
Abstract
Nonhuman primates (NHPs) have been and remain a highly valuable animal model with an essential role in translational research and pharmaceutical drug development. Based on current regulatory guidelines, the nonclinical safety of novel therapeutics should be evaluated in relevant nonclinical species, which commonly includes NHPs for biotherapeutics. Given the practical and ethical limitations on availability and/or use of NHPs and in line with the widely accepted guiding “3Rs” (replace, reduce, and refine) principles, many approaches have been considered to optimize toxicity study designs to meaningfully reduce the number of NHPs used. Standard general toxicity studies usually include four groups of equal size, including one group of vehicle control animals. Here, we describe an approach to achieve an overall significant reduction in control animal use, while also resolving many of the issues that may limit application of fully virtual control animals. We propose in Good Laboratory Practice (GLP)-compliant toxicity studies to maintain concurrent control group animals for the in-life phase of the studies, but to limit euthanasia to a subset of control animals. The nonterminated control animals can then be returned to the facility colony for reuse in subsequent studies. The proposed study design could lead to a 15% to 20% reduction in NHP usage. The scientific, logistical, and animal welfare considerations associated with such an approach and suggested solutions are discussed in detail.
This “Points to Consider” article is a product of a working group of the Scientific and Regulatory Policy Committee (SRPC) of the Society of Toxicologic Pathology (STP). It has been reviewed and approved by the SRPC and Executive Committee of the STP, but it does not represent a formal Best Practice recommendation of the Society; rather, it is intended to provide key “Points to Consider” in designing nonclinical studies or interpreting data from nonclinical toxicity and safety studies intended to support regulatory submissions. The points expressed in this document are those of the authors and do not reflect the views or policies of their employing institutions. Readers of Toxicologic Pathology are encouraged to send their thoughts on these articles or ideas for new topics to the Editor.
Introduction—The Responsible Use of Nonhuman Primates in Biomedical Research, Limitations, and Replace, Reduce, and Refine Considerations
Nonhuman primates (NHPs) have been and remain an invaluable animal model for pharmaceutical drug development, particularly for biotherapeutics where there is commonly no other pharmacologically relevant nonclinical species. However, the use of an NHP species, rather than other test systems (including in vitro or transgenic animal alternatives), must be scientifically justified due to ethical considerations and practical limitations on NHP availability.10,44 The COVID-19 pandemic brought the fragility of the NHP supply chain into sharp relief due to the immediate and increased NHP need to support COVID-19-directed research programs coupled with an NHP export ban from China.54,55 This acute disruption in NHP availability spurred the publication of a Food and Drug Administration (FDA) guidance in February 2022 to reduce the demand on NHPs. 23 While this guidance has since been rescinded with the expiration of the COVID-19 Public Health Emergency, there remains industry-wide focus to further replace, reduce, and refine (3Rs) NHP use.1,10
As NHPs are commonly used in general toxicity studies, 10 opportunities to optimize and streamline toxicity study designs could have a meaningful impact on NHP use. Several approaches have been considered, including use of a single sex, exclusion of vehicle control groups, omission of recovery groups, and reduction in number of dose groups. 1 Regarding minimization of control animals, one proposed approach is to completely eliminate concurrent control groups (CCGs) in toxicity studies and replace them with virtual control groups (VCGs) that are generated using data from vehicle control animals that were evaluated in prior studies.26,29,53,58 This approach has the potential to reduce the number of animals in a standard general toxicity study by about 25%. However, there are several noteworthy issues that may hinder the implementation of the VCG approach.3,49 Even for relatively homogeneous rodent strains, VCGs must be assembled from recent (within 2-5 years) control animal data to be relevant for comparison with current studies, meaning complete replacement of CCGs is not feasible.27,29 In addition, even with the pre-selection of “comparable” animals to build VCGs, a considerable reduction in sensitivity for the identification of test article-related changes in clinical pathology parameters has been noted when using VCGs.3,28,29 Furthermore, the logistical challenges of assembling and reporting clinical pathology and anatomic pathology data from a virtual control database have yet to be solved. 27 For these reasons, other possible approaches to refine study design, such as minimizing the use of recovery groups, may be more tenable in the short term. 50
In this article, we describe an alternative approach to achieve a significant reduction in control NHP use that resolves many of the issues related to VCGs. The proposal is to maintain CCG animals for the in-life phase of the studies, but to limit euthanasia to a subset of control animals and to return the remaining noneuthanized control animals to the laboratory animal colony at completion of the in-life phase for reuse in a subsequent study. With the inclusion of CCG animals for the in-life phase, all nonterminal endpoints consistently have concurrent control data for evaluation of test article-related effects. The euthanasia of a subset of control animals will not only provide terminal endpoints (such as anatomic pathology data) from concurrent control animals, but also contribute to the maintenance of the historical control pathology database. The terminal data points from the euthanized concurrent controls, although limited, will increase confidence in the pathologist’s interpretation and overall study conclusions. The scientific, logistical, and animal welfare considerations associated with such an approach and suggested solutions are discussed in detail. Finally, the design principles of a validation study to demonstrate the adequacy of the approach to characterize test article-related toxicities and assess human risk are outlined. The validation study will be published in a separate manuscript.
Proposal to Limit Euthanasia of Control Animals in Good Laboratory Practice Safety Evaluation Studies
Based on current recommendations, the standard experimental design for Good Laboratory Practice (GLP) toxicity studies in NHPs generally includes four groups (one vehicle control group and three test article-dosed groups) of 3 to 6 animals/sex/group, for a total of 24 to 48 animals in each study (Table 1). The variability in the number of animals per study depends on several parameters, including the duration of the toxicity study, the potential inclusion of a recovery arm, and/or the clinical phase that the toxicity study is intended to support. However, it is important to emphasize that the absolute number of groups and animals per group is not clearly defined by regulatory guidelines,33,34 but driven by established norms based on best scientific practices6,14 or regulatory requests.
Number of NHPs generally used in GLP safety evaluation studies.
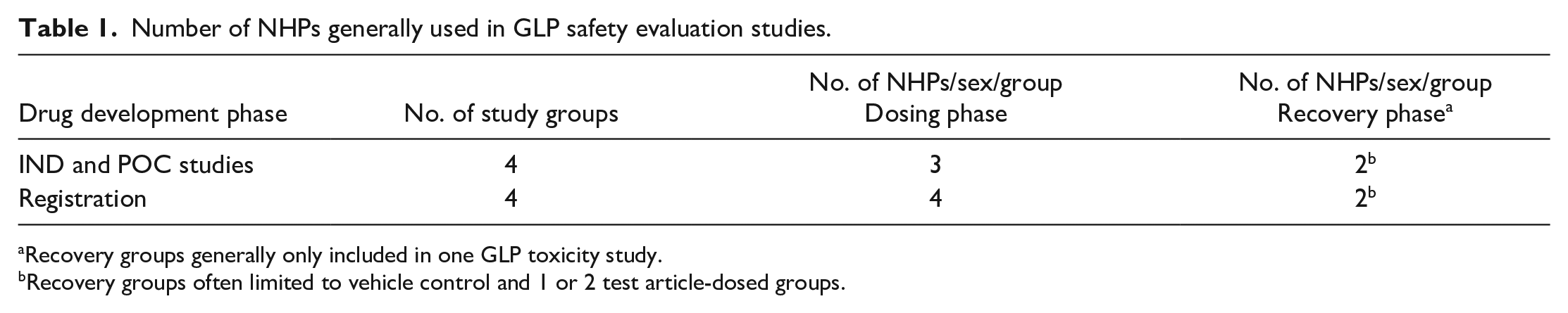
Recovery groups generally only included in one GLP toxicity study.
Recovery groups often limited to vehicle control and 1 or 2 test article-dosed groups.
Pertaining to the standard study design, there are at least 25% of animals in each study assigned to the control group, which usually corresponds to 6 to 12 control animals per GLP study. Control animals are generally dosed with the pharmaceutical vehicle, and they are subjected to rigorously identical experimental conditions and procedures as the test article-dosed animals. These control animals enable appropriate characterization of findings related to the vehicle, specific experimental procedures, husbandry practices, environmental conditions, and even intercurrent diseases/pathologies. When assessing findings in test article-dosed animals, control animals may enable better delineation of test article-related findings versus background/incidental changes and contribute to thorough characterization of the toxicity profile of the test article under evaluation.
As part of the global effort to reduce animal use for pharmaceutical drug testing and considering the limited supply and high cost of NHPs, our proposal is to reduce the euthanasia of control animals in GLP-compliant toxicity studies. Specifically, we are proposing that the standard number of in-study concurrent control animals be maintained per company and best scientific practices, but that the euthanasia of control animals be limited to one animal/sex in each study. This proposed study design could lead to approximately 15% to 20% reduction in NHP usage.
The Standard Number of Control Animals Is Maintained for the In-Life Phase(s) of the Studies—Rationale and Benefits
The availability of a full complement of control animals during the in-life phase(s) of the study enables comprehensive documentation of all nonterminal endpoints (including in-life observations, clinical pathology, electrocardiography, ophthalmology, circulating biomarkers, immunophenotyping parameters, etc.) to assist in the characterization of test article-related findings. These nonterminal endpoints in control animals provide valuable data, particularly for the monitoring of animal health and the evaluation of vehicle and procedural effects, as illustrated hereafter. Control animal data can indeed provide critical support to the identification of intercurrent pathologies in NHP studies (e.g., spikes in liver enzymes due to viral infection, gastrointestinal disease due to infectious agents) that, although infrequent, may represent a real interpretation challenge in the absence of concurrent control animals.36,47 Identification of test article-related clinical pathology findings often also requires availability of clinical pathology data from concurrent controls, particularly in the case of novel biomarkers that do not have established reference ranges and/or may not be available when selecting for virtual control animals. Although clinical pathology results are primarily interpreted in NHPs by comparing dosing phase data with individual baseline (pretest) values, the assessment also takes into consideration the changes in control animals. This additional comparison with control animal values enables identification of procedural, environmental, and age-related changes that could appear to be test article related without confirmation of a similar change in concurrent control animals. Furthermore, reference intervals and historical control data (HCD) alone should not be used to assess test article relationship of clinical pathology results in safety assessment studies since they do not reflect the specific conditions of the study of interest.30,31,57 The assessment of clinical pathology data in concurrent control animals is the best way to appreciate the influence of the vast number of intercurrent factors on results in a given study, and to conclude whether or not changes are likely to be test article related.4,5,22,28,56 Evaluation of other endpoints, such as electrocardiography and ophthalmology, are also greatly facilitated by the availability of concurrent control animal data.
Euthanasia of Control Animals Is Limited by Default to One Animal/Sex in Each Study
Our proposal of including euthanasia of one control animal/sex by default in each study enables availability of anatomic pathology data from a limited number of concurrent control animals. This limited anatomic pathology data set from concurrent controls serves a dual purpose:
1. Serve as in-study concurrent control data to increase confidence in evaluation and interpretation of the pathology data.
In-study concurrent control anatomic pathology data facilitates identification of changes related to extraneous variables, such as the vehicle, the dosing procedure, and also tissue processing (e.g., tissue collection, sampling, and staining variations).8,46 This limited anatomic pathology data set may be complemented by access to historical control databases that preferentially include digital whole slide images (WSIs). This will be further discussed below (see the “Scientific and Regulatory Considerations: Study Conduct, Interpretation, and Reporting” section, point 4).
2. Contribute to the maintenance of a historical control database that necessitates a continuous stream of new data.
Several guidelines recommend a narrow window of HCD, usually in the order of a 2- to 5-year period, to be considered for toxicological evaluations, whether in the context of general toxicity studies, carcinogenicity studies, or reproductive toxicity studies. 39 The euthanasia and evaluation of one control animal/sex for anatomic pathology endpoints will contribute to the maintenance of a relevant historical control database.
Furthermore, due to the considerable genetic heterogeneity of NHPs, potential differences in animal husbandry and pathogen exposure prior to initiation of toxicity studies, and the relatively low number of animals per dose group in toxicity studies, it is important to bear in mind that it is not possible for a single CCG to include the full spectrum of potential background lesions.13,15,37 As a result, anatomic pathologists often rely on their experience, HCD, and published background data in addition to the data from a CCG to reach a conclusion regarding the test article relationship of microscopic findings and to render the final pathology interpretation.12,13,16,18,39
In addition, the exclusion of control NHPs from non-GLP studies, such as dose-range finding toxicity studies and investigative studies, is already an established practice at some Testing Facilities, where such studies may be conducted without concurrent control animals or with nonterminal control animals. Although these studies do not have the same objectives as regulated studies and may not require the same stringent evaluation criteria, they still provide the opportunity for anatomic pathologists to comment on their experience and confidence in evaluating toxicity studies without concurrent controls. An informal survey of anatomic pathologists involved in these studies indicated that they are comfortable with the anatomic pathology interpretation, relying on their personal experience and available historical data (personal communications from authors of this article). Pathologists also indicated that the advent of digital pathology and readily available digital WSIs allows for review of a greater number of control tissues that greatly enhance their confidence in interpretations made without concurrent controls (see the “Scientific and Regulatory Considerations: Study Conduct, Interpretation, and Reporting” section, point 4).
Points to Consider for Effective Implementation of the Proposal
Scientific and Regulatory Considerations: Study Conduct, Interpretation, and Reporting
Point 1: The control animals scheduled for necropsy are allocated during protocol development
The current standard toxicity study design includes necropsy and anatomic pathology evaluation (organ weights, macroscopic findings, and microscopic observations) of all in-study concurrent controls. Our proposal is to reduce euthanasia of in-study concurrent controls per protocol to one animal per sex in each study, even if there are multiple necropsy time points, such as interim, terminal, and recovery necropsies. In that situation, the preferred option will be to euthanize one control animal/sex at the end of the dosing phase (terminal necropsy), which is usually the pivotal phase. Further support for our proposal is provided by a recent article from a diverse panel of drug development scientists expressing the opinion that there is not a need to euthanize control animals in the recovery arms of nonrodent toxicity studies. 35
We recommend that one control animal per sex be identified prior to the initiation of dosing that will be necropsied at the end of the dosing phase (or another time point if considered more relevant). Specifically, the selection of control animals for necropsy should not be influenced by clinical observations or clinical pathology findings during the in-life phase(s).
Implementation of this recommendation will help prevent selection bias of the control animals identified for necropsy and aligns with randomization guidelines for the design of toxicity studies. 25
This default proposal of limiting euthanasia to one control animal per sex can be adjusted, if needed, based on specific study and/or testing facility considerations, such as unusual vehicle/formulation, atypical route of administration, and/or unavailability of relevant HCD, which may justify euthanasia of additional control animals to enable confidence in the interpretation.
Point 2: Unexpected findings during the course of a study may lead to the decision to increase the number of euthanized control animals
In-life observations (clinical signs, body weight, and food consumption) and other nonterminal endpoints, such as interim clinical pathology data should be reviewed as feasible prior to necropsy to enable a final decision regarding control animal euthanasia. Unexpected findings potentially related to the health status of the animals, the tolerability of the experimental procedures, or other extraneous variables may justify the need to necropsy more than one control animal per sex. As an example, one author of this article shared their experience from a recently conducted GLP-compliant NHP study in which there were gastrointestinal clinical signs and moribundity that led to euthanasia of individual NHPs in both the control and test article-dosed groups. The in-life findings were strongly suggestive of intercurrent gastrointestinal disease and the recommendation in such a case would be to euthanize all the vehicle control animals to enable appropriate characterization and interpretation of incidental versus test article-related findings and to prevent potential further transmission of the disease. To enable amendment of the study protocol in case of unexpected findings, we recommend that a pre-necropsy meeting involving the study team be consistently held a few days prior to necropsy to review available data and confirm the total number of control animals to be euthanized and necropsied.
Point 3. Organ weights are collected from the control animals that are euthanized
Although evaluation of organ weights is considered an integral component of toxicity studies, 52 organ weights are most valuable in rodent studies where there are commonly at least 10 animals/sex/group that are phenotypically fairly homogeneous. There are recognized challenges and limitations to organ weight interpretation in NHPs that arise from the low number of animals/sex/group, the inter-animal variability (related to age, body weight, and intrinsic elements), 2 and additional confounding factors, such as variable onset of puberty and thymic involution. Because of these factors, the value of organ weight data in toxicity studies in NHPs is recognized as limited relative to the information obtained from the microscopic examination.
We recommend that organ weights are still collected in the vehicle control animals that are euthanized. It is acknowledged that the lower number of concurrent control animals further limits the ability of the study pathologist to make comparisons, but the recorded values can be included in the testing facility historical control database. This database of organ weight values could be helpful in identifying test article-related organ weight changes when filtered for relevant variables.
Point 4: The limited in-study control anatomic pathology data are complemented by access to historical control databases and histology glass slides/digital images as needed
As alluded to previously, the availability of anatomic pathology data and specifically microscopic pathology data from one concurrent control animal per sex will provide valuable assistance to the pathologist for the evaluation, but will limit the range of background and incidental findings documented in control animals in the context of a given study.
This limited data set may be complemented by HCD from the testing facility. HCD are tabulated, pooled control data from studies conducted under similar conditions that are used to establish reference background data on spontaneous microscopic findings (including incidence and severity). Pathologists already commonly consult HCD when, for example, a rare or unusual finding only occurs in a test article-dosed animal. In this case, the reference to a larger historical control group is often beneficial for the interpretation of the finding. 38
We cannot stress enough the importance of robust HCD with defined criteria for consistent recording of background changes for the successful implementation of the proposal described in this article. It is imperative that testing facilities continue to improve the quality of their HCD. This includes exploring processes that can increase the consistency of pathologists’ thresholding, scoring, and diagnostic terminology of spontaneous findings. Of note is that most historical control database queries are filtered based on study duration. However, to compensate for the reduction of animals included in such databases, an improved approach may be to filter data based on the age of the animals at necropsy (i.e., 1-3 years, 3-5 years, and ≥ 5 years) rather than study duration. Indeed, such a practice could increase the amount of relevant data and provide more robust HCD. These concepts will be further discussed in an article that will report the results of the validation study described below (see the “Prospective Validation of the Proposal” section).
Alternatively, pathologists may favor reviewing histology slides from control animals from previous studies rather than relying only on the diagnoses provided in the HCD, given that, histopathology is by nature a largely descriptive and interpretive science. 17 Histology slide review from previous studies is greatly facilitated by the ongoing digital transformation of the pathology discipline, which has resulted in high-resolution scanning of glass slides to generate digital WSIs that can be stored and viewed on a computer screen. 51 This digital transition has indeed gained significant momentum over the last few years, thanks to remarkable advancements in scanner throughput, computer graphic processing power, display resolution, and storage capability. So now, in addition to access to HCD and glass slides from previous studies, pathologists in some laboratories can also review WSIs from control animals from previous studies, significantly increasing the pool of control tissues readily accessible and available for a confident identification (enabling side-by-side morphologic comparisons of tissues) and interpretation (setting perspective from reference/historical control animals) of study findings.17,32
In addition, WSIs facilitate consultations with colleagues on site or in other geographical locations and improve access to subject matter experts.
Therefore, a well maintained and regularly updated control data repository including digital WSIs will optimally support and enable the proposed study design changes that promise to reduce the number of control group NHPs that are euthanized in toxicology studies.
Point 5: The microscopic evaluation and/or peer review is performed by a qualified and experienced toxicologic pathologist
Microscopic evaluation is a very critical component of the toxicity characterization of new drugs to enable accurate assessment of potential risks to human health. Appropriate training and experience of study pathologists, along with integrity, have long been recognized as “vitally important in the assurance of high-quality histopathology” 17 and there are published global standards for proficiency assessment and recognition of qualified toxicologic pathologists.7,19 Studies with limited numbers of euthanized control animals might lead to additional interpretation challenges and will specifically benefit from the expertise of pathologists who are not only qualified, but also experienced with toxicity studies in NHPs. To not be too restrictive regarding study pathologist assignment, our recommendation is that either the study pathologist or the peer-review pathologist has at least 3 years of experience at a testing facility or test site involved in the pathology evaluation and interpretation of GLP NHP toxicity studies before being assigned to a study with a reduced number of euthanized control animals.
Point 6: The microscopic evaluation of GLP studies with reduced number of euthanized control animals is peer reviewed
The pathology peer review consists of the review of a subset of histology slides and pathology data by a second pathologist not directly involved with the primary evaluation. Although not a requirement from GLP regulations, pathology peer review is a best practice meant to ensure the accuracy and quality of the pathology diagnoses and interpretations. 43 Given that the current proposal of euthanasia of only one control monkey per sex in each study will significantly reduce the concurrent control pathology data available for interpretation, our recommendation is that these studies are consistently peer reviewed to strengthen the confidence in the overall interpretation. The peer review should follow current best practice recommendations 41 and FDA guidance document, 24 with contemporaneous peer reviews documented in the study protocol and peer-review statements included in the final study report.
Point 7: The pathology report only tabulates and includes anatomic pathology data from concurrent control animals
To facilitate pathology reporting and prevent re-evaluation of histology slides/WSIs from the control database, our proposal is that the report only includes anatomic pathology data from the concurrent study control animals. Control glass slides/WSIs from the historical repository reviewed by the study pathologist during the microscopic evaluation serve the same purpose as an informal consultation with a fellow pathologist, which is to enable confidence and accuracy in the interpretation. The specific control slides reviewed are not identified in the report and these informal “consultations” are not documented.
Point 8: HCD are referenced and included in an appendix of the pathology report if used for the interpretation
HCD provide information on spontaneous and incidental findings in control animals of the same species under similar experimental conditions. As discussed in the guidance documents of several agencies, HCD should preferably originate from the laboratory conducting the study and should come from studies conducted within two to five years prior to or after the study being evaluated.20,48 Since HCD quality is influenced by rigor, consistency, and accuracy in the recording of background changes, 38 we recommend that historical control histopathology data are peer reviewed to increase confidence in the usefulness of HCD for appropriate interpretation of study data. While HCD are usually well maintained and readily available at leading Contract Research Organizations (CROs), smaller organizations conducting GLP studies in NHPs may want to proactively develop comprehensive and searchable historical control databases to further enable implementation of the current proposal in their organizations. If HCD are used to support the interpretation of a study, the subset of HCD (for an organ and/or a specific finding) should be included in an appendix of the pathology report as is already common practice when referencing HCD. In addition, slight variation in terminology for similar findings among various studies can be combined for interpretation.
Ethical and Operational Considerations for the Reuse of NHPs
The proposal in this article to reduce euthanasia of control animals will only be successful if control animals are reused effectively with thoughtful considerations of ethical aspects and logistical concerns.
Although reuse of NHPs is already an integral component of some drug development studies, such as nonterminal pharmacokinetic/pharmacodynamic (PK/PD) studies, it is important to highlight the specific directives from different nations that guide or regulate animal reuse.
Point 9. Reuse of animals is covered in EU and US policies
The European Union Directive 2010/63/EU
21
on the “Protection of Animals used for Scientific Purposes” allows the reuse of animals as an acceptable approach to reduce the total number of laboratory animals used. The Directive states that the number of animals used could be reduced by performing procedures on animals more than once (. . .) However, the benefit of reusing animals should be balanced against any adverse effects on their welfare, taking into account the lifetime experience of the individual animal.
To enable appropriate assessment of the potential impact of procedures on the welfare of the animals, the Directive provides a framework for the severity classification of procedures (mild, moderate, and severe) performed in animals based on the degree of pain, distress, or lasting harm in individuals, and further includes specific examples of different types of procedures assigned to each of the severity categories. 21 Procedures usually performed in vehicle control animals in toxicity studies, such as vehicle administration by standard routes, administration of anesthesia, electrocardiogram (ECG) monitoring with noninvasive techniques, short-term (< 24 hours) restraint in metabolic cages, and so on, are all considered mild severity procedures with no significant impairment of the well-being or general condition of the animals. Based on the procedures performed in control animals in toxicity studies, there is no barrier to the reuse of the control animals from a large majority of studies. There are, however, toxicity studies where the routes of administration (e.g., continuous intravenous infusion, intraocular or intrathecal/intraspinal administration), the nature of the vehicle (e.g., cyclodextrin-based formulation), or even the duration of the study may justify euthanasia of the animals and preclude reuse.
In the United States, the Guide for the Care and Use of Laboratory Animals, or Guide, by the Institute for Laboratory Animal Research (ILAR) of the National Research Council is the most authoritative source of information on animal care and use. 45 Regarding animal reuse, the Guide specifically indicates that “reduction should not be a rationale for reusing an animal or animals that have already undergone experimental procedures especially if the well-being of the animals would be compromised.” The Guide primarily objects to the reuse of animals that have undergone procedures associated with impairment of well-being or general condition. This is in line with Directive 2010/63/EU and consistent with the welfare recommendations provided above.
Several professional organizations, including the American College of Toxicology, Innovation and Quality (IQ) in Pharmaceutical Development, and BioSafe, have also evaluated options for reuse of NHPs, specifically protein non-naïve animals or animals in PK/PD studies.11,40,42
Point 10. The final decision regarding reuse of animals belongs to the testing facility Institutional Animal Care and Use Committee and/or appropriate animal welfare body
The Institutional Animal Care and Use Committees (IACUCs) in the United States or other similar committees in other countries should discuss the merit of animal reuse from an animal welfare centric perspective.
The IACUCs oversee and evaluate all aspects of the institution’s animal care and use program to ensure ethical research and compliance with regulations and policies. Essential elements in IACUC adherence include protocol review, minimizing harm, and a benefit-to-burden analysis. The reuse of a control animal, which had previously been included in a toxicity study, will have to be reviewed by the IACUC in consultation with the attending veterinarian. The decision to reuse will be based on the health status of the animal and will take into consideration the cumulative harm from the previous procedures and the additional burden of reuse, per Directive 2010/63/EU and AALAS-FELASA report of harm-benefit analysis of animal experiments. 9
The recommendation would then be that when control animals, which fulfill the conditions for reuse, are reused in a subsequent toxicity study, they should be scheduled for euthanasia at termination of the study. Given the number of experiments that require euthanasia, there is no requirement to reuse the animals more than once to achieve the reduction goal, since other animals in subsequent studies can be identified for reuse. Control animals identified for reuse could also potentially be assigned to an internal pharmacokinetic colony instead of being enrolled in another toxicity study.
Point 11. An NHP reuse program is in place at the testing facility
For testing facilities performing NHP studies, it will be imperative to discuss and communicate the reuse proposal, ensure support from facility management, and determine capacity for NHP reuse. Clear lines of responsibilities will be needed to ensure adherence to the proposal and appropriate reuse of NHPs where feasible. The reuse program at each testing facility will require the involvement of the appropriate stakeholders (including NHP behavior expert, veterinary and animal care staff, and primate colony manager) and compliance with animal welfare guidelines as outlined previously.
Reuse programs may be easier to implement by companies that perform studies internally or that own dedicated NHP colonies at CROs. For others, reuse programs will benefit from the commitment and cooperation between sponsors and CROs, where, for example, control animals rotating off studies at a CRO could be proposed for use in a subsequent study for a different sponsor. This would likely require the sponsor to be willing to share information, such as vehicle exposure, dosing route, dosing regimen, and any other relevant animal health and use records from the reused control animals, while the CRO could be expected to split the animal cost between the 2 sponsors.
Of importance is that social grouping of NHPs should be planned prior to the start of the study to ensure compatible animals are regrouped at the end of the study and animals are closely monitored until group stability is confirmed. There are additional practical considerations that will need to be taken into account, such as, for example, the cost associated with holding animals and the potential need for additional health screening of animals between studies.
In conclusion, with the acknowledgment of animal welfare and operational considerations, we believe that the proposal to limit euthanasia and reuse control animals would achieve greater alignment with the principles of 3Rs of ethical animal use.
Prospective Validation of the Proposal
To demonstrate the scientific reliability of the approach, we have defined a framework for prospective validation of the proposal. The specifics and outcome of the validation exercise will be published in a separate manuscript.
The validation will be performed using toxicity studies in NHPs that include the current standard number of control animals. A wide range of studies based on duration, route of administration, control vehicle and test article characteristics, as well as the experience level of the study pathologists and/or peer-review pathologists, will be included in the validation exercise. Additional criteria, such as frequency of dosing and origin of NHPs will also be considered.
For studies included in the validation, the protocol may need to be amended to reflect the new workflow. The wording of the amendment could be as follows: To support reduction of control nonhuman primate termination (3Rs goal), a preliminary microscopic evaluation of the study will include only one control animal/sex along with the full set of animals dosed with the test article. A preliminary interpretation will be shared with the peer review pathologist. The additional terminal control animals will then be evaluated and a draft pathology report will be issued and sent to the peer review pathologist. Any differences in the interpretation between the two evaluations will be communicated to the peer review pathologist and discussed during the pathology peer review process. The objective of this two-step evaluation is to evaluate the impact that the reduction in control animal termination could have on the interpretation of the study.
The single control animal/sex selected for the preliminary evaluation will be identified prior to the first day of dosing, as discussed above. All in-life and clinical pathology data from all study animals will be provided to the study pathologist prior to the preliminary evaluation. Following the completion of the microscopic evaluation, an abbreviated memo style report will be issued by the study pathologist and sent to the peer-review pathologist. The study pathologist will then evaluate the remaining controls. Once the microscopic evaluation is complete, a draft pathology report will be prepared and sent to the peer-review pathologist along with the glass slides (or WSIs).
The consensus meeting between the study pathologist and the peer-review pathologist will address any differences in interpretation between the preliminary evaluation and the evaluation including the full set of control animals. This process will limit bias for both the study pathologist and the peer-review pathologist. Following the consensus, the study pathologist and peer-review pathologist will complete a survey addressing the outcomes of the 2-step evaluation and any changes to the study interpretation that were identified. The focus will be to identify any differences in target organ identification and adversity determination. Additional endpoints to be discussed will be nonsubstantive differences regarding background, incidental, and/or minor microscopic changes and severity grades.
The results from these studies will be collated, tabulated and analyzed to determine how reliable studies with only one terminal control animal/sex are to identify risk assessment endpoints.
Conclusion
This proposal offers a viable compromise between traditional study designs in which all CCG animals are euthanized at the conclusion of the dosing period and replacement of CCGs by virtual control animals. We have tried to gather a comprehensive list of points to consider in implementing such a proposal and have provided practical recommendations aimed at increasing the likelihood of technical success and reduction in the overall number of NHPs used in nonclinical safety assessment. The authors will test the validity of these recommendations in the ongoing prospective validation of the proposal. We encourage continued discussion and debate within the scientific community about opportunities to optimize animal use in biomedical research.
Footnotes
Acknowledgements
The authors thank Ronald Wange and Kevin Snyder from the FDA for participating in the working group and contributing to the creation and refinement of this proposal.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.