silica gel was reacted with 2, 4-toluene diisocyanate (TDI) in dry hexane for 4 h at room temperature to obtain Si-TDI. Upon completion of the reaction, the product was filtered, rinsed thoroughly by hexane and dried under reduced pressure. The immobilization of β-CD-DIMOTs onto Si-TDI was then carried out by stirring Si-TDI (5 g) in anhydrous hexane (200 ml) under nitrogen atmosphere. After 30 min, a solution of β-CD-DIMOTs (1.8 g) in anhydrous hexane was added. Stirring was continued for 24 h. The synthesized solid was filtered and washed with toluene, acetone and distilled water to obtain a purified product. The product was characterized using elemental analysis, FT-IR and TGA. The aforementioned procedure was also applied to immobilize the native β-CD onto the Si-TDI.
FT-IR/KBr, cm–1: 3297 (OH), 2922 (C–H), 1652 (C=C), 1152 (C–N). 1H NMR, DMSO-d6: Hl (7.68, s), Hk (7.61, s), Hb-Hj (1.23-1.28, t), Ha (0.85, t), H8 (7.46, d), H9 (7.11, d), OH-2–OH-3 (5.64–5.79, m), H1 (4,83, s), OH-6 (4.44–4.54, m), H6* (3.91), H3, H5, H6 (3.54–3.63), H2–H4 (3.20–3.34, m), H11 (2.28, s). 13C NMR, DMSO-d6: Ca (16.13), Cb (19.79), Cc (28.62), Cd (22.48), Cg (22.48), Ch (21.38), Ci(22.48), Cj (31.37), Ck (126.42), Cl (128.75), Cm (14.40), Cn (129.84), C9 (128.17), C8 (126.06), C1 (102.38), C4 (81.95), C2 (73.49), C3 (72.43), C5 (70.74), C6 (60.36), C6* (45.66). CHNS (%): C (40.45), H (6.35), N (1.71), S (1.61).
Column packing approach
The CSPs (2.5 g) were suspended in approximately 15 ml HPLC-grade hexane and poured into a stainless steel column (250 mm × 4.6 mm). Thereafter, the CSPs were packed under 35 MPa with hexane for about 24 h.
HPLC analysis instrumentation and conditions
The newly packed column was flushed with 100% hexane at a flow rate of 0.2 ml/min for 24 h. The flow rate was increased to 0.5 ml/min to obtain a stable baseline. The NSAIDs were prepared at a concentration of 500 mg/l in MeOH. The injection volume was 20 μl. The flow rate was fixed at 0.5 ml/min. The reversed separation mode of mobile phase consisted of ACN/water and MeOH/water, whereas, polar organic consisted of various volume fraction mixture of ACN and MeOH.
Calculations of chromatographic data
The retention factor



The dead time
Preparation of β-CD-BIMOTs/NSAIDs complexes
The complex of β-CD-BIMOTs with NSAIDs was prepared using the conventional kneading method (Cwiertnia et al., 1999; Daruházi et al., 2008). β-CD-BIMOTs and NSAIDs (with the ratio of 1:1) were triturated with mortar and pestle in small amount of ethanol to form homogenous paste. The slurry was kneaded for 30 min and dried to a constant mass. The final product was characterized using 1H NMR and NOESY. The prepared samples were dissolved in DMSO-d6. A 700 μl of the resulting solution was introduced into standard 5 mm NMR tubes, and the spectra of 1H NMR and NOESY were recorded at 27℃. For NOESY experiments, the spectra were recorded with a mixing time of 700 ms with 256 increments and 40 scans.
Determination of the absorption spectra of β-CD-BIMOTs/NSAIDs complexes
A 2.0 mL of 0.01 mM NSAIDs aliquot and 3.2 ml of 0.0032 M β-CD-BIMOTs solution was transferred accurately into a 10.0 ml standard volumetric flask and diluted to the mark with ultra-pure water. The absorption spectra of β-CD-BIMOTs/NSAIDs complexes were recorded against a blank reagent which was prepared with the same reagent concentration but without the addition of NSAIDs. In addition, absorption spectra of NSAIDs and β-CD-BIMOTs were also recorded. All the absorbance was measured at 200–800 nm separately against blank reagent.
Result and discussion
FTIR characterization
The spectra of β-CD, β-CD-BIMOTs and β-CD-DIMOTs are shown in Figure 5. The broad O-H stretching band around 3200–3300 cm−1 for β-CD, β-CD-BIMOTs and β-CD-DIMOTs is corresponded to the OH group in the β-CD molecules. The intense band at 1657 cm−1 in IR spectra of β-CD-BIMOTs was attributed to C = C of the aromatic ring of 1-BzlIm moieties (Figure 5(b)). The weak bands known as overtones at 1665–2000 cm−1 were also attributed to the aromatic ring (Socrates, 2004) of 1-BzlIm moieties. The C-H band occurred at ∼2900 cm−1 of β-CD-BIMOTs and β-CD-DIMOTs spectra (Figure 5(b) and (c)) were found to be more intense than that of β-CD (Figure 5(a)). These results indicate that β-CD was successfully functionalized with 1-BzlIm and C10MIm.
FT-IR spectra of (a) β-CD (b) β-CD-BIMOTs (c) β-CD-DIMOTs.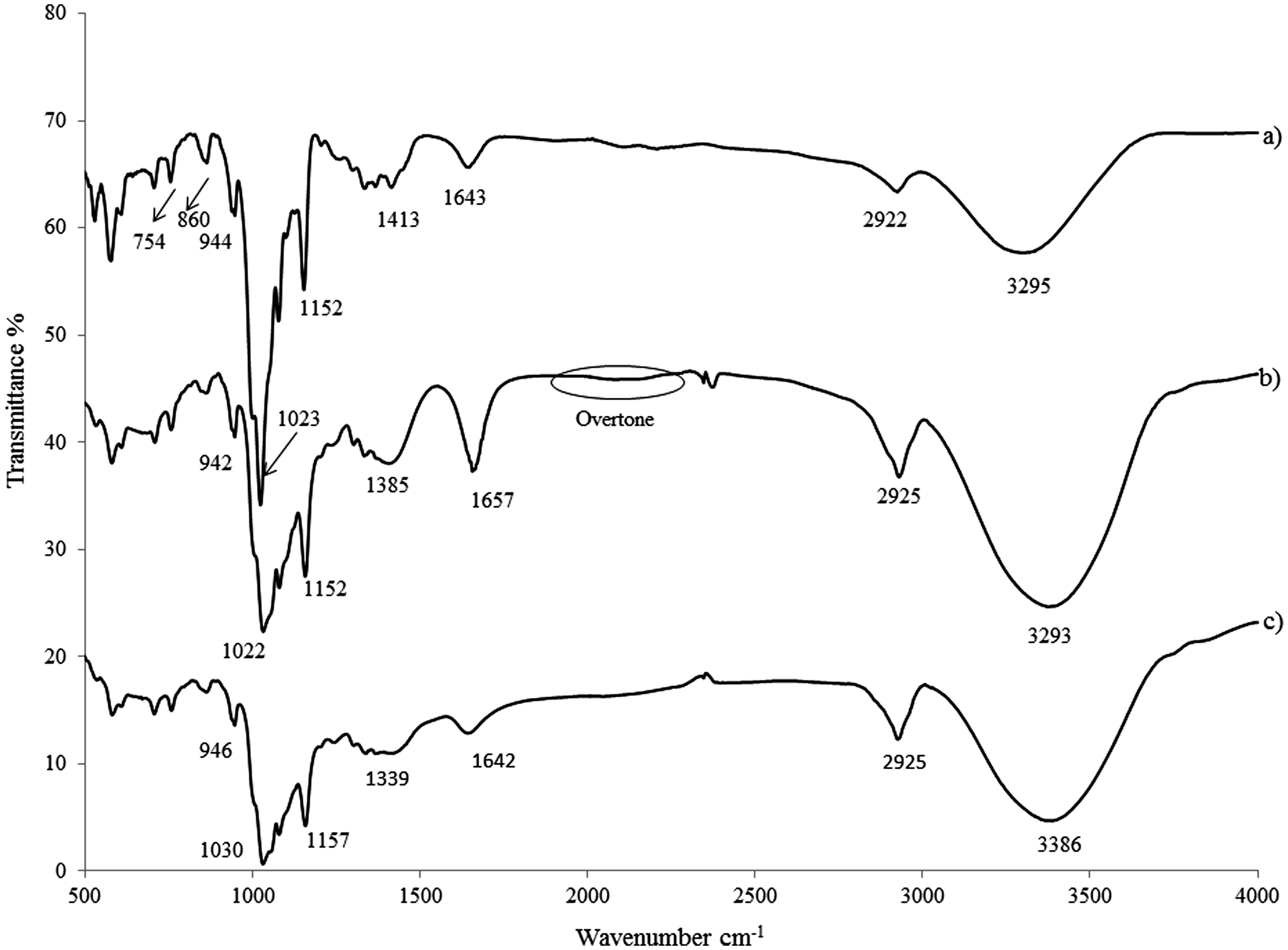
The spectra of Si-TDI, native β-CD-CSP, β-CD-BIMOTs-CSP and β-CD-DIMOTs-CSP are shown in Figure 6. Spectra of Si-TDI (a) show the presence of the isocyanate (O = C = N-) group at 2280 cm−1. The para position isocyanate group is expected to react with OH groups on the surface of silica to form Si-TDI (Arnold, 1957; Rahim et al., 2016a, 2016b). The remaining isocyanate group at ortho-position would react with secondary OH group of β-CD or β-CD functionalized IL. Therefore, the isocyanate peak disappeared after immobilization of native β-CD, β-CD-BIMOTs and β-CD-DIMOTs onto Si-TDI (Figure 6(b) and (d)).
FT-IR spectra of (a) Si-TDI (b) native β-CD-CSP (c) β-CD-BIMOTs-CSP (d) β-CD-DIMOTs-CSP.
Thermal analysis
TGA analyses were performed on the Si-TDI, native β-CD-CSP, β-CD-BIMOTs-CSP and β-CD-DIMOTs-CSP in the temperature range of 50 to 900℃. Based on the thermogram shown in Figure 7, there was an initial loss of weight at temperature below 100℃ for all samples. This was attributed to the removal of physically adsorbed water and/or remaining solvent residues. Physically adsorbed water was further removed completely by heating to around 200℃. TDI attached to the silica surface decomposed in the region between 125 and 250℃ (Guo et al., 2005). In addition, Si-TDI showed a small but noticeable weight loss in the region 250–600℃, caused by the dehydration of the silica surface (Poole et al., 2003). The thermogram of β-CD-BIMOTs-CSP and β-CD-DIMOTs-CSP showed two very distinct weight losses. The weight loss occurred at the range of 210–357℃ can be attributed to the decomposition of organic moieties at the surface. The weight loss takes place at 400–600℃ might be due to the decomposition of the residual methoxy groups on silica (Antochshuk and Jaroniec, 2000). The incessant decrease in weight of native β-CD-CSP, β-CD-BIMOTs-CSP and β-CD-DIMOTs-CSP between 600 and 900℃ can be assigned to the decomposition of the β-CD. Overall, the thermogram of β-CD-BIMOTs-CSP showed more pronounced weight loss than β-CD-DIMOTs-CSP at all isothermal temperatures. This is because the long alkyl chain of β-CD-DIMOTs-CSP prevents it from becoming volatile at high temperatures (Lu et al., 2002).
Thermogram of (a) Si-TDI (b) native β-CD-CSP (c) β-CD-BIMOTs-CSP (d) β-CD-DIMOTs-CSP.
Elemental analysis
The elemental composition of β-CD-DIMOTs-CSP was C: 15.56%, H: 2.33%, N: 4.72%, S: 1.42%. The degree of surface coverage for β-CD-DIMOTs-CSP was calculated from the following equation (Hongdeng et al., 2014)

Screening performance of β-CD functionalized ILs
The effect of different groups attached to imidazolium cation (present in β-CD functionalized IL) on the separation of chiral compounds was studied. The performance of β-CD-BIMOTs-CSP and β-CD-DIMOTs-CSP was compared with native β-CD-based CSP for the enantioseparation of NSAIDs. The chromatogram in Figure 8(a) showed that ibuprofen achieved baseline separation while the other NSAIDs (Figure 8(b) and (c)) were poorly enantioseparated using β-CD-BIMOTs-CSP. It is obvious that β-CD-BIMOTs-CSP showed better chromatographic performance as compared to that of β-CD-DIMOTs-CSP and native β-CD based CSP. These results suggest that β-CD-BIMOTs-CSP might provide additional interaction with NSAIDs thus enhanced the enantioseparation. The planar aromatic of 1-BzlIm attached to β-CD-BIMOTs-CSP is approached by planar analytes in preference, forming π–π interaction (Wang et al., 2012) that contributed to better enantioseparation. The long alkyl chain of β-CD-DIMOTs-CSP is able to cover the partial cavity of β-CD (Meier-Augenstein et al., 1992) resulting in decreased its chiral selectivity. Thus, the optimization of mobile phase for the enantioseparation of NSAIDs was further investigated using β-CD-BIMOTs-CSP. Additionally, the interactions of the enantioseparation on β-CD-BIMOTs-CSP were evaluated.
The chromatograms for the enantioseparation of selected NSAIDs on (a) β-CD-BIMOTs-CSP (b) β-CD-DIMOTs-CSP (c) native β-CD-CSP condition: (i) 90/10 ACN/water (ii) 50/50 ACN/water and (iii) 30/70 ACN/water.
Chromatographic data and evaluation on the interactions of enantioseparation on β-CD-BIMOTs-CSP
Chiral separation data for the NSAIDs on β-CD-BIMOTs CSP.


The chromatograms of fenoprofen, ibuprofen, indoprofen and ketoprofen responding to different pH of mobile phase.
As can be seen from Table 1, ibuprofen was completely resolved with Rs value of 2.51, meanwhile, indoprofen showed partial separation with Rs value of 1.09. Ketoprofen and fenoprofen were also partially enantioseparated and fenoprofen attained the lowest Rs value (0.54). The relatively low Rs values of ketoprofen and fenoprofen were because of the substituent in the meta position that made their orientation in an unfavorable way to fit into the β-CD-BIMOTs cavity (Fanali and Aturki, 1995). The higher Rs values of ibuprofen and indoprofen are probably due to the para position of the substituent (containing the chiral center) on the aromatic ring (Fanali and Aturki, 1995). This is in good agreement with previous studies which also proved that para-substituted aromatic rings can fit properly into the CD cavity forming inclusion complex, but the extent of the penetration mode is dependent on the polarity and feature structure of analytes (Fanali and Aturki, 1995; Núñez-Agüero et al., 2006). It can be concluded that the less polar ibuprofen achieved better enantioseparation than polar indoprofen (Velkov et al., 2007).
Even though the polarity of fenoprofen and ibuprofen are close to each other (log Pfenoprofen = 3.8, log Pibuprofen = 3.7) (Velkov et al., 2007), ibuprofen achieved higher Rs value when high organic solvent content (90% ACN) was used. This is because ibuprofen can be fitted into β-CD-BIMOTs cavity, whereas fenoprofen with two aromatic rings was less favorable to be fitted into β-CD-BIMOTs cavity due to steric hindrance effect. According to previous simulation study (Núñez-Agüero et al., 2006), there was also moderate and weak hydrogen bonding between the carboxyl group of ibuprofen and hydroxyl groups of β-CD during the complexation. Ketoprofen which is composed of a similar structure (two aromatic rings) as fenoprofen, achieved better enantioseparation than fenoprofen. This is due to the presence of carbonyl group in ketoprofen which enhanced the formation of hydrogen bonding with β-CD-BIMOTs rather than ether linkage in fenoprofen (Lommerse et al., 1997). Therefore, it can be said that, apart from the inclusion complex formation, hydrogen bonding also played an important role in enhancing the enantioseparation of NSAIDs.
Chemical shifts corresponding toβ-CD-BIMOTs in the presence of NSAID.

Note: Values in bold refer to the highest induced shift of that particular proton.
Induced shifts corresponding to NSAID in the presence of β-CD-BIMOTs.

Note: Values in bold refer to the highest induced shift of that particular proton.

The deduced structure of β-CD-BIMOTs.

The deduced structure of NSAID/β-CD-BIMOTs complexes: (a) (i) ibuprofen (ii) β-CD-BIMOTs/ibuprofen, (b) (i) indoprofen (ii) β-CD-BIMOTs/indoprofen (c) (i) ketoprofen (ii) β-CD-BIMOTs/ketoprofen, (d) (i) fenoprofen (ii) β-CD-BIMOTs/fenoprofen.
Additionally, the UV/Vis absorption spectra of β-CD-BIMOTs/NSAIDs complexes were further investigated to acquire more information on the interactions between NSAIDs and β-CD-BIMOTs. The plots of UV/Vis absorption for β-CD-BIMOTs, NSAIDs and β-CD-BIMOTs/NSAIDs complexes are presented in Figure 12. The results obtained revealed that β-CD-BIMOTs had λmax in the range of 230–260 nm. The λmax of β-CD-BIMOTs/ibuprofen, β-CD-BIMOTs/indoprofen and β-CD-BIMOTs/fenoprofen complexes appeared at 262, 256 and 256 nm, respectively, referring to β-CD-BIMOTs. The absorbance of β-CD-BIMOTs/ibuprofen, β-CD-BIMOTs/indoprofen and β-CD-BIMOTs/fenoprofen underwent the hyperchromic effect (increase in absorbance), while the absorbance of β-CD-BIMOTs/ketoprofen experienced the hypochromic effect (decrease in absorbance). Both the hyperchromic and hypochromic effects observed were due to the π–π* transition of dipole moments of the aromatic ring. The transition dipole moment of this chromophore interacts with the induced dipoles of the neighboring chromophores, depending on their relative orientation. If the dipoles are along the same axis and one behind the other, then the intensity of the absorption band will increase and hyperchromic effect is observed. Conversely, if the dipoles are parallel and adjacent, a decrease in intensity of the absorption band occurs, and hypochromic effect is observed (Peral and Gallego, 2000). The hypochromic effect on β-CD-BIMOTs/ketoprofen can also be attributed to the limitation for π–π* transition because of hydrogen bonding (Peral and Gallego, 2000) at carbonyl group between aromatic rings of ketoprofen. The variations that occurred in the UV/Vis spectra were a consequence of complexation of NSAIDs with β-CD-BIMOTs accompanied by π–π interaction and hydrogen bonding. These results clearly prove the ability of IL to form π–π interaction in addition to the existing superposition of inclusion complex and hydrogen bond for the enantioseparation of NSAIDs.
Absorption spectra of (a) β-CD-BIMOTs/ibuprofen, (b) β-CD-BIMOTs/indoprofen, (c) β-CD-BIMOTs/ketoprofen and (d) β-CD-BIMOTs/fenoprofen with (β-CD-BIMOTs): 0.032 mM (NSAIDs): 0.01 mM; T = 25℃.
Conclusions
In this study, β-CD-BIMOTs-CSP and β-CD-DIMOTs-CSP were successfully synthesized and compared for enantioseparation of NSAIDs. The β-CD-BIMOTs-CSP performed better than β-CD-DIMOTs-CSP and β-CD-CSP due to the additional π-π interaction which was possible with β-CD-BIMOTs-CSP. Furthermore, a better enantioseparation of ibuprofen, fenoprofen, indoprofen and ketoprofen using β-CD-BIMOTs-CSP were observed due to the superposition of hydrogen bonding, hydrophobic and also π-π interactions. From 1H NMR, NOESY and UV/Vis studies, NSAIDs were proven to form inclusion complexes with β-CD-BIMOTs-CSP.
Footnotes
Acknowledgements
The authors acknowledge Ministry of Higher Education (MOHE) for providing fellowship to one of the authors-cum-researchers, Ms. NurulYani Rahim.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors would like to seize this opportunity to express their gratitude to the University Malaya for the IPPP grant PG027/2013A and UMRG grant (RP006A-13SUS and RP011B-14SUS).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.