
Abstract
Existing nervous system sampling and processing “best practices” for nonclinical general toxicity studies (GTS) were designed to assess test articles with unknown, no known, or well-known neurotoxic potential. Similar practices are applicable to juvenile animal studies (JAS). In GTS and JAS, the recommended baseline sampling for all species includes brain (7 sections), spinal cord (cervical and lumbar divisions [cross and longitudinal sections for each]), and 1 nerve (sciatic or tibial [cross and longitudinal sections]) in hematoxylin and eosin–stained sections. Extra sampling and processing (ie, an “expanded neurohistopathology evaluation” [ENHP]) are used for agents with anticipated neuroactivity (toxic ± therapeutic) of incompletely characterized location and degree. Expanded sampling incorporates additional brain (usually 8-15 sections total), spinal cord (thoracic ± sacral divisions), ganglia (somatic ± autonomic, often 2-8 total), and/or nerves (2-6 total) depending on the species and study objectives. Expanded processing typically adds special neurohistological procedures (usually 1-4 for selected samples) to characterize glial reactions, myelin integrity, and/or neuroaxonal damage. In my view, GTS and JAS designs should sample neural tissues at necropsy as if ENHP will be needed eventually, and when warranted ENHP may incorporate expanded sampling and/or expanded processing depending on the study objective(s).
Keywords
This article is an opinion piece submitted to the Toxicologic Pathology Forum (TPF). This perspective is the particular view of the author and does not represent an official position of the Society of Toxicologic Pathology, British Society of Toxicological Pathology, or European Society of Toxicologic Pathology, nor should it be considered to reflect the opinions, policies, or positions of regulatory agencies. The TPF is designed to stimulate discussion of topics relevant to regulatory issues in toxicologic pathology. Readers of Toxicologic Pathology are encouraged to send their thoughts on TPF opinion articles or ideas for new discussion topics to toxicologicpathologyforum@toxpath.org.
Introduction
Neurotoxicity is a principal concern when developing new products to treat neural and non-neural diseases. This concern is highlighted by the extensive regulatory guidance for evaluating the neurotoxic potential of test articles (TA) during nonclinical toxicity testing;10,11,30,46 wide-ranging recommendations for sampling, processing, analysis, and/or interpretation of microscopic findings in the central (CNS) and peripheral (PNS) nervous systems of common laboratory animal species commonly used in nonclinical safety testing;7,8,16,17,36,53 -56,59,60 and ongoing efforts to develop globally harmonized diagnostic nomenclature for microscopic findings that occur in neural tissues of common laboratory animals.19,20,27,50,63,67 Therefore, toxicologic pathologists must be ready to identify and characterize microscopic findings in a constellation of CNS and PNS organs as well as define their likely interpretation (ie, by differentiating TA-related vs procedure-associated vs incidental background vs artifactual changes from normal microanatomic features) and their translational implications for human risk assessment.34,35,37,57
The appropriate approach to neuropathology evaluation during nonclinical toxicity studies depends on the situation. Existing “best practice” recommendations for nervous system sampling, processing, and evaluation during nonclinical general toxicity studies (GTS) are designed to provide a baseline survey of regional anatomy and cell features in selected major sites in the CNS and PNS that are targeted by recognized neurotoxicants.16,17 The brain is screened in 7 levels; full coronal sections are assessed for rodents while coronal hemi-sections are used for non-rodents. 16 Spinal cord is examined at two sites minimally (cervical and lumbar segments 17 ) but ideally at three sites (adding the thoracic segment 16 ) in cross and longitudinal (commonly oblique 8 ) orientations. Sciatic nerve (or sometimes tibial nerve, especially in nonhuman primates where intramuscular injections of chemical restraint agents may traumatize the sciatic nerve 26 ) in cross and longitudinal orientations is generally the sole PNS specimen viewed during nonclinical GTS.16,17 Processing for these CNS and PNS samples employs routine fixation (immersion in neutral-buffered 10% formalin containing ~1% methanol as a stabilizer), embedding (paraffin), and staining (hematoxylin and eosin [H&E]). Similar baseline sampling and processing practices for neuropathology evaluation are followed for juvenile animal studies (JAS),13,31,45 which in essence are a GTS in which the subjects are immature animals rather than adults. Recent publications4,7,13,32,38,58 acknowledge that the baseline practices used during nonclinical GTS and JAS (where the TA has unknown, no known, or very well-known neurotoxicity) may be insufficient for scenarios where (1) the TA is designed to be neuroactive by intent (eg, TA targeting a neurological disease) or (2) the TA is expected or known to be neurotoxic based on prior experience with the TA or other agents of the same product class but the spectrum of effects (including microscopic findings) is uncertain or incompletely characterized.
In contrast to the baseline practices noted above, an “expanded neurohistopathology evaluation” (ENHP, alternatively rendered as “expanded neuropathology” or “enhanced neuropathology”) is designed to provide a wider ranging but still focused nervous system analysis.4,13,24,32,38,58 In general, the approach to ENHP involves sampling more CNS regions and/or PNS organs, processing serial sections of selected neural structures to examine key anatomic features or cell type–specific biomarkers not captured during the baseline evaluation, or both. These adjustments beyond current “best practices” recommended for GTS can improve the sensitivity of the neuropathology safety assessment for a relatively modest increase in cost, labor, and time. Importantly, technically laborious, low-throughput, and more expensive analytical approaches included in dedicated neurotoxicity studies, such as perfusion fixation (to best preserve the dense lipid-rich CNS parenchyma 12 ), plastic embedding (to optimize myelin stabilization in PNS specimens 17 ), cytoarchitectural analysis (transmission electron microscopy 52 ), or quantitative measurements of regional neuroanatomy dimensions or neural cell populations (eg, morphometry14,40 and stereology15,21), are generally not included—or necessary—for ENHP in GTS and JAS.
In my experience, an ENHP is used most often in three situations. First, an ENHP may be included in the pathology assessment for GTS in which in-life neurological signs and/or an unexpected neuropathological finding suggest that the TA has a neuroactive effect that requires further characterization. Second, an ENHP may be incorporated in JAS in which any potential toxicity to the immature nervous system represents a key (but not the sole) concern of interest. 13 Finally, an ENHP is frequently added by sponsors to nonclinical studies on a proactive basis for TA that are either intended to interact with target cells in the CNS and/or PNS or that are certain to elicit neural (biochemical, functional, and/or structural) effects as indicated by prior internal or published experience with the TA or similar molecules. Indeed, the last scenario is commonly used by experienced sponsors to proactively address the first two situations as a means for minimizing delays in the product development program.
No single approach to ENHP is suitable for all situations that might be encountered during nonclinical toxicity testing, and to date no proposal has been published that describes rational options for incorporating ENHP principles into nonclinical GTS or JAS. This opinion article addresses this gap by describing flexible and practical approaches for implementing ENHP—when warranted—in nonclinical GTS and JAS. In doing so, this article emphasizes practices for microscopic evaluation that may be either incorporated as options when initially writing the study protocol or easily added during an ongoing study by a protocol amendment. These concepts are not relevant to the design of dedicated neurotoxicity studies because ENHP (specifically more sampling and additional special neurohistological processing [eg, perfusion fixation, plastic embedding of nerve] and procedures [eg, assessing biomarkers for glial, myelin, and neuron responses and/or quantitative measurements]) already constitutes an integral element of the morphological analysis for such neuro-focused undertakings.14,17,30,40
Situations Benefiting From an ENHP
Multiple factors may warrant adoption of an ENHP approach to screen the nervous system during a nonclinical GTS or JAS. Three major parameters in this regard include the nature of the TA, the route of administration (ROA), and findings observed during the initial evaluation of H&E-stained neural tissue sections.
The nature of certain TA classes predicts that they will produce changes (functional and/or structural) in the nervous system. Recognized neuroactive small molecule TA that readily enter the CNS and PNS and produce changes in neural function include anesthetics, anticonvulsants, antidepressants, antineoplastic chemotherapeutics, antipsychotics, anxiolytics, hypnotics, mood stabilizers, and pain relievers, to name a few. The fact that a neuropharmacologically active TA alters nervous system function does not mean that the structural changes will incontrovertibly be visible in neural tissues. 1 However, ENHP may be warranted for TA from product classes that are known to be neuroactive and/or neurotoxic (eg, antineoplastic chemotherapeutics, neuroprotective gene therapies delivered using an adeno-associated virus [AAV] vector) to more fully characterize the impact on nervous system structures. 6
The ROA may be associated with neural damage, usually to the brain or spinal cord. Some TA (eg, AAV vectors, cell grafts) need to be delivered directly into the CNS by stereotactic intraparenchymal (IPa) injection into a brain nucleus or by intrathecal (IT) infusion into the cerebrospinal fluid. In such cases, insertion of the cannula or needle produces some degree of unavoidable physical trauma to the delicate neural tissue, which must be discriminated from any effects elicited by exposure to the TA.4,25,66 The limited range of tissue responses in the CNS means that findings related to the procedure and to TA exposure are often discriminated based on differences in the lesion degree rather than the kind of reaction. For such situations, inclusion of suitable control groups (especially vehicle-treated and sometimes sham-operated animals) coupled with ENHP provides the most straightforward means for distinguishing TA-associated effects.
Finally, the observation of unexpected findings in H&E-stained tissues during the initial microscopic evaluation may warrant addition of a post hoc ENHP component to investigate the nature of the changes. In particular, an ENHP evaluation may be requested by health authorities when neural findings affect bilaterally symmetrical structures (even if the degree of the effect is not symmetrical) or if the incidence and severity of the finding increase as the dose rises.
Practical Approaches to Implementing Expanded Neurohistopathology
The appropriate means for implementing an ENHP evaluation will be selected on a case-by-case basis. In general, a pathologist is the proper individual to organize the ENHP design. Ideally, the pathologist will have prior experience in neuropathology evaluations to help guide the ENHP analysis. Some pathologists feel intimidated when faced with complex neuropathology questions, in particular those posed by newer generations of innovative TA (biologics, cell and gene therapies, gene editing, etc).2 -4,18 In such cases, consultation with a seasoned subject matter expert in toxicologic neuropathology represents a prudent strategy for selecting the most suitable approach to neuropathology evaluation, including the design of any ENHP. Accordingly, ENHP designs often represent a collaboration among pathologists from both the sponsor and an external test facility (eg, contract research organization), on occasion supplemented with an independent consulting neuropathologist. This team approach is especially helpful for small sponsors (which often lack an experienced staff pathologist, let alone a neuropathologist) and is fairly common for larger sponsors (which often include ENHP proactively in designing GTS and JAS for TA that are anticipated or likely to engage targets in the CNS and/or PNS).
Study protocols for GTS and JAS commonly include language that permits implementation of an ENHP evaluation. For instance, the list of protocol-specified tissues typically includes “Brain” and “Spinal Cord” and “Nerve” for collection, processing, and microscopic evaluation, sometimes also including particular sites to assess (eg, “Spinal Cord [cervical, thoracic, and lumbar segments]” or “Nerve [sciatic]”) and/or a footnote stipulating that the sampling and processing will be performed according to current best practices (ie, reference numbers 16 and 17). Similarly, study protocols often include a statement that “Special stains may be used at the discretion of the study pathologist” to characterize findings observed during the initial routine examination. If necessary, the ENHP may be initiated by a protocol amendment that details the extra neural tissues and special methods to be included in the expanded evaluation.
Expanded Sampling
Two simple alternatives exist for expanding neural tissue sampling during nonclinical GTS and JAS. The first approach is to collect more neural specimens at necropsy, and the second is to prepare and evaluate additional tissue sections.7,16,17,24,53 -56,60
Expanded sampling at necropsy
Minor adjustments to neural tissue sampling at necropsy represent the most important component of proactively addressing the occasional need to incorporate ENHP evaluation into nonclinical GTS and JAS. 24 The rationale for this emphasis is that tissues not collected at necropsy are discarded permanently and thus unavailable for later ENHP analysis. An infrequent but real hazard of not collecting a complete complement of neural tissues at necropsy is that an entire Investigational New Drug (IND)–enabling nonclinical study may need to be repeated so that a gap in the data set related to missing neural tissues might be filled.
Conventional practice for nonclinical GTS and JAS already collects and preserves the majority of critical CNS and PNS tissues, though not necessarily in a systematic manner across all studies. The logical means for adapting the current cost-effective approach for pathology evaluation to support ENHP are to standardize collection of neural tissues from all animals across all species for all nonclinical GTS and JAS, as follows. The brain should be extracted from the cranial vault (although the olfactory bulbs of rodents are often left in situ, especially for studies with inhaled TA 16 ). The spinal cord may be removed from the vertebral canal either whole or as short segments, or short lengths of vertebral column (2-3 vertebrae) with the spinal cord left in situ may be isolated. 22 Collection of sciatic and tibial nerves is recommended with existing best practices for GTS (ie, Situation 1) suggesting bilateral collection of both nerves. 17 Ganglia might be covered by collecting 1 or more dorsal root ganglia (DRG), with extant best practices recommending that at least one lumbar DRG be retained for GTS. 17 Ganglia can also be harvested speedily by isolating a short length of vertebral column that includes at least two intervertebral disks, leaving the DRG in situ for possible removal later. Given the usual design of IND-enabling GTS and JAS, the principal albeit minor adjustments to neural tissue sampling would be to add DRG and tibial nerve to the standard list of protocol-specified tissues collected at necropsy. Inclusion of additional ganglia (eg, trigeminal ganglia, autonomic ganglia) and nerves should be considered for TA from classes with known neurotoxicity affecting one or more PNS elements (eg, AAV-based vectors),5,6 although the choice regarding whether or not to collect the additional elements remains with the sponsor and study design team. 17 As noted above, tissues not collected at necropsy are lost forever, 51 so the default approach for nonclinical GTS and JAS is to sample neural organs extensively (Table 1) but process selectively. Fixed but unprocessed tissues and blocks of embedded tissue are retained in accordance with current regulatory guidance.28,29
Examples of additional sampling for expanded neurohistopathology during nonclinical general toxicity studies and juvenile animal studies.
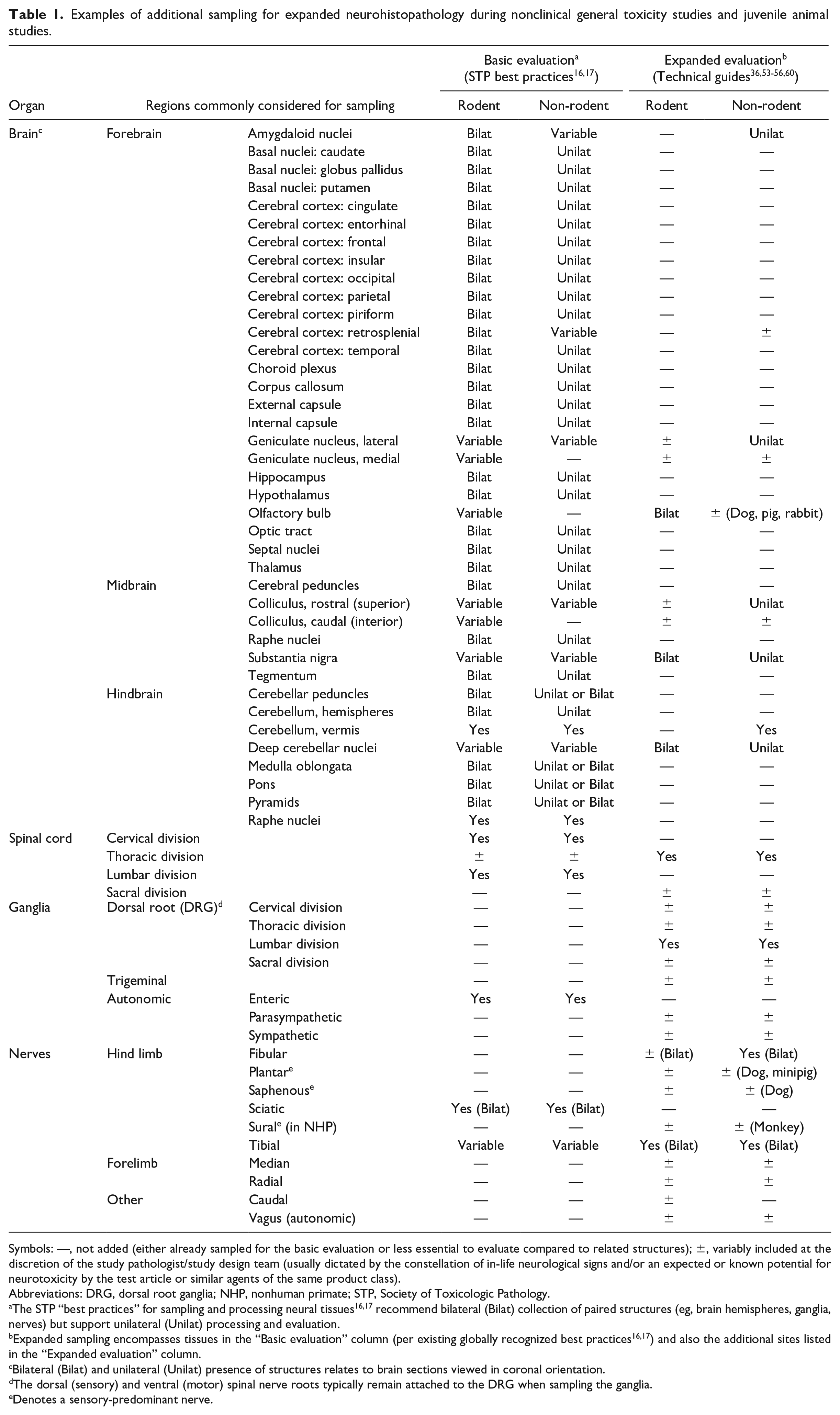
Symbols: —, not added (either already sampled for the basic evaluation or less essential to evaluate compared to related structures); ±, variably included at the discretion of the study pathologist/study design team (usually dictated by the constellation of in-life neurological signs and/or an expected or known potential for neurotoxicity by the test article or similar agents of the same product class).
Abbreviations: DRG, dorsal root ganglia; NHP, nonhuman primate; STP, Society of Toxicologic Pathology.
The STP “best practices” for sampling and processing neural tissues16,17 recommend bilateral (Bilat) collection of paired structures (eg, brain hemispheres, ganglia, nerves) but support unilateral (Unilat) processing and evaluation.
Expanded sampling encompasses tissues in the “Basic evaluation” column (per existing globally recognized best practices16,17) and also the additional sites listed in the “Expanded evaluation” column.
Bilateral (Bilat) and unilateral (Unilat) presence of structures relates to brain sections viewed in coronal orientation.
The dorsal (sensory) and ventral (motor) spinal nerve roots typically remain attached to the DRG when sampling the ganglia.
Denotes a sensory-predominant nerve.
Existing regulatory guidance does not prescribe a rigid design for the neuropathology portion of nonclinical safety studies.30,31,42 -45 Accordingly, sponsors retain considerable flexibility in determining the appropriate samples to process for microscopic evaluation. Thus, the expanded sampling approach outlined above does not necessitate that the extra neural tissues slated for collection (ie, DRG and tibial nerve) be processed and analyzed. If a decision is made later to initiate an ENHP evaluation of these additional sites, neural tissues from all animals should ideally be processed to block at the same time to limit the potential for systematic procedural artifacts (eg, reduced immunolabeling or staining) that tends to develop in samples from intermediate dose groups due to longer fixation while stored in the wet tissue archive. An additional reason for processing neural tissues from all dose groups at the same time is that direct TA delivery to the CNS typically causes procedure-related changes in some animals from all cohorts including vehicle-treated controls; in general, these procedural effects are characterized across all dose groups so that they can be effectively distinguished from TA-associated findings. In practice, neural tissues from intermediate groups of rodent studies are sometimes retained in fixative until ENHP is completed for the high-dose and control groups. This decision represents an acceptable compromise that balances the substantial costs incurred in processing large treatment groups from standard rodent studies with the fairly frequent situation where assessment of tissues from the high-dose group is sufficient to complete the ENHP.
Expanded sampling by additional trimming/embedding
Current best practice for microscopic evaluation of neural tissues during nonclinical GTS for all test species is streamlined. For IND-enabling studies, the assessment incorporates brain (7 sections), spinal cord (at least cervical and lumbar divisions [cross and longitudinal sections for each]), and 1 nerve (sciatic or tibial [cross and longitudinal sections]).16,17 Therefore, a simple approach to ENHP is to assess “more” for some or all of these tissues, in some cases also adding one or more ganglia. 24
Expanded tissue trimming varies to some extent depending on several factors. One is the size of the organ. For brain, a common expansion used in rodents (mainly rats due to their larger brain size) is to physically trim the organ to produce 1 or occasionally 2 more slices (for a total of 8-9 slices per animal, thus essentially trimming and embedding the entire brain). Alternatively, additional rodent brain levels may be prepared by step-sectioning through the block to acquire supplementary sections at a distance from the usual trimming level. The choice of rodent brain levels to assess for ENHP may be defined based on known functional correlates for various brain regions. 60 Expanded brain trimming in non-rodents usually involves preparing a greater number of coronal hemi-sections from one hemisphere; commonly 3 to 8 additional hemi-sections are added (for a total of 10-15 brain hemi-sections per animal). Annotated atlases suggesting possible options for expanded brain sampling in non-rodents have been published recently for rabbits, 56 dogs, 53 and minipigs, 55 and a similar expanded sampling approach has been suggested for cynomolgus monkeys 7 based on a prior annotated brain atlas prepared for this species. 54 A variant used in some cases (if the contralateral side is not destined for biodistribution or molecular analyses) is to generate an increased number of hemi-sections for the first brain hemisphere and then generate the identical complement of hemi-sections from the opposite hemisphere as well. This latter expansion often yields 20 to 30 brain sections per animal for non-rodents and offers the twin benefits of more comprehensive screening 65 and permitting evaluation of lesion symmetry.
A second factor influencing expanded tissue trimming is the nature of the TA. For example, embedding of more DRG (including sacral DRG, which are not mentioned for collection and evaluation in current best practices 17 ) may be warranted for AAV-based vectors since these agents are known to cause ganglionic pathology and tend to produce greater effects in caudal (lumbar and sacral) ganglia. 5 In this regard, the number of DRG assessed in GTS for AAV products tends to range from 2 to 8 total ganglia, often supplemented by inclusion of a trigeminal ganglion (a DRG equivalent associated with cranial nerve V), even though current best practice for GTS (Situation 1) is to collect only 1 lumbar DRG. 17 If available, more nerves (2-6) may be added for TA that exhibit a substantial impact on DRG structure or function.
A third factor that may dictate expanded trimming involves the dimensions of the injection site. A simple IT injection in the lumbar cistern, even if repeated, is associated with a relatively small region of tissue, so a single section may be sufficient for screening. In contrast, insertion of a cannula in the vertebral canal results in contact between the device and the spinal cord over a larger expanse of tissue, so it is likely that several sections may be needed to assess the scope of findings over the entire cannula site. Similarly, characterization of procedural effects associated with IPa injection usually will require multiple sections to encompass not only the administration site (ie, location of TA deposition) but also the injection track left when the cannula was advanced to reach the administration site.
A final factor that may impact the tissue trimming scheme is the set of neural regions to be evaluated. For example, the rationale for expanded brain sampling is to cover more anatomic regions with known functional correlates (as outlined in the Appendix of the article that outlines current industry “best practice” recommendations for neural sampling in nonclinical GTS 16 ). Current baseline best practice recommendations for neural tissue sampling during GTS16,17 cover an impressive range of target sites in the CNS and PNS known to be affected by various neurotoxic agents.57,65 Importantly, best practices permit the selection of alternate trimming levels for brain to capture particular domains rather than adding more trimming levels, at the discretion of the sponsor. 16 Nonetheless, inclusion of additional sections, especially for the large brains of non-rodents, is likely to be the most effective approach for consistently sampling many brain regions of interest (Table 1).
Similarly, deliberate embedding of additional DRG, and perhaps other ganglia (eg, trigeminal ganglia and/or autonomic ganglia) and nerves (Table 1), affords the simplest means for the systematic evaluation of more PNS tissue—providing, of course, that the extra ganglia and nerves were collected during the necropsy. The baseline best practice recommendations for PNS sampling during GTS (Situation 1 17 ) specify that collection and evaluation of sciatic nerve (with retention but no immediate evaluation of 1 lumbar DRG) are an acceptable screen for PNS toxicity of TA with no or unknown neurotoxicity.16,17 In practical terms, embedding and microscopic evaluation of 1 DRG during a GTS (with or without other ganglia and nerves) are already an ENHP approach relative to conventional practices. That said, collection and embedding additional DRG and at least 1 more distal nerve branch (often the tibial or fibular) are a recommended practice to employ for TA with suspected or known PNS effects to ensure that adequate neural tissue has been surveyed. 17
A practical approach for expanded PNS sampling for GTS and JAS is to modify the necropsy procedure to collect a few extra neural tissues, as follows. First, short vertebral column segments from the cervical (C1-C3) and lumbar (L3-L6) regions may be isolated. These vertebral column segments will harbor at least two DRG pairs for each region. Second, acquire one trigeminal ganglion from the pair located on the skull base on either side of the pituitary gland.5,41 Finally, harvest both sciatic trunks and both tibial nerves. Instead of immediate embedding, the DRG may be left in situ in the vertebral segments while the trigeminal ganglion and all but one sciatic nerve may be retained in fixative (in cassettes or attached to card stock) until an ENHP is initiated. In the hands of an experienced technician, this expanded sampling approach can be implemented easily at necropsy for all nonclinical species for an added dissection time per animal of 3 to 5 minutes for non-rodents and 2 to 3 minutes for rodents.
Expanded Processing
In nonclinical GTS and JAS, routine screening is limited to H&E-stained sections for all neural organs.13,16,17 Special neurohistological procedures may be added if necessary to characterize changes observed in the initial analysis of the H&E-stained sections.13,16,17,24 Study protocols for GTS and JAS typically include language that permits special methods to be used at the discretion of the pathologist.
Common neurohistological processing procedures that may be implemented when warranted for ENHP in nonclinical GTS and JAS are given in Table 2. These methods may be performed on routinely processed (formalin-fixed, paraffin-embedded) tissues, with one exception: amino-cupric-silver for detecting neuronal degeneration in the CNS is performed on frozen sections. The special methods are deployed in tiers, where H&E (to evaluate general cell and tissue features) represents Tier 1.13,16,17,24 This choice is practical since H&E is the standard stain for all tissues evaluated in nonclinical GTS and JAS, which permits the neural organs to be processed in bulk along with all other protocol-specified tissues. Several special methods may be included in higher tiers,5,16,17 with the choice of procedures to include in each tier depending on the preference of the study pathologist. Moreover, the composition of the higher tiers may shift with such factors as the length of the study and the neural organ that is being assessed. For example, Fluoro Jade (FJ) is an effective stain for detecting neuron degeneration in the CNS 16 but usually is not helpful for identifying neuronal degeneration in DRG. 17
Conventional examples of processing for expanded neurohistopathology evaluation during nonclinical general toxicity studies and juvenile animal studies. a
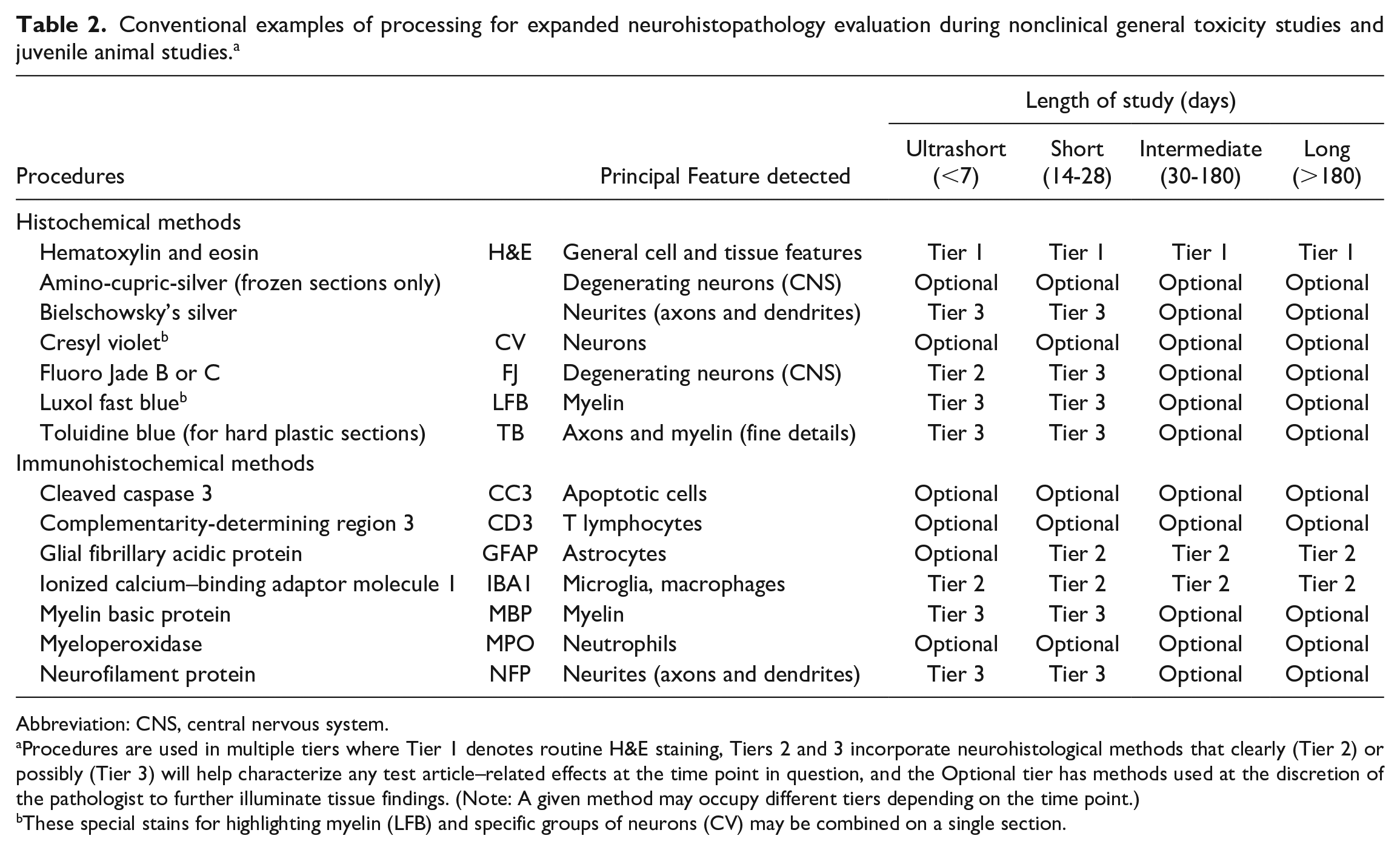
Abbreviation: CNS, central nervous system.
Procedures are used in multiple tiers where Tier 1 denotes routine H&E staining, Tiers 2 and 3 incorporate neurohistological methods that clearly (Tier 2) or possibly (Tier 3) will help characterize any test article–related effects at the time point in question, and the Optional tier has methods used at the discretion of the pathologist to further illuminate tissue findings. (Note: A given method may occupy different tiers depending on the time point.)
These special stains for highlighting myelin (LFB) and specific groups of neurons (CV) may be combined on a single section.
The rest of this section gives the organization of Tier 2 and Tier 3 batteries of special procedures and explains the rationale for this approach to ENHP. When designing ENHP for GTS and JAS, I rely on Tiers 1 and 2 and seldom invoke Tier 3 except in those instances where a sponsor seeks to evaluate the Tier 3 battery as a due diligence exercise when assembling a neuropathology data set destined for regulatory review. (Other procedures listed in Table 2 are useful when available, but they are usually employed as part of an investigational toxicity study rather than as components of a general toxicity screen. Therefore, these methods are listed for completeness but are not further discussed.)
My standard special stain for neuronal degeneration in ENHP is FJ B or FJ C. Both FJ variants work well, but FJ C may miss some degenerating neurons depending on the test system. 62 In general, FJ is most relevant for short-term studies since neuronal damage leading to neuronal death typically occurs in the first few (2-4) days following TA exposure (at least for small molecules);24,65 in my experience, persistence of neuronal degeneration is rare in FJ-stained sections of GTS or JAS over 28 days in length. Accordingly, I include FJ as a Tier 2 stain for studies up to 7 days in length but consider it to be a Tier 3 procedure for longer studies. Degenerating neurons may also be highlighted if H&E-stained sections are evaluated using fluorescent light with a fluorescein isothiocyanate (FITC) filter,47,61 although some neuropathologists have had inconsistent success in identifying dead cells using this method perhaps stemming from differences in the light wavelengths passed by various fluorescent filters (R.H. Garman, personal communication, October 2023). In the future, comparisons confirming the concordance between epifluorescent H&E and FJ signals may permit H&E to subsume the role of FJ as a screening method for neuronal necrosis.
My preferences for evaluating glial responses to TA-associated or procedure-related parenchymal injury are two well-characterized immunohistochemical (IHC) methods that showcase reactive glia: glial fibrillary acidic protein (GFAP) for astrocytes and ionized calcium–binding adaptor molecule 1 (IBA1) for microglia and infiltrating macrophages. These two procedures are appropriate for all nonclinical species. In the CNS, reactive microglia arise sooner (hours to 7 days after a stimulus) compared to reactive astrocytes (approximately 7-14 days after stimulus; Figure 1, left column) while reactive astrocytes persist longer than reactive microglia (Figure 1, right column). 9 Moreover, IBA1-labeled sections generally exhibit a uniformly pale background (ie, high signal-to-noise ratio) while GFAP-labeled sections often possess a patchy, variably intense background. Therefore, I employ IBA1 as the anchor of a Tier 2 ENHP battery at all time points while I consider GFAP to be an ancillary ENHP procedure most relevant for studies 14 days in length or greater.
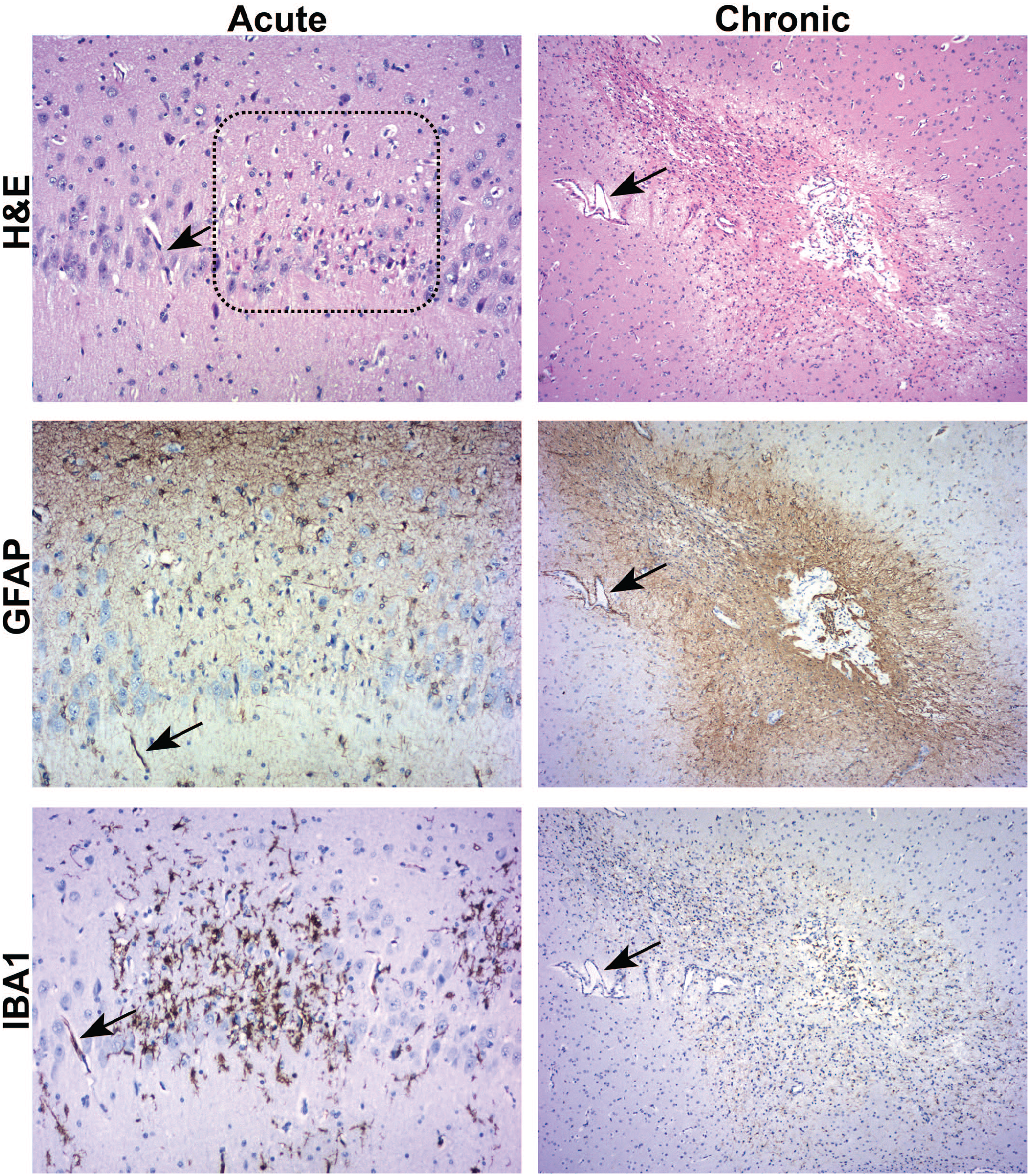
Representative acute and chronic glial responses to parenchymal injury in the cynomolgus monkey brain after direct central administration of a biologic test article. Acute responses (left column, shown here for 3 DPI in the hippocampus) commonly appear as increased numbers of reactive microglia expressing IBA1 with very few or no reactive astrocytes expressing GFAP; the dotted box (H&E panel) encompassing a focus of acute neuronal necrosis—indicated by many “red dead” cells with hypereosinophilic cytoplasm and pyknotic nuclei—is co-localized with the larger aggregate of reactive microglia (IBA1 panel). Chronic responses (right column, shown here for 365 DPI near a central focus of parenchymal necrosis in the cerebral cortex) are characterized by enhanced GFAP expression (ie, a glial “scar,” evident as a wide field of labeled reactive astrocyte processes around an irregular cell-depleted core) with limited IBA1 expression. Processing: immersion fixation in neutral buffered 10% formalin, paraffin embedding, serial 5-µm-thick sections for H&E or IHC with either anti-GFAP or anti-IBA1. For the IHC sections, the chromogen is DAB and the counterstain is hematoxylin. For each time point, the arrows denote profiles of the same blood vessel visible in all serial sections. DAB indicates 3,3′-diaminobenzidine; DPI, days post-injection; GFAP, glial fibrillary acidic protein; H&E, hematoxylin and eosin; IBA1, ionized calcium–binding adaptor molecule 1; IHC, immunohistochemistry.
Assessment of nerve fiber integrity for ENHP is occasionally helpful for GTS and JAS. The rationale is that the appropriate combination of cell type–specific methods may be able to identify the primary target for neurotoxic injury (ie, neuronal processes vs myelinating cells). The workhorse methods for this purpose are silver degeneration stains (eg, Bielschowsky’s and Bodian’s) or pan-neurofilament protein (an IHC method) to evaluate axons and either Luxol fast blue (LFB, a histochemical stain) or myelin basic protein (MBP, an IHC method) to examine myelin sheaths. If desired, LFB may be combined on the same section with cresyl violet (CV, a stain that highlights nuclei, nucleoli, and Nissl substance chiefly in neurons but also in reactive glial cells). When warranted, I use Bielschowsky’s silver stain and LFB (with or without CV) or rarely MBP as Tier 3 stains for ENHP in GTS and JAS; in general, I usually employ these procedures only for early time points (eg, 7 days or earlier after initial exposure since axonal debris is removed in this time frame 64 ) as long-term nerve fiber degeneration involves disintegration of both axons and myelin. Thin (1-µm-thick) resin (“hard plastic”) sections of nerve stained with toluidine blue provide exquisite detail of axon organelles and especially the lamellar arrangement in myelin sheaths,17,22 but this procedure typically is not used for GTS and JAS but rather is reserved for situations in which a TA is suspected or known to induce neurotoxicity. I do not suggest using glycol methacrylate (GMA [“soft plastic”]) for embedding immersion-fixed nerves as structural resolution in GMA is no better than the nerve fiber preservation offered by paraffin.14,17
In summary, my usual ENHP approach for enhanced processing in nonclinical GTS and JAS is to begin with H&E (Tier 1) for both CNS and PNS and then add IBA1, GFAP, and for early time points FJ (Tier 2) to further characterize findings in the CNS. I implement these special procedures on a case-by-case basis. For non-rodents, I often select a subset of brain sections (typically about 30%-40% of the levels available for evaluation) or spinal cord sections (the cervical and lumbar segments) for processing and evaluation. For rodents, the entire brain is generally embedded in 2 or 3 cassettes, so all brain levels will be processed for these special procedures even if only a subset is chosen for evaluation. If findings in H&E-stained sections or in-life neurological signs suggest that a particular brain domain is involved, I will typically implement ENHP using one (IBA1) or more (IBA1, GFAP, and sometimes others) of the Tier 2 special procedures to the affected level(s) as well as the sections that contain any neural nuclei or tracts that connect to the involved regions, for all blocks of rodent brain and 30% to 40% of blocks for non-rodent brain. The rationale for limiting additional processing is that this approach supports a more complete characterization of any neural finding while limiting the ENHP cost in terms of labor, money, and time.
Expanded Analysis
In dedicated neurotoxicity studies, nonclinical safety assessment for biomedical products incorporates both functional testing and neuropathological evaluation.30,31,45 The foundations of the neuropathology evaluation for dedicated neurotoxicity studies (as for GTS and JAS) are macroscopic observations, brain weights, and microscopic evaluation focused on semi-quantitative severity grading of findings. However, because detailed characterization of neurotoxicity is the focus of dedicated neurotoxicity studies, special processing (eg, perfusion fixation,12,17,33,39,48 plastic embedding of selected PNS samples17,39) is necessary to optimize neural tissue preservation and minimize artifacts associated with fixation.34,49 Moreover, advanced analytical methods such as morphometric14,40 and/or stereological21,23 measurements are often included in dedicated neurotoxicity studies to quantify TA-related morphological alterations. Such special processing and analytical procedures are expensive, laborious, and slow, so dedicated neurotoxicity studies are conducted only when TAs are suspected or known to impact the CNS and/or PNS.
The ENHP approach is a rational compromise designed to maximize the amount of neuropathology data that can be obtained from routine safety bioassays (eg, GTS and JAS) in which neurotoxicity is a possible concern but not the primary focus of the screen, and for which special neurohistological processing and neuropathological techniques would provide little useful data relative to the greatly heightened expense. Accordingly, an ENHP is grounded on conventional processing conditions (immersion fixation and paraffin embedding) and neuropathology endpoints (macroscopic observations, brain weights, and microscopic evaluation based on semi-quantitative scoring) for the initial analysis, after which additional H&E-stained tissue sections and/or specially processed tissue sections (eg, cell type– or process-specific markers [Table 2]) may be added as warranted to extend the neuropathology data set to address particular questions. I generally do not incorporate quantitative measurements (eg, morphometry, stereology) for ENHP as such methods require perfusion fixation and special processing (eg, highly homologous tissue sections, systematic uniform random sampling) that are not suitable for implementation in GTS and JAS.
Conclusion and Recommendations
ENHP in nonclinical GTS and JAS is occasionally requested by health authorities tasked with assessing the risk of novel TA for human patients. That said, the design of ENHP is not codified, so sponsors must decide what adjustments are necessary to the baseline pathology evaluation to address the regulatory questions underlying the ENHP request. Such decisions are made on a case-by-case basis (ideally by a study pathologist with some experience in neuropathology evaluation) in which the weight of evidence provided by findings on H&E-stained neural tissue sections as well as related study data (brain weights, gross findings in neural organs, in-life neurological signs, etc) and any known class effects for similar TA are integrated to provide a rational scientific justification for the chosen ENHP design.
I approach ENHP as a multifactorial problem with a menu of alternatives that may be used singly or in combination. To me, the most critical design consideration for GTS and JAS is that the original study protocol should be designed to include expanded sampling at necropsy of all the main neural tissues needed to support an ENHP. This adjustment is essential as any tissues not taken at necropsy will be permanently lost. Therefore, I recommend that this modification be adopted for all GTS and JAS to avoid the rare but real possibility that a study might need to be repeated because a missing neural tissue was not available for evaluation to answer questions posed by health authorities. Very minor adjustments to study designs for both GTS and JAS are needed to achieve this objective, to wit collection of at least 1 DRG and tibial nerve in addition to the usual neural organs (brain, spinal cord, and sciatic nerve). This expanded sampling should not be difficult since the DRG are easily collected by isolating a short segment of lumbar vertebral column (eg, L3-L6), leaving the DRG in situ to save time during the necropsy, while the tibial nerve arises from the distal sciatic nerve and so is readily identified and harvested with little additional dissection. For additional assurance, I recommend that neural sampling at necropsy also includes collection of a short cervical vertebral column segment (eg, C1-C3, to obtain cervical DRG) and one trigeminal ganglion. These additional neural specimens may be retained in fixative since expanded processing is not undertaken until an ENHP is actually initiated.
The other design parameters that support ENHP are already inherent in the study protocols for conventional GTS and JAS. Typically, study protocols indicate what neural organs will be evaluated microscopically without specifying how the organs will be trimmed. In addition, protocols commonly provide the study pathologist with the discretion to use special procedures to further characterize findings observed in routine H&E-stained sections. Accordingly, no adjustments beyond additional sampling at necropsy should be necessary to support ENHP for the average GTS and JAS. If desired, a subject matter expert in toxicologic neuropathology may be consulted to provide additional confidence that the final ENHP design will effectively address any questions or concerns raised by regulatory reviewers.
Footnotes
Acknowledgements
The author gratefully thanks his colleagues Dr Mark Butt, Dr James Fikes, Dr Robert Garman, and Dr Deepa Rao for their critical conceptual review, and Ms Beth Mahler for her assistance in optimizing the layout and resolution of the figure.
Author Contributions
The author is solely responsible for the contents and crafting of this article.
Declaration of Conflicting Interests
The author declares no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.