
Abstract
Over the last decade, Mycobacterium tuberculosis has mutated into a putative ‘superbug’, as treatments against it have failed due to increasing antimicrobial resistance. As a result, the rising incidence of multidrug-resistant tuberculosis (MDR-TB) is posing a significant public health threat, thus, the need to develop effective drugs for MDR-TB has become an urgent priority. To identify new drug candidates for the treatment of MDR-TB, the present study was based on mycobacterial shikimate kinase (MtSK) as the pharmacological target. One hundred potential MtSK inhibitors were identified from literature and database searches to identify compounds that were designed to specifically function as MtSK antagonists. The ADME properties of these compounds were evaluated by using the SwissADME web tool. ProTox-II software was also used to investigate any potential endocrine disrupting effects, mediated through their interaction with oestrogenic and/or androgenic receptors. This study also aimed to predict LD50 values of potential drug candidates that would be active against the standard H37Rv strain of M. tuberculosis, by using the ProTox-II in silico tool. The molecules for which no structural hazard alerts were identified with these software tools were further subjected to molecular docking analyses and molecular dynamic simulations to estimate their ability to interact with the MtSK enzyme. Preliminary results from SwissADME indicated that 30 molecules were drug-like, due to their physicochemical and pharmacokinetic properties. However, subsequent analysis with ToxTree and ProTox-II indicated that only three of these 30 drug-like molecules were suitable for taking forward into further in vitro experiments. This study, which is based on the use of commonly used open-source in silico tools, identified new MtSK ligands for potential use in the development of new drugs for the therapeutic management of tuberculosis. An initial prediction of their safety profile was also generated.
Keywords
Introduction
Tuberculosis (TB) is the second most common infectious disease in the world, behind COVID-19 (and above HIV/AIDS); it is the 13th largest cause of mortality worldwide. 1 As per a recent World Health Organisation (WHO) report, an estimated 10 million people fell ill with TB worldwide in 2020, of which 1.1 million were children, and the mortality was estimated to be 1.5 million. 1 Over the last decade, Mycobacterium tuberculosis has mutated into a putative ‘superbug’, as treatment methods and regimens have failed in the face of increasing antimicrobial resistance (AMR). 2 Multidrug-resistant tuberculosis (MDR-TB) continues to be a public health emergency, and patients with MDR-TB have limited treatment options for good recovery. Thus, the need to develop drugs for MDR-TB and find new pharmacological targets has become urgent in recent years.
Shikimate kinase (MtSK), an enzyme that belongs to the nucleoside monophosphate (NMP) kinase family, has been identified as an important target in the mycobacterial proteome, due to its novel presence within the tubercle pathogen and its absence in mammalian cells. 3 It plays a significant role in the development and maintenance of the atypical mycobacterial cell wall, which is one of the preliminary defence systems that M. tuberculosis employs against therapeutic drugs. 4 In M. tuberculosis, MtSK is the fifth enzyme in the shikimate pathway cascade, which catalyses a phosphate transfer from ATP to the shikimate anion to produce shikimate-3-phosphate (S3P) and ADP. 5 It is responsible for the synthesis of the aromatic amino acids phenylalanine, tyrosine and tryptophan, which play an important role in the maintenance of the integrity and structure of the mycobacterial cell wall. Strategically designing molecules to inhibit the bioactivity of MtSK is an approach currently under investigation for the treatment of TB. 6 A specific set of structural scaffolds could be identified as being probable pharmacophores, and then molecular docking analyses could be carried out to assess any potential antagonistic effects of these ligands on MtSK.
Several tools are available to evaluate the Absorption, Distribution, Metabolism and Elimination (ADME) characteristics of thousands of drug candidates, to select the few that show promise for further development through the drug discovery process. The SwissADME tool provides key characteristics of compounds, such as molecular weight, number of rotatable bonds, hydrogen bond acceptor and donor potential, and total polar surface area. 7 It also predicts lipophilicity, water solubility and pharmacokinetic properties, such as gastrointestinal absorption, blood–brain barrier (BBB) permeation and interaction with the P-glycoprotein (P-gp) efflux pump. SwissADME can screen large chemical libraries by using multiple filters, including Lipinski, Ghose, Veber, Muegge and Egan, to evaluate lead-likeness and determine their druggability (the likelihood of a drug-like compound to modulate or inhibit an interaction between two proteins). 8
ToxTree is an in silico tool that can identify potential hazards, using information based on molecular structure. 9 Proposed lead molecules can be toxicologically profiled by using ToxTree, which uses a decision-tree approach to examine the potential toxicological implications of the structural moieties in the query compounds. The lead molecules are evaluated against alerts for properties such as mutagenicity, genotoxic and non-genotoxic carcinogenicity, micronucleation, chromosomal aberrations, and the ability to bind DNA and protein.
Molecular docking analyses present still images of complexed protein–ligand co-crystals, whereas molecular dynamic simulations display physical movements and chemical interactions within a protein–ligand co-crystal over a fixed time-period. Molecular docking studies prove useful in predicting the extent of the interaction and complexation between different biomolecules, and to estimate the stability of these complexes. In contrast, molecular dynamic simulations provide precise information on the chemical structures, conformational changes, and dynamics of biological molecules and their complexes. Therefore, the combination of molecular docking analysis and molecular dynamic simulation permits quick screening of large libraries, and the simultaneous calculation of interaction energies to accelerate drug discovery processes.10,11
In the current study, a literature search in PubMed was performed, in order to identify compounds that were designed to specifically function as MtSK inhibitors. One hundred molecules were randomly selected from the 767 compounds obtained from the literature search. These compounds were known molecular scaffolds — such as benzothiazole, benzimidazole, imidazole, triazole, phenylpiperidine, morpholino-piperazine, oxazole, benzoxazole, naphthalene-sulphonic acid and xanthene. The molecules were then evaluated for their pharmacokinetic and drug-like properties by using the SwissADME tool.
The thirty most promising molecules were then subjected to toxicological profiling with ToxTree software. The MtSK inhibitors that had no structural alerts identified in ToxTree were then screened for any potential endocrine-disrupting effects (mediated via their potential interaction with oestrogenic and/or androgenic receptors), by using ProTox-II.
An attempt to predict LD50 values of potential anti-TB drug candidates was also made with ProTox-II, which employs a random forest algorithm for predicting the toxicities of small molecules, by annotating structures that may be potentially hazardous.
The 30 drug-like molecules were also subjected to molecular docking analyses, in order to predict the extent of their interaction and complexation with the co-crystallised structure of the MtSK enzyme. For the lead-like molecules that were indicated to be potent antagonists of the MtSK enzyme, molecular dynamic simulations were then undertaken to estimate the stabilities of the molecule–target complexes.
Methods
Identification of possible lead molecules
A literature survey was performed to identify compounds that were designed to specifically function as MtSK inhibitors by using the terms “MtSK inhibitors”, “Mycobacterium tuberculosis shikimate kinase inhibitors” in PubMed; their ZINC IDs 13 and PubChem IDs 14 were retrieved. The literature search identified 767 compounds, of which 100 compounds were randomly selected. The structures were downloaded in 2-D format from the respective websites, and their SMILES (Simplified Molecular Input Line Entry System) were generated by using the online tool SMILES translator (https://cactus.nci.nih.gov/translate/). SMILES is an easy-to-use chemical notation that allows the user to input a chemical structure in a way that can be processed by software. 15
Prediction of pharmacokinetic properties and drug-likeness by using SwissADME
The Swiss Institute for Bioinformatics’ online tool, SwisADME (http://www.swissadme.ch/), was used to predict the PK and drug-likeness properties of the molecules and to analyse their druggability and lead-likeness by creating a bioavailability radar for the compounds. The SMILES were entered into the tool’s input box on the web page, and the tool generated the results independently.
The prediction of toxicological potential and threat aversion by using ToxTree
The toxicity of a molecule/ligand can be determined by its structural characteristics, and the ToxTree software forecasts specific toxicity potential according to the different structural moieties within the molecule. Structure–activity relationships, incorporated within the software, were used to identify possible toxicological outcomes. The following decision trees were chosen:
Kroes TTC (Threshold for Toxicological Concern)
The prediction of the threshold for toxicological concern of a molecule provides a pragmatic approach to assess the potential of a molecule/ligand to manifest toxicity in a physiological system above a certain amount of regulated intake. 16
Carcinogenicity assessment (genotoxicity, non-genotoxicity, mutagenicity)
The potential of a molecule to induce cancer in a physiological system is a functional attribute of its structural components. The decision tree identifies the structural moieties within a molecule that could be a possible concern with regard to carcinogenicity or mutagenicity within a physiological system, indicating the possibility that such moieties could induce toxicities via genotoxic or non-genotoxic pathways.17,18
In vitro mutagenicity assay (Ames Assay) alerts, as per the Istituto Superiore di Sanità (ISS) rule base
This rule base provides structural alerts based on the presence of structural moieties that may be positive for inducing mutations in Salmonella typhimurium. 19
Structural alerts for the micronucleus assay in rodents
The ability of a ligand to induce micronuclei is considered to be a manifestation of toxicity, as it indicates the possibility of chromosomal aberrations. This rule base identifies structural alert moieties present in ligands that may possess the potential to induce micronucleus formation. 20
DNA binding alerts
The structural attributes of the lead molecules were assessed by using the DNA binding rule base, in order to identify whether the ligands have the potential to form covalent bonds with DNA within a physiological system. Such covalent interactions between a ligand and DNA are irreversible in nature, hampering the development of an effective therapeutic agent. 21
Protein binding alerts
The structural attributes of the lead molecules were also assessed by using the protein binding rule base, in order to identify the potential to form covalent bonds with proteins within a physiological system — such covalent interactions would be unfavourable. The SMILES generated were inputted into the software, and the results were generated by manually selecting the decision tree to be applied. 22
Prediction of endocrine disruption potential
The 30 molecules deemed most promising following the initial analyses were further screened in silico for their potential to interact with oestrogenic and/or androgenic receptors and cause functional endocrine disruptions within a physiological system. The online tool, ProTox-II (https://tox-new.charite.de/protox_II/), was employed for this purpose. SMILES were inputted into the input box, and the receptors were chosen sequentially to estimate the bioactive potential of the lead molecules and to interpret their impact on the endocrine proteome. The targeted endocrine receptors included: the androgen receptor (AR), androgen receptor-ligand binding domain (AR-LBD), oestrogen receptor-α (ER-α), oestrogen receptor-ligand binding domain (ER-LBD) and peroxisome proliferator-activated receptor-γ (PPAR-γ).12,23
Prediction of LD50 values
Toxicological classification for LD50 values.

Molecular docking analyses and molecular dynamic simulations
Selection and processing of the macromolecule
The 3-D structure of the M. tuberculosis MtSK target protein, complexed with shikimic acid, was procured from the public database, RCSB PDB (https://www.rcsb.org/). After a careful evaluation of the selection parameters (i.e. resolution of the protein structure and Ramachandran outliers), 2IYQ (PDB DOI: 10.2210/pdb2IYQ/pdb) was selected as the appropriate macromolecule. It had a crystallographic resolution of 1.8 Å and was complexed with shikimic acid and ADP. 24 The macromolecule was then processed by using the Discovery Studio tool. The addition of H atoms was carried out and was followed by the equitable distribution of respective charges of the amino acid groups across the structure of the macromolecule. Energy minimisation of the macromolecule was then carried out with Chimera software. 24 The macromolecule was then submitted to CASTp (http://sts.bioe.uic.edu/castp/index.html?1bxw) for the prediction of the active site of the enzyme, and the final processed file of the macromolecule 2IYQ was used for the molecular docking analyses.
Retrieval of ligands
Public libraries such as the ZINC database (https://zinc.docking.org/) and the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) were used to procure the structures of the 30 ligands (out of the initial 100 ligands) that were considered the most promising following the preliminary pharmacokinetic and toxicological screening. The 3-D structures of the 30 ligands were downloaded in ‘.sdf’ format and the ligand structures were optimised by using the energy minimisation module on the open-source molecular docking analysis tool, PyRx. Further, the validation of the structures of the 30 ligands was carried out by stereochemical modifications, such as the addition of valence hydrogen, and optimisation of the bond lengths and bond angles. The ligands were subsequently converted to ‘.pdbqt’ format for use in the molecular docking analyses.
Molecular docking analyses
Molecular docking analyses were carried out by using the AutoDock Vina Module 1.1.2. implemented on the PyRx software. 25 The macromolecule 2IYQ was similarly uploaded to the AutoDock Vina, designated as the macromolecule, and converted to the ‘.pdbqt’ format. Docking parameters, such as the algorithm for docking and grid space for docking ligands onto the macromolecule, were finalised and the docking analyses were carried out for the 30 ligands that could potentially act as MtSK antagonists. Molecular docking analyses were carried out by using the Lamarckian Genetic Algorithm integrated within the AutoDock Vina module.25,26 Analyses for the visualisation of ligand–protein interactions, both in 2-D and 3-D, were carried out with the Discovery Studio software (see online Supplemental Material).
Molecular dynamic simulations
After performing a virtual screening for suitable binding affinity in PyRx, and for potential toxicity in ToxTree and ProTox-II, three promising lead molecules were identified — namely, ZINC15707201 (M1), ZINC11790367 (M2) and ZINC588497 (M23). These ligands were tested for co-crystallisation stability by performing a 10-nanosecond simulation and then a high-throughput 100-nanosecond simulation with the latest version of GROMACS software (2023 series), which gives fast and reliable results for biomolecules (see online Supplemental Material). These simulations were subsequently analysed by using xmgrace and UCSF Chimera software to investigate whether these ligands would diffuse from the MtSK target protein over time. By visualising the whole trajectory of the interaction in UCSF Chimera, it was possible to confirm that, theoretically, these molecules were able to substantially interact with the MtSK target protein.6,10,11
Results and discussion
Prediction of pharmacokinetic properties and drug-likeness by using SwissADME
To identify the molecules most likely to be safe and druggable, each of the initial 100 molecules was subjected to an evaluation of its pharmacokinetic and drug-like properties, including the bioavailability profile, Log P value, ability to breach the BBB, GI absorption and ability of P-gp to eliminate the substance from the cranial environment. Of these 100 molecules, 30 were found to have physicochemical properties falling within the pink area of the bioavailability radar in Figure 1 — i.e. within a suitable physicochemical space for oral bioavailability. The pink area represents the optimal range for each property, namely: — lipophilicity (LIPO): a XLOGP3 value of between −0.7 and +5.0; — size (SIZE): a molecular weight (MW) of between 150 and 500 g/mol; — polarity (POLAR): a total polar surface area (TPSA) of between 20 and 130 Å2; — solubility (INSOLU): a log S value not higher than 6; — saturation (INSATU): the fraction of sp3 hybridised carbons not less than 0.25; and — flexibility (FLEX): no more than 9 rotatable bonds.
7
Selection of drug-like molecules after the pharmacokinetic analysis. Analysis of the 100 potential MtSK inhibitors, by using SwissADME.
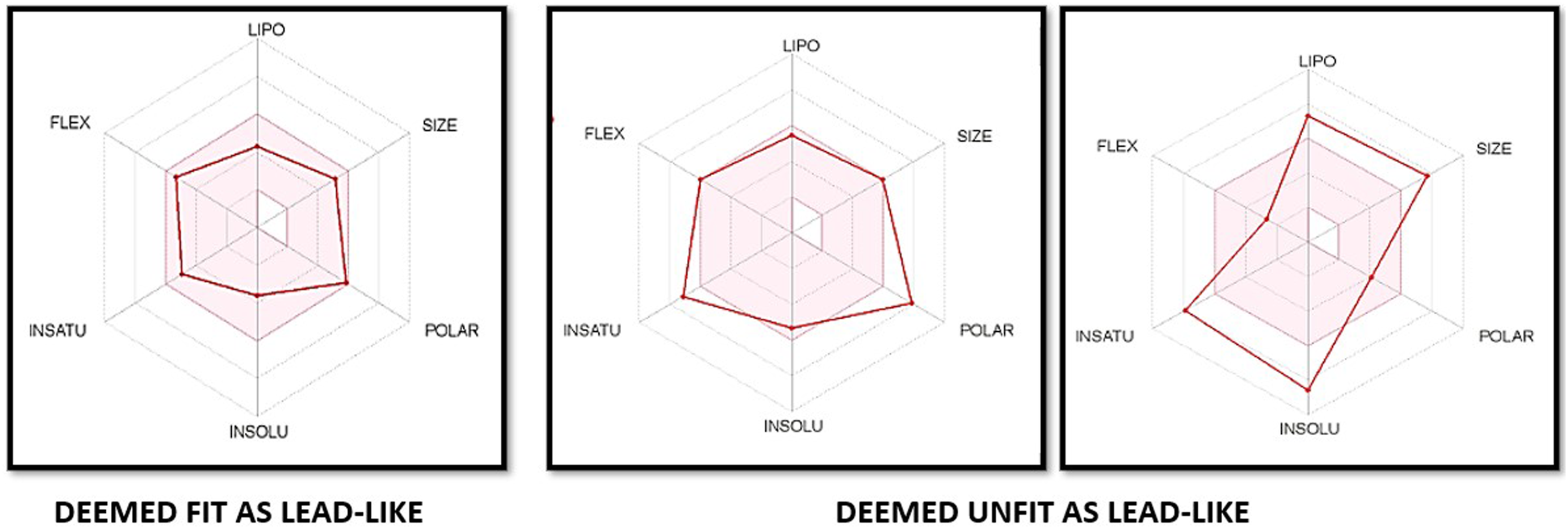
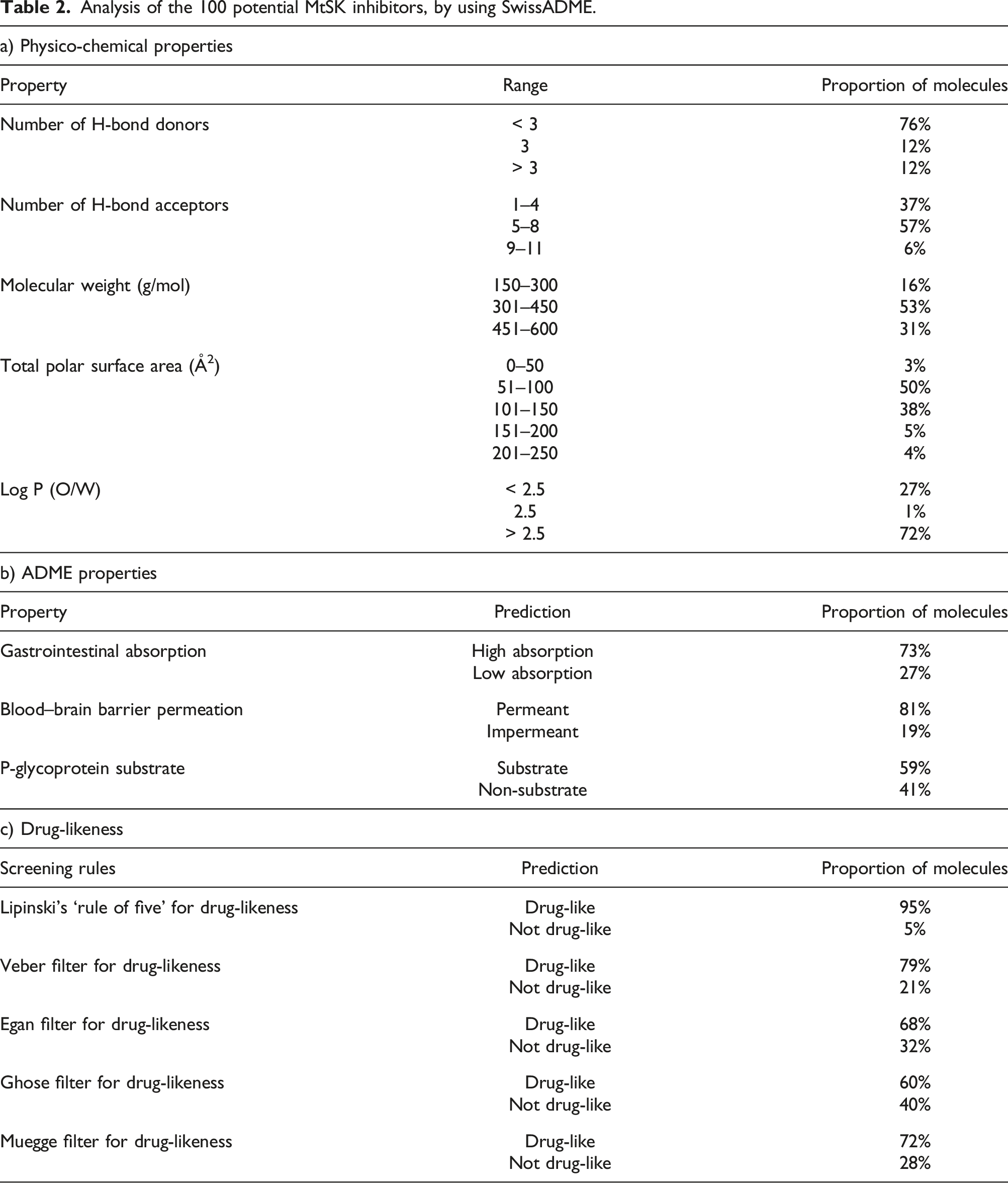
The permeability of a compound, and thus its bioavailability, is related to its molecular weight. High molecular weight compounds tend to have poor bioavailability, due to their limited permeability across membranes. It is notable that the molecular weight of every analysed compound was less than 600 Da, and was skewed toward 301–450 Da — 53% of the compounds fell within this range. Only 16% of the compounds studied had a low molecular weight (i.e. between 150–300 Da), whereas the remaining 31% were between 451–600 Da.
The Total Polar Surface Area (TPSA) is a measure of a compound’s surface that is represented by polar atoms. It correlates with passive diffusion, and thus the permeability and bioavailability of a particular compound. As mentioned above, the optimal range for the TPSA, according to the SwissADME, is between 20 and 130 Å2. TPSA prediction data suggested that 50% of the compounds analysed in the current study had a TPSA of between 51–100 Å2, and 38% of them had TPSA values within the 101–150 Å2 range.
The lipophilicity of a compound is determined by its partition coefficient and is expressed as a Log P value. This is the logarithm of the ratio of solubilities of a particular compound in water and octanol. The higher the Log P value, the higher the lipophilicity of a compound, and the desirable range (as per SwissADME) is between −0.7 and +5.0. The Log P value of a compound indicates its absorption, transportation and distribution potential, and thus its overall bioavailability. A low water–octanol lipophilicity coefficient (i.e. 1.5–2.5) was predicted for 27% of the molecules analysed, and a value above 2.5 was predicted for 72% of the compounds.
The predicted ADME properties (Table 2b) showed that 73 of the 100 molecules analysed would have high GI absorption, and that 81% would be able to penetrate the BBB; 59% were predicted to be potential P-gp substrates. Understanding whether or not compounds are substrates of P-gp is crucial for evaluating their active efflux across biological membranes. This is especially important for processes such as transport from the gastrointestinal wall to the lumen, or across the BBB. A primary function of P-gp is to safeguard the central nervous system (CNS) from foreign substances.
Lipinski’s ‘rule of five’ showed that 95% of the analysed compounds were drug-like. The Veber filter indicated that 79% of the compounds had drug-like structural attributes, while the Egan filter showed 68% of compounds to be drug-like (Table 2c). The Ghose and Muegge filters indicated that 60% and 72% of the molecules, respectively, were drug-like.
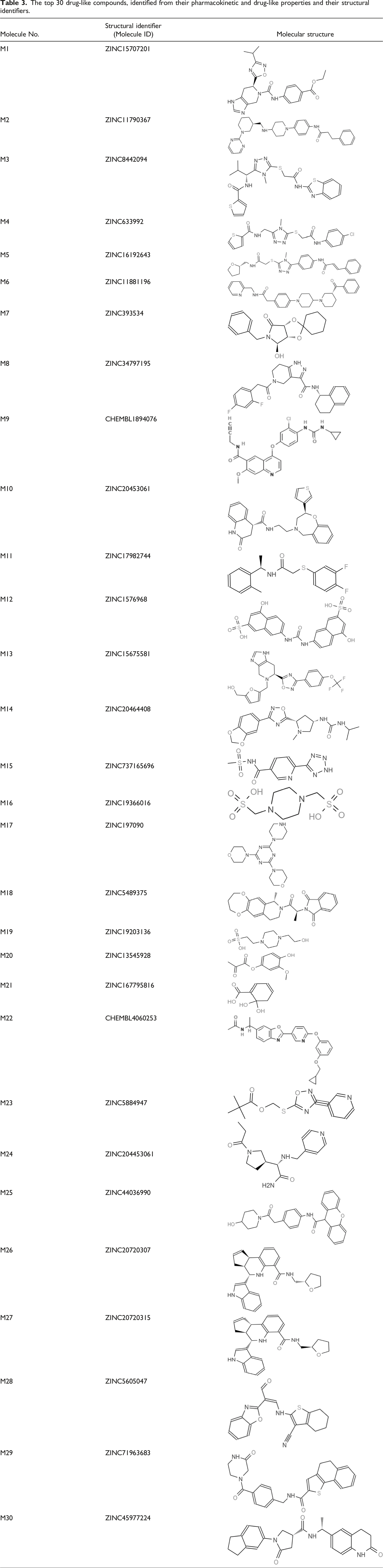
The top 30 drug-like compounds, identified from their pharmacokinetic and drug-like properties and their structural identifiers.
Prediction of toxicological potential and threat aversion by using ToxTREE
Summary of the ToxTree analysis for the top 30 drug-like compounds.
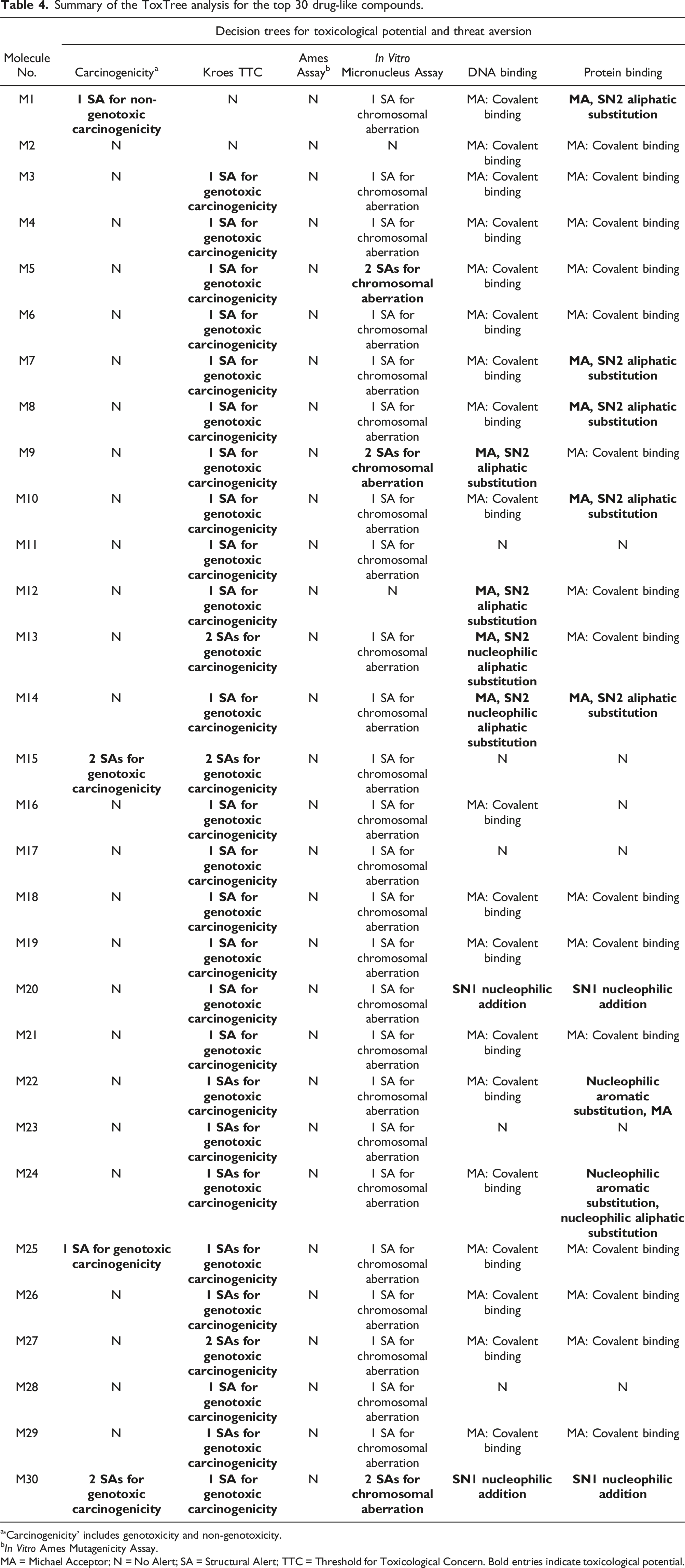
a‘Carcinogenicity’ includes genotoxicity and non-genotoxicity.
bIn Vitro Ames Mutagenicity Assay.
MA = Michael Acceptor; N = No Alert; SA = Structural Alert; TTC = Threshold for Toxicological Concern. Bold entries indicate toxicological potential.
The highest probability of binding to DNA and interfering with its structure and function applied to six of the 30 drug-like molecules, while 19 molecules showed lower potential for interacting with DNA; five compounds were predicted to be devoid of structural moieties likely to interact with DNA and hamper physiological function. The protein binding decision tree indicated that nine molecules had two structural alerts for interaction with skin proteins, while 15 molecules elicited one structural alert for protein interaction. The 24 molecules that triggered at least one structural alert were all Michael acceptors and were capable of binding covalently to skin proteins. In contrast, six molecules were predicted to be free of structural moieties involved in the formation of such covalent bonds.
Prediction of endocrine disruption potential and hepatotoxicity
The ProTox-II analysis for the prediction of hepatotoxicity and endocrine disruption potential.

aThe values in parentheses in this column indicate the probability of incidence of the classified effect — either ‘Inactive’ in terms of hepatotoxicity, or ‘Active’. A value of < 0.7 represents a ‘low’ probability of incidence; a value of > 0.7 represents a ‘high’ probability. The bold entries in this column indicate a potential for hepatotoxic activity (i.e. drug-induced liver injury; DILI).
bThe values in parentheses in this column indicate the probability of incidence of the classified effect — i.e. ‘Inactive’ with regard to their interaction with the oestrogen receptor-α. A value of < 0.7 represents a ‘low’ probability of incidence; a value of > 0.7 represents a ‘high’ probability of incidence. Hence, the bold entry indicates that this ‘Inactive’ classification has a low probability of incidence (< 0.7).
Interactions with the androgenic receptor, androgenic receptor-ligand binding domain, oestrogenic receptor-ligand binding domain, and PPAR-γ were also analysed and deemed to be insignificant — i.e. all were classified as ‘Inactive’ with regard to their interactions, with (high) probabilities of incidence of between 0.75 and 1.0; thus these data are not shown in the Table.
The same tool, ProTox-II, was also used to predict the potential for hepatotoxicity (i.e. drug-induced liver injury; DILI). Two molecules (M9 and M16) were highlighted by ProTox-II as having the potential to cause DILI, albeit at a low probability of incidence (< 0.7); these are indicated in bold in Table 5. The remaining 28 molecules were classified as ‘Inactive’ with regard to their potential hepatotoxic activity — 17 with a high probability of incidence (> 0.7) and 11 with a low probability (< 0.7).
Prediction of LD50
Predicted LD50 values of the 30 compounds deemed to be drug-like.

Molecular docking analyses
The binding affinity scores for the 30 drug-like molecules, derived from molecular docking analysis on PyRx.

The assessment was performed against the target enzyme, MtSK (2IYQ protein structure).
The molecular docking scores of the 30 drug-like molecules were compared, and those with the top three scores in terms of the calculated binding energy (i.e. < –7 kcal/mol) were subsequently chosen for the molecular dynamic simulations — namely, M1, M2 and M23.
Molecular dynamic simulations
From the molecular dynamic simulation data, the complex between M1 and 2IYQ appeared to be stable (Figure 2a and b). However, the lead molecule showed some deviation from the original binding site to a possible allosteric site, as noted from the rise in root-mean-square deviation delta values (δ-RMSD values). The complex formed by M2 and 2IYQ (Figure 2c and d) also seemed significantly energetic, as the interactions between the lead molecule and the crystalline structure of the MtSK enzyme show significantly deviating δ-RMSD values. RMSD and δ-RMSD plots of the interactions between the three most promising lead molecules and the MtSK target enzyme (2IYQ prostein structure).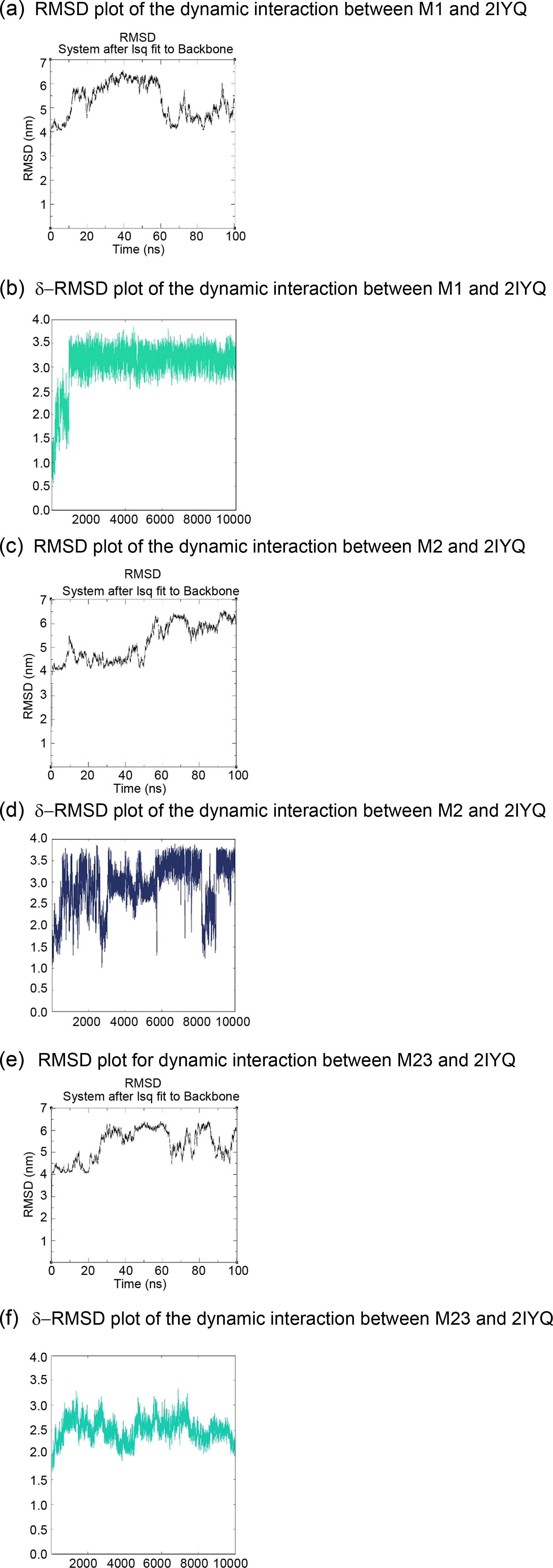
The complex between M23 and 2IYQ (Figure 2e and f) was reportedly more stable than that formed with M1 and M2, as evidenced by the lower drift in δ-RMSD values. Despite some drifts observed in the δ-RMSD values, the complexes formed remained stable enough to ensure that the crystalline structure of the enzyme would not denature, as the degree of gyration was maintained for all of the structures.
Thus, the present study finally proposed three molecules to be the most promising candidates in terms of predicted safety profile and potency of MtSK inhibition — namely, M1, M2 and M23 — which could potentially be taken forward into further in vitro experiments.
Synthesis scheme for two of the proposed ligands
A proof-of-concept attempt was made to predict a scheme of synthesis for M1 and M2 by using a retrograde synthon approach to aid in vitro studies in the future.
The retrosynthesis prediction of M1 was performed with IBM RXN for chemistry webserver. The proposed chemical pathway for the synthesis of M1, deduced from the retrosynthesis prediction, is depicted in Figure 3. Step 1 in Figure 3a includes three different reactions. The first reaction is protection of the N-1 and N-7 of spinacine using isobutyrlchloroformate in tetrahydrofuran (THF) with the aid of diisopropylethylamine (DIPEA) at 40°C to form the N-1,N-7 protected (N-carbamoyl) spinacine derivative (not shown). The second reaction is heterocyclisation of the resulting N-1,N-7 protected spinacine derivative by N-hydoxyisobuyramidine with DIPEA in THF at 120°C to N-1,N-7-protected 1,2,4-oxadiazolylspinacine derivative (not shown). The third reaction is N-1,N-7-deprotection (removal of carbamoyl group) from N-1,N-7-protected 1,2,4-oxadiazolylspinacine derivative using trifluoracetic acid (TFA) in dichloromethane (DCM) to yield 6-[3-(propan-2-yl)-1,2,4-oxadiazol-5-yl]spinacine.
27
Step 2, shown in Figure 3b, is the condensation of oxadiazolylspinacine derivative obtained from Step 1 (Figure 3a) with ethyl 4-isocyanatobenzoate in acetonitrile (ACN) by using microwaves (MW, 200W) at 70°C to give the desired compound, i.e. M1.
28
Retrosynthesis pathway prediction for M1.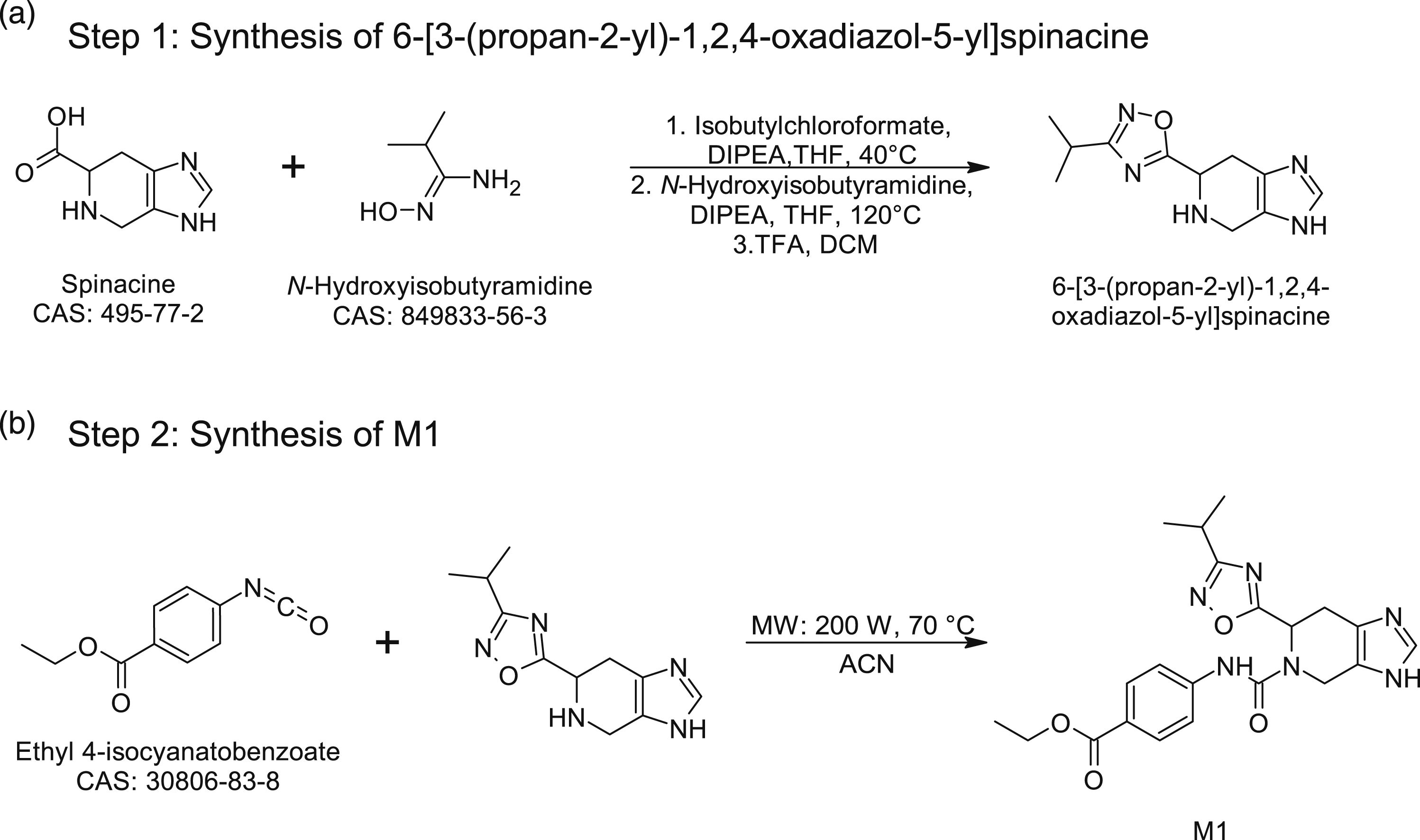
The retrosynthesis pathway prediction for M2 was carried out based on a literature survey of known chemical reactions, and the proposed synthetic scheme obtained from the literature review is shown in Figure 4. Step 1 (Figure 4a) is the condensation of 3-pheylpropanoic acid with 1-(4-aminophenyl)piperidin-4-ol by using N,N´-dicyclohexylcarbodiimide (DCC) and dimethylaminopyridine (DMAP) in dimethyl sulphoxide (DMSO) at 0°C, to give the phenylpropanamide derivative.
29
Step 2 (Figure 4b) is the N-alkylation of (1-(pyrimidin-2-yl)piperidin-3-yl)methanamine with the phenylpropanamide derivative obtained in Step 1 with the use of a manganese catalyst complex, MnBr(CO)2[NH(CH2CH2P(CH(CH3)2)2)2], and potassium tert-butoxide (t-BuOK) in toluene to give the desired compound, i.e. M2.
30
Retrosynthesis pathway prediction for M2.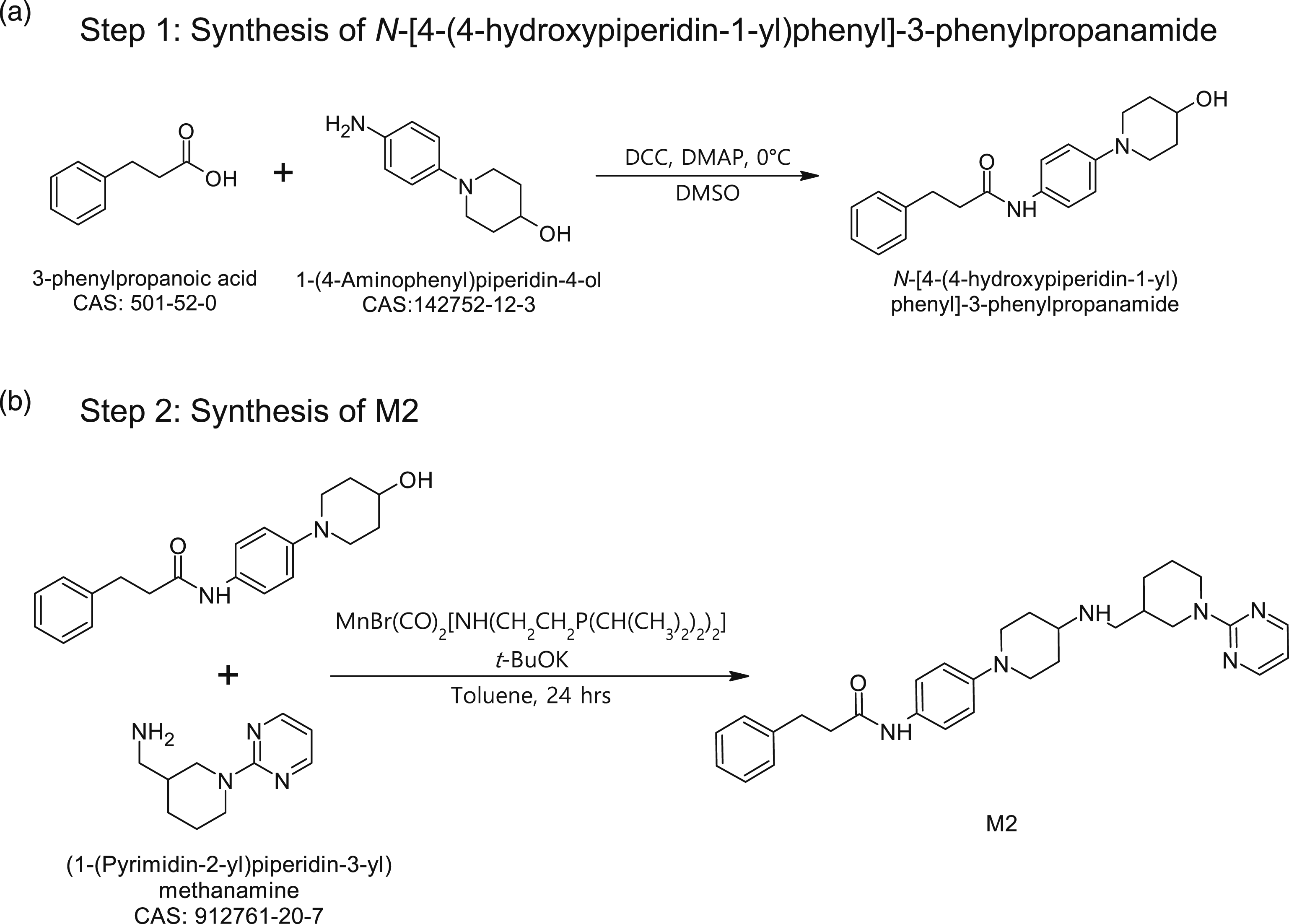
Conclusions
Strategies that target MtSK as new anti-TB therapies show potential but remain under-explored. Toxicoinformatics is an important element in the drug development process, and the emergence of structure-based predictions and read-across studies, as well as the integration of artificial intelligence in such investigations, has considerably accelerated drug discovery and development. Moreover, these new approaches confer economic, time-saving and ethical benefits, as they can be used for the initial evaluation of compounds and thus ensure that conventional in vivo studies are carried out only if the initial in silico results are promising. The present study, which employed a range of commonly used open-source in silico tools to predict the safety and efficacy of potential MtSK inhibitors, has led to the identification of three new MtSK ligands as possible drugs for the therapeutic management of TB. More in-depth analyses of these potential MtSK inhibitors are warranted in future studies.
Supplemental Material
Supplemental Material - Toxicological Profiling of Potential Shikimate Kinase Inhibitors Against Mycobacterium tuberculosis
Supplemental Material for Toxicological Profiling of Potential Shikimate Kinase Inhibitors Against Mycobacterium tuberculosis by Ashish Jhangiani, Vandana Panda, Aanchal Sukheja, Sneha Thomas, Piyush Dusseja, Siddhartha Pandya and Anand Chintakrindi in Alternatives to Laboratory Animals
Supplemental Material
Supplemental Material
Supplemental Material
Footnotes
Acknowledgment
The authors thank Prof. Urmila Joshi for her valuable advice to help improve the manuscript.
Author contributions
AJ contributed to the conceptualisation of the idea, literature review, drafting of the manuscript, and generation and procurement of the raw data. VP contributed to the conceptualisation of the idea, inferencing of the raw data, and writing and revision of the manuscript. AC contributed to the prediction of synthesis schemes for the molecules. SP contributed toward the molecular dynamic simulations performed for the three molecules and molecular docking analyses performed for the 30 molecules. AS, ST and PD contributed toward the procurement of raw data and literature review. All authors reviewed and approved the final manuscript.
Declaration of conflicting interests
The author(s) declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.