
Abstract
The 2025 joint British and European Society of Toxicologic Pathology Congress commenced with a thought-provoking keynote lecture detailing the history, learnings, and limitations of the 2-year rodent bioassay. Animal carcinogenicity studies and the implementation of the Ames test for mutagenicity have enhanced our understanding of carcinogenesis, toxicokinetics, metabolism, and carcinogenic modes of action. These learnings have challenged the relevance of the 2-year bioassay for the determination of human risk and have raised questions about the need for its continued use. The first session was dedicated to carcinogenesis and Best Practice concepts with three presentations providing an introduction and general overview of rodent carcinogenicity studies: a toxicologist’s perspective on the complexities of carcinogenicity studies pertaining to study design, data interpretation, and determination of human safety relevance; an experienced pathologist’s overview regarding the intricacies and challenges of histopathological evaluation of carcinogenicity studies, the harmonization of nomenclature, and the data interpretation challenges; and a CRO (contract research organization) pathologist’s perspective on the evolution of carcinogenicity studies with an emphasis on peer-review nuances. A case study presentation on hepatocellular foci in rodents advocated for the need to record all foci of cellular alterations with recommended implementation of size/lower threshold criteria for small lesions.
The 2-Year Bioassay: What We Have Learned
Keynote Speaker: Samuel M. Cohen, MD, PhD
The 2-year rodent bioassay evolved from animal studies in the 1950s and 1960s, with standardization in the 1970s and continuing to the present. Standardization has included husbandry issues, diet, and statistical approaches, but most importantly, histopathology classification. The results of the bioassay have greatly enhanced our understanding of carcinogenesis and have led to the identification of several key molecular events, including the identification of several nuclear receptors involved with carcinogenesis, at least in rodents, such as constitutive androstane receptor (CAR), peroxisome proliferator-activated receptor alpha (PPARα), aryl hydrocarbon receptor (AHR), estrogen receptor (ER), progesterone receptor (PR), and androgen receptor (AR). Delineation of carcinogenesis as a multi-step process also occurred from animal studies. The distinction between genotoxic and non-genotoxic carcinogens began with studies by Ames in the 1970s. Non-genotoxic carcinogens rely on increased cell proliferation as their mode of action. Insights into metabolism and toxicokinetics have also been gained from animal studies. The 2-year bioassay uses the maximum tolerated dose (MTD) as the highest dose. However, this results in toxicity, which can be important in contributing to the development of tumors, which may not be relevant to human exposures. To avoid this issue, incorporation of toxicokinetics is increasingly being utilized to establish a kinetically based maximum dose (KMD). Detailed mechanistic research has identified numerous modes of action responsible for carcinogenesis in rodents, many that are not relevant to human cancer risk. The 2-year bioassay has provided the foundation for considerable progress in establishing best practices for performing all animal studies, not just 2-year bioassays, and in our understanding of carcinogenesis. Nevertheless, it is time to discontinue its use and utilize what has been learned.
Navigating the Complexities of Carcinogenicity Testing: A Toxicologist’s Perspective
Helen-Marie Dunmore, MS, MRSB, ERT
The objectives of nonclinical carcinogenicity testing are to identify the tumorigenic potential in animals and to assess the translational risk to humans. The practice of requiring carcinogenicity studies in rodents was instituted for pharmaceuticals and other products that were expected to be administered continuously for at least 6 months or where dosing in an intermittent manner leads to total lifetime exposure of at least 6 months. As per the revised International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) S1B guidance for medicines development, the value of conducting in vivo carcinogenicity studies, typically for small molecules, derived from nonclinical investigations (key biologic, pharmacologic, and toxicologic information) may lead to the need for 2-year carcinogenicity studies in mice and/or rats. Alternatively, a waiver request based on a weight of evidence (WoE) approach, for one or both in vivo studies, may be acceptable to regulatory authorities. More recently, a 6-month study in transgenic mice for pharmaceuticals may be conducted instead of the 2-year mouse bioassay. The nonclinical carcinogenicity assessment strategy (CAS) in the revised ICH S1B Addendum is important not only in considering whether to perform carcinogenicity studies but also for interpreting study outcomes with respect to relevance for human safety.
Practical aspects of designing GLP-compliant carcinogenicity assays in rodents to meet regulatory expectations (medicines and chemicals) for product approval, Food and Drug Administration (FDA) and European Medicines Agency (EMA) regulatory interactions for study protocol approval, and when carcinogenicity study waivers may be appropriate were discussed.
A case study was also presented that highlighted the importance of setting up an expert pathology working group (PWG) and how to navigate discordance in Benefit:Risk assessment between the FDA and EMA. The example discussed potential carcinogenicity of lorcaserin (a selective serotonin 5-HT2C receptor agonist that reduced body weight) for a weight loss indication New Drug Administration (NDA) in the United States and Marketing Authorization Application (MAA) in the European Union.
Case Study: Hepatocellular Foci in Rodents: Lowering the Threshold for Recording
Leslie Bosseler, Dipl ECVP, DVM
Hepatocellular foci of cellular alteration (FCA) in rodents are putative preneoplastic lesions that are sporadically encountered in (sub)chronic toxicology studies. The morphology of FCA can vary depending on their predominant staining characteristic (basophilic, eosinophilic, or clear cell) on H&E staining, their reactivity to IHC cell markers (eg, γGT, GSTP), and their size. While large FCA are easily recognizable, smaller ones tend to be overlooked or not recorded. Also, recording criteria, especially for the lower threshold are not defined. Yet, not recording FCA in short-term studies may lead to missed opportunities to flag potentially carcinogenetic compounds early.
We presented 3 cases where small FCA were present in 1- to 3-month rat studies. In the first case, very small foci were seen after 1 month, which developed to numerous foci after 6 months and carcinomas after 1 year. In the second case, small foci were seen after 3 months, which developed to a 100% incidence of foci after 6 months. In both these cases, the FCA were originally not recorded by the study pathologist during the 1- or 3-month study. The third case showed a spectrum of FCA morphologies during a 3-month study of a known carcinogen (TCDD).
Using these cases as examples, we advocated for the recording of all FCA, even in studies <6 months and the implementation of size/lower threshold criteria for small lesions. In case of diagnostic uncertainty or when no consensus can be reached during the peer review, the use of an additional IHC marker such as GSTP is strongly recommended.
Just Another Study Type? Challenges and Pitfalls with the Histopathological Evaluation of Carcinogenicity Studies
Matthias Rinke, DVM (Pathology), FIATP
Despite the fact that carcinogenicity studies are considered to provide only little value in the prediction of human risk, these 2-year bioassays are still performed in laboratory rodents to determine whether the “life-long” exposure to a substance, usually a chemical or drug, bears a carcinogenic potential for humans. Various international guidelines regulate their execution, but there are still distinct differences between the assessment of agrochemicals and pharmaceuticals, for example, concerning study duration and international acceptance. While the Tg rasH2 mouse model is now accepted as a second species for pharmaceuticals, genetically engineered mice are not yet accepted for other chemicals. As an example for an outdated practice, Dr Rinke pointed also out to the “Delaney Clause” of the FDA which since 1958 bans food additives found to cause or induce cancer in humans or animals as indicated by testing and has been criticized since decades. 2 In general, carcinogenicity studies are performed at the end of a development phase and might be conducted after the registration of a compound. Thus, the outcome of their assessment is often on the critical path to registration.
While in former years many companies performed and evaluated their studies in-house, they are now, especially in the pharmaceutical world, mostly entirely sourced out to a CRO (Contract Research Organization). A poll among the audience during the session revealed that approximately 50% of the 85 voters had evaluated one or more carcinogenicity studies by themselves, 30% of which had also performed a peer-review. Interestingly, 18% of the responders have performed a peer-review on a carcinogenicity study without the experience of evaluating a carcinogenicity study as the primary pathologist. Finally, nearly a third of the voters (32%) answered that they have no experience with this type of study.
These results show that there might be a lack of experience of the pharmaceutical companies’ pathologists with reliance on the qualifications of CRO pathologist colleagues. A thorough peer-review by a company’s pathologist therefore is highly recommended not only to prove the quality, but also to learn and gain experience on species and strain-specific lesions. Education Seminars as provided, for example, by the RITA group (https://reni.item.fraunhofer.de/reni/public/rita/) might help in this. Moreover, good communication between the company and CRO pathologists with knowledge of the compound profile is imperative.
Harmonization progress has been made with the introduction of commonly accepted organ trimming guides and internationally standardized nomenclature, with the use of common diagnostic criteria (INHAND). Nevertheless, the distinction between hyperplastic and neoplastic lesions, benign or malignant classifications, and appropriate grading systems can be difficult. Special techniques such as immunohistochemistry may be necessary for neoplasm classification to avoid the use of terms such as carcinoma, sarcoma, or even tumor NOS (not otherwise specified). The application of an appropriate grading system for preneoplastic lesions is another topic to be discussed. For example, a focal hyperplasia of the pars distalis of the pituitary gland which, according to the criteria, is close to an adenoma should never be graded as a mild or moderate finding. Moreover, higher grades will allow the peer-reviewing pathologist to identify such “borderline” cases more easily. Reading a carcinogenicity study can be a daunting task because of the large number of animals. There are no common recommendations on how to evaluate such a study except the more general guideline on toxicologic histopathology. 1 Tissues may be evaluated animal-by-animal, which affords an encompassing overview of an animal’s complete health status. An organ-by-organ technique allows more focused attention to changes and helps with grading consistency. Making both methods applicable requires special preparation. Diagnostic drift is possible, and alternating evaluation of animals from different treatment groups is therefore necessary. Decedent animals may be examined in advance to determine the causes of death. An example how to prepare and evaluate a carcinogenicity study is given in Table 1.
Recommendations for preparation and evaluation of a carcinogenicity study.

Assessment and interpretation of the results from a carcinogenicity study can be challenging for both the study and peer review pathologist. Quality of slides starts with careful necropsy, and histotechnique is crucial. Missing parts of an organ (eg, adrenal medulla or smashed pituitary glands) might negatively influence statistics (Figure 1). A recurring discussion is given to the extent of the evaluation: Some guidelines require only the investigation of animals from the control and high dose groups, decedent animals of the interim groups as well as all masses observed at necropsy. To Dr Rinke’s opinion this approach saves money and resources only on the first view. Later necessary work, for example, due to unexpected findings or additional requests from authorities bears the risk of even higher expenses and delays in the registration process.
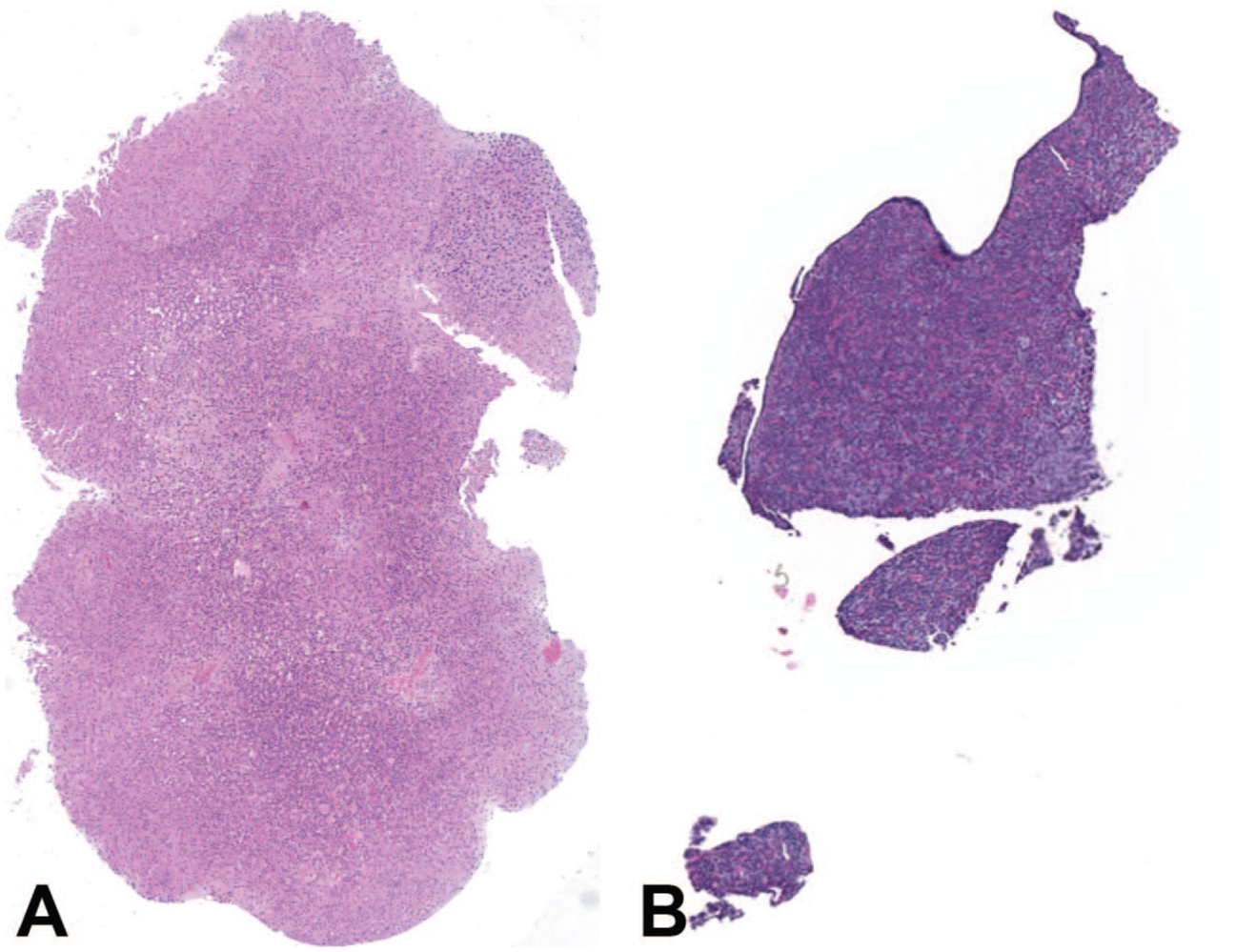
Examples of bad slide quality: (A) adrenal gland without medulla left and (B) damaged pituitary gland right with missing pars intermedia and pars nervosa.
Statistical analysis is typically not available prior to the peer review but may be essential for data interpretation. Historical control data may be fundamental for the justification of data interpretation but misuse has to be strictly avoided.
Not only these, but several other factors that may have an impact on the histopathologic evaluation of a carcinogenicity study were discussed.
Carcinogenicity Studies: A CRO’s Unique Perspective of Peer Review and Beyond
Dr Torrie A. Crabbs, DVM, DACVP
Dr Crabbs provided a unique perspective on the evolving landscape of carcinogenicity studies focusing on pathology peer review. Drawing from decades of Contract Research Organization (CRO) experience, Dr Crabbs provided a basic overview on the historical development of the pathology peer review (PPR) process, to include EPL’s original involvement with standardization and implementation of peer reviews and the emergence of pathology working groups (PWGs), particularly through its early collaboration with the National Cancer Institute and the 2-year carcinogenesis bioassay testing program. These efforts ultimately contributed to the development of a thorough approach to assess the quality of the pathology review process and served as the foundation for the Pathology Quality Assurance Program.
PPRs are specialized reviews that typically take place following completion of the initial review by the study pathologist (SP) and involve examination of a subset of tissues and diagnoses by a reviewing pathologic (RP).
The purpose of a PPR is to improve the accuracy and quality of the pathology data and narrative and to increase confidence in that data by confirming target organs, identifying any potentially missed targets, ensuring use of consistent diagnostic terms within a study and, possibly, across studies, ensuring harmonization of nomenclature and diagnostic criteria, both intra-organizationally and globally through initiatives like INHAND/SEND, confirming NOELs/NOAELs, and ensuring that data meets regulatory agency requirements, when applicable.
It is important to note that PPRs are not intended to be a complete re-read of a study; they include a concentrated review of a defined subset of slides/diagnoses. PPRs should not be blinded re-examinations; it is critical the RP be aware of all original diagnoses and dose groups, in addition to any relevant animal and histopathologic data, such as clinical observations, body/organs weights, clinical pathology changes, and histopathologic findings from shorter term studies that could impact interpretation of the study. PPRs are not intended to serve as a performance review of the SP; they are to improve the quality and confidence of the study data. PPRs should be handled in a professional manner and used as opportunities for training and education, not belittlement. In addition, PPRs do not generate a second set of data; any data generated during the peer review is considered notes with the signed pathology report serving as the raw data.
Informal PPRs, second opinions from colleague(s) on one of more slides/diagnoses, are extremely common and typically conducted without formal documentation, whereas formal PPRs are documented reviews that should appear within the study protocol/amendments. Protocol requirements depend on when the decision was made to conduct the PPR If made prior to the start of the study (ie, during protocol design), the plan should be included in the protocol with specifics added later as an amendment. If the protocol was already finalized, a protocol amendment is required.
Ideally, PPRs are contemporaneously conducted prior to signing the final pathology report, in which case changes to the data/narrative can be directly incorporated prior to signing. If the pathology report has already been finalized, a retrospective PPR is conducted, and the report amended to reflect any post-PPR changes in data; conduct of the PPR needs to be added to the protocol as an amendment.
In general, PPRs are performed on a case-by-case basis taking into consideration not only the importance of the decisions that will be made based on the study, but also the skill/experience of the pathologist and any possible regulatory requirements. Generally speaking, PPRs are not required by Federal Agencies but, they are considered a best practice standard and are highly endorsed, especially by the FDA. There are two instances when PPRs are required by Federal Agencies. The Environmental Protection Agency (EPA) requires a PPR and PWG of a compound when it is being re-registered and the EMA—European Medicines Agency requires complete review of 10% of each dose group has and at least 10% of neoplasms.
Despite the lack of federal requirements for PPRs for most studies, increased consideration should be given to carcinogenicity studies due to their large size, extended length of review time, increased possibility for diagnostic drift and inconsistencies in terminology, and general lack of experience/familiarity of pathologists with findings in aged rodents.
An RP can be from within the sponsor's organization (ie, an internal reviewer) or from outside the sponsor's organization (ie, an external reviewer) Internal reviewers likely have more extensive knowledge of the compound but could have a potential for bias, whereas external reviewers may be able to provide more objectivity, but will likely be less familiar with the compound, it’s mechanism of action, and any findings from earlier studies or associated with similar compounds.
PPRs can be conducted onsite at the SP's test facility or remotely at the RPs facility or home office. Traditionally, on-site PPRs were preferred, as all study materials were readily available, and differences of opinion could be directly discussed with the SP and immediately reconciled. In addition, reduced distractions can result in a more focused and expedited review. Remote PPRs have increased in popularity, particularly since the Covid-19 pandemic eliminating the need for travel. However, shipment of glass slides or conversion of slides to digital whole slide images (WSI) is necessary, creating additional risks and complications, such as loss or breakage of glass slides or difficulties implementing digital imaging processes.
What is reviewed during a PPR and how the review is conducted will vary from organization to organization and study to study but should be clearly defined in SOPs. At EPL, PPRs typically include: review of any identified or suspected target organs from all animals in all dose groups to confirm targets and assure appropriate use of terminology; review of all tissues from a specified percentage of control and high-dose animals (complete review animals) to establish baseline and threshold levels and identify any potentially missed target organs; proliferative lesions from all animals in all dose groups, to include neoplasms, hyperplastic findings, and foci of alteration; and all tissues from at least a subset of early death animals to determine any relationship with test article exposure.
Diagnostic differences between the SP and RP can occur for multiple reasons. The most common are diagnostic drift and duplication of diagnoses, particularly in carcinogenicity studies. Diagnostic drift occurs when there is “drifting” of the SP's original criteria for a particular diagnosis over time, whereas duplication occurs when multiple terms are used to identify the same finding. Additional sources for diagnostic differences include unfamiliarity with a lesion, differing criteria for tumor classifications, and variations in threshold levels, particularly in non-neoplastic aging findings.
Upon completion of the review, it is essential that the SP and RP reconcile any and all differences. While typically this is easily accomplished, additional expert opinions and/or pathology working groups (PWGs) may be necessary.
Key differences between peer review of carcinogenicity and subchronic studies, common pitfalls encountered during the review process, the shift from traditional to digital review formats, and lessons learned over time were briefly highlighted with insight into how peer review practices have adapted to adjust to technological advancements and regulatory expectations.
Conclusion
The use of animal carcinogenicity studies has resulted in enhanced understanding of carcinogenesis; however, genotoxicity studies, toxicokinetics, and mode of action studies are now routinely applied in preclinical safety assessment. These enhanced learnings, in addition to challenges with resource-intensive study designs, complex histopathological evaluations, and difficult data interpretation, question the need for continued mandatory use of animal carcinogenicity studies to identify tumorigenic translational human risk.
Footnotes
Acknowledgements
The authors thank the joint British Society of Toxicologic Pathology (BSTP), European Society of Toxicologic Pathology (ESTP) Scientific Organizing Committee, and the organizer of the Session 1, Jonathan Carter, Ute Bach, Laetitia Elies, Heike Antje Marxfeld, Antonia Morey-Matamalas, Shelley Patrick (chair of the session), Dirk Schaudien, and Jacqui Stewart, for their support of this program. They would also like to thank the staff of BSTP and ESTP for organizational support in producing the materials and organizing the meeting space.
Author Contributions
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.