
Abstract
Adequate tissue sampling is known to reduce the likelihood that the toxicity of novel biomolecules, chemicals, and drugs might go undetected. Each organ, and often specific structurally and functionally distinct regions within it, must be assessed to detect potential site-specific toxicity. Adequate sampling of the brain requires particular consideration because of the many major substructures and more than 600 subpopulations of generally irreplaceable cells with unique functions and vulnerabilities. All known neurotoxicants affect specific subpopulations (usually neurons) rather than damaging a certain percentage of cells throughout the brain; thus, all populations should be independently assessed for lesions. Historically, the affected neural cell subpopulation has not been predictable, but it is now clear that sampling selected populations (e.g., cerebral cortex, hippocampus, cerebellar folia) cannot forecast the health of other populations. This article reviews the neuroanatomical domains affected by several model neurotoxicants to illustrate the need for more comprehensive neurohistological evaluation during nonclinical development of novel compounds. The article also describes an easily executed, cost-effective method that uses a set number of evenly spaced coronal (cross) sections to accomplish this comprehensive brain assessment during nonclinical safety studies performed in rodents, dogs, and nonhuman primates.
Introduction
There is much discussion and debate about the appropriate level of sampling that is advisable or that should be required during nonclinical neurotoxicology studies. Regulations of the United States Environmental Protection Agency (EPA), United States Food and Drug Administration (FDA), and other regulatory agencies (Bolon et al. 2010), as well as the guidelines published by the World Health Organization (WHO) and other organizations (Harry et al., 2001), vary in their discussions and recommendations on this topic, but the typical regulatory “directive” is to avoid giving any specific instructions for sampling. Rather, it is explicitly stated or implied that investigators should use the best scientific methods available at the time of the study (FDA 2000), and this may in fact be the wisest approach. The neurotoxicology community is constantly developing new methods of evaluation and increasing our communal understanding of potential neurotoxic profiles. Past approaches lacked the methods and data that we have acquired in the past few decades regarding the specificity and unique profiles that neurotoxicants may have in the brain; future studies should have the flexibility to shift methods and approaches as our understanding shifts. This document will explore existing neuroanatomic sampling methods and data as they apply to the selection of sampling schemes for brain evaluation. Specifically, this review will discuss established principles of neurotoxicity, highlighted by known examples, and then describe potential sampling approaches and their potential efficacy with regard to the principles and examples.
There appear to be three basic schools of thought in devising a sampling approach to detect neurotoxicity. The first approach is a routine or standard screening approach in which specific and predefined coronal levels of the brain are sampled. The goal of this approach is to screen for neurotoxicity in what is perceived to be the most important regions of the brain. In the rodent, often only three or four levels are taken, positioned to reveal those regions that have been shown to be most likely to develop neurotoxicant-induced structural changes (e.g., cerebral cortex, basal ganglia, hippocampus, cerebellum, pons). Benefits of this approach include consistency in sampling locations across compounds and laboratories as well as minimization of histology and pathology efforts and costs. The potential weakness of this approach is missing a neurotoxic event if the affected region was not one that was selected for sampling. The second approach is a targeted study in which an area of potential neurotoxicity has been previously identified and is now subjected to a detailed examination. This approach is often performed in addition to acquisition of the routine or standard screening sections. Benefits of this approach include examination of a known potential target, and minimization of histology and pathology efforts and costs. As with the routine sampling, the potential weakness of this approach is missing a neurotoxic event if the impaired region was not selected for sampling. The third approach is to devise a comprehensive strategy that gathers more brain sections, thus sampling all or most of the over 600 subpopulations of the brain. The number of levels sampled in “comprehensive” neurotoxicity studies seems (anecdotally) to vary among pathologists, but usually involves substantially more levels (often ten to twenty-plus in the rodent) than are taken in the standard or routine approach. The primary benefit of this more in-depth approach is the potential to evaluate more regions of the brain. The potential weakness of this approach is the additional effort and cost required to perform the increased sampling, and the requirement for a pathologist who has a substantial amount of additional neuroanatomical training to be able to evaluate the many discrete neural cell subpopulations that are now present in the increased number of sections.
Materials and Methods
Neurotoxicity can take many forms, ranging from a temporary change in brain neurochemistry to enduring structural alterations. Cell death was chosen as the basis for the analysis undertaken for this article because this end point is a permanent expression of neurotoxicity that can be readily detected and quantified using a variety of histological markers. The data set consisted of the anatomical regions in which cell death was witnessed to occur using nuclear-cell body (e.g., hematoxylin and eosin) or degeneration (amino cupric silver or Fluoro-Jade) stains.
The number of known neurotoxicants is quite large, though many of them cause similar lesions to a relatively small number of major brain structures. The subset of neurotoxicants chosen as examples for this review was selected from among those that have been documented in peer reviewed journals to illustrate some key trends and concepts. Most of the neurotoxicants that received the type of evaluation and research needed for this review were drugs of abuse or other high-profile compounds discovered owing to mass human neurointoxication. Although the examples include some failed pharmaceuticals, in most cases the lesions induced by a failed pharmaceutical compound are not published, nor is additional effort expended in understanding the precise scope of the neurotoxic impact once the product has been dropped from development.
For each compound in this review, the locations in the brain documented to have experienced cell death in one or more studies were combined to create the anatomical profile of site-specific vulnerability for each compound. In the studies being reviewed to create these profiles, factors such as the timing of necropsy following exposure and the methods used to detect cell death could have an impact on the regions and extent to which cell death was witnessed. Since the intent of this study was to create a comprehensive map of brain regions affected by each compound, all regions found to be affected by a given agent from one or more studies were combined into one anatomic map. The affected regions were superimposed on the structures shown in a well-recognized rodent brain atlas (Paxinos and Watson 2007) to permit more ready understanding of these detailed neurotoxicity profiles by individuals who lack significant advanced training in neuroanatomy.
Results
The following list enumerates the sites at which cell death has been reported for a selected set of well-known neurotoxicants. For convenience, the neurotoxic agents are listed in alphabetical order. 3-Acetyl pyridine (3AP): hippocampus and inferior olive nucleus (Desclin and Escubi 1974). Alcohol: olfactory bulb, caudal (posterior) piriform cortex, entorhinal cortex, dentate gyrus (Crews et al. 2000; Han et al. 2005; Ikegami et al. 2003). Amphetamine: parietal cortex, barrel field of primary somatosensory cortex, frontal cortex, hippocampus, tenia tecta, piriform cortex, septum, caudate/ putamen, thalamic nuclei (Belcher et al. 2005; Bowyer 2000; Bowyer et al. 1998; Bowyer et al. 2004; Carlson et al. 2000; Ellison 2002; Jakab and Bowyer 2002; Jakab and Bowyer 2003). Carbonyl sulfide: caudal (inferior) colliculus (Morrison et al. 2009). Domoic acid: olfactory bulb, anterior olfactory nucleus, dorsal tenia tecta, lateral septal nucleus, reuniens thalamic nuclei, and hippocampus (Colman et al. 2005). Kainic acid: hippocampal fields CA1 and CA3, polymorphic layer of dentate gyrus, parasubiculum (Benkovic et al. 2006; Benkovic et al. 2004). Methamphetamine: parietal cortex, barrel field of primary somatosensory cortex (Belcher et al. 2005; Ellison 2002; Schmued and Bowyer 1997). 3,4-Methylenedioxymethamphetamine (MDMA): frontoparietal region of neocortex (Carlson et al. 2000; Ellison 2002; Jensen et al. 1993; Johnson, Shvedova et al. 2002; Johnson, O’Callaghan et al. 2002; O’Shea et al. 1998). 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): ventral tegmental area (VTA); substantia nigra (Luellen et al. 2003). MK-801 (also known as dizocilpine): retrosplenial cortex; dentate gyrus; piriform cortex; tenia tecta; amygdala; entorhinal cortex (Creeley, Wozniak, Nardi et al. 2006; Creeley, Wozniak, Labruyere et al. 2006; Ellison 1995; Fix et al. 1996; Fix et al. 1993; Fix et al. 1994; Fix et al. 1995; Horvath et al. 1997; Maas et al. 2005; Olney 2003; Olney et al. 2004; Olney et al. 2000; Olney et al. 1987; Olney, Ikonomidou et al. 1989; Olney, Labruyere et al. 1989; Jevtovic-Todorovic, Hartman et al. 2003; Jevtovic-Todorovic, Beals et al. 2003; Jevtovic-Todorovic et al. 2000; Wozniak et al. 1998). 3-Nitropropionic acid (3NPA): caudate/putamen, prefrontal cortex, insular cortex, entorhinal cortex, parietal and sensory cortex, CA1, CA3 and dentate gyrus of hippocampus (Miller and Zoborsky 1997). 2′-NH2-MPTP: dorsal raphe (Luellen et al. 2003).
p-Chloroamphetamine (PCA): raphe nuclei (B-7 and B-8), B-9 serotonergic cell group, ventral midbrain tegmentum (Belcher et al. 2005; Harvey et al. 1975; Wilson and Molliver 1994). Phencyclidine (PCP): entorhinal cortex, dentate gyrus in ventral hippocampus, cingulate and retrosplenial cortex (Carlson et al. 2000; Ellison 1995).
Schematic representations of the affected brain regions are given for each agent below.
Discussion
The results for each of the compounds in this manuscript highlight the variability in what specific regions are affected by different neurotoxicants. Upon closer examination, these variations illustrate several contemporary principles of neurotoxicity that will have direct relevance in selecting a proper approach to neuroanatomic sampling.
Principle #1
The location in the brain affected by a neurotoxicant can be very small, yet the seemingly minor damage may have a significant functional impact. The anatomical profiles affected by 3AP, methamphetamine, MPTP, and 2′NH2-MPTP are all quite small relative to some other compounds in the study. Perhaps 2′-NH2-MPTP, the smallest profile of these compounds, may best illustrate this point. The only region vulnerable to cell death from this agent is the raphe nuclear group, which occupies approximately 2-mm3 in the rat brain (Figure 1). Although the directly affected region is quite small, the effect on the brain is a large one. Nearly all serotonergic cell bodies in the brain lie in the raphe nuclei, which in turn project axons widely throughout the brain. Therefore, losing these cells might be expected to cause profound long-term negative effects, as serotonin is involved in regulating normal functions as well as several diseases (e.g., depression, anxiety, stress, sleep, vomiting). Its importance is emphasized by the large number of pharmaceutical agents that therapeutically regulate brain serotonin levels (e.g., Paxil, Prozac, Zoloft).
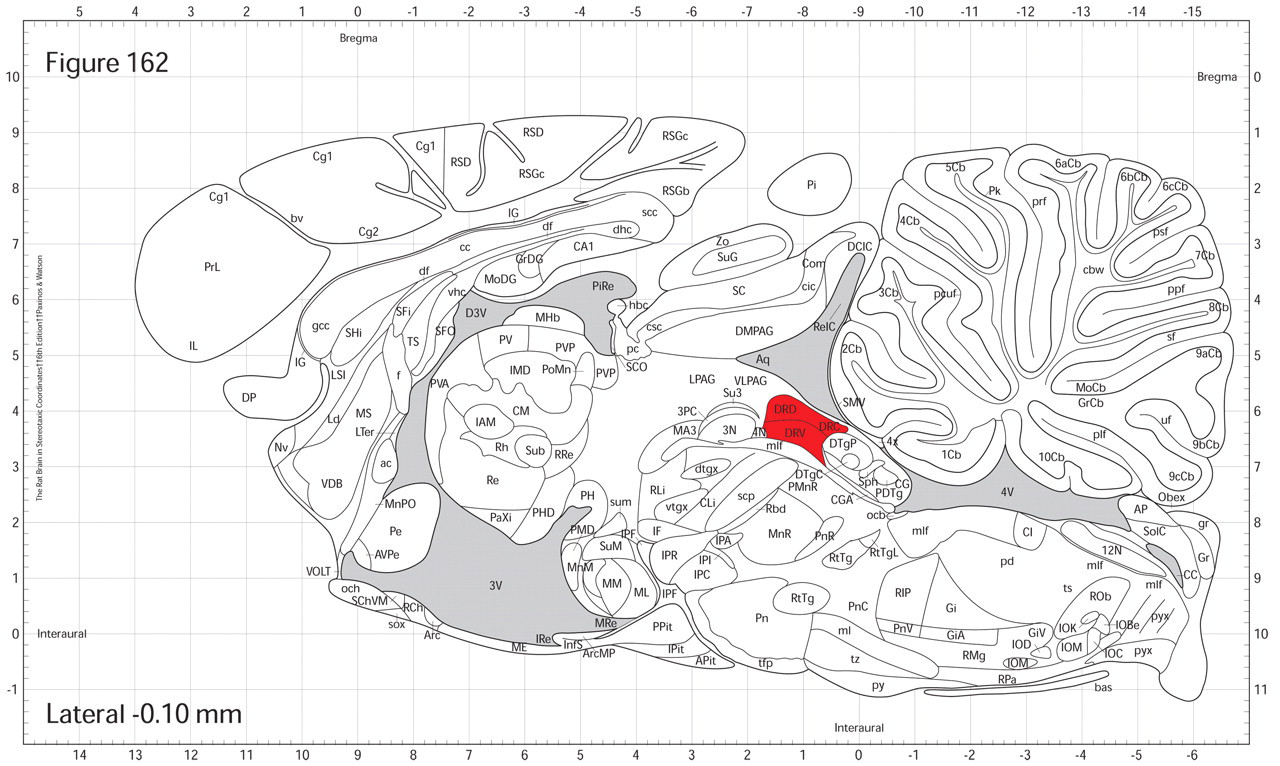
The brain region affected by 2′-NH2-MPTP (dorsal raphe) is shaded in red. Although this region occupies a small portion of the brain, its functional significance is large because of the broad intracerebral distribution of pathways arising from this site.
Principle #2
The brain location(s) vulnerable to a given neurotoxicant are unpredictable. One of the most common flaws in safety assessments is to look only in damage-prone areas where lesions are expected. This flaw occurs predominantly in three ways. First, the locations chosen for evaluation are based on generic areas of the brain that are assessed for all compounds (e.g., cerebral cortex, hippocampus, cerebellum/pons). Second, the locations may be based on known neurotoxic profiles of compounds of the same or a similar class, thereby inviting a sampling approach that is too focused on the regions of projected vulnerability. Finally, only the regions thought to be susceptible based on behavioral and functional responses to a compound are assessed for potential neurotoxicity. All of these three abbreviated schemes will lead to incomplete sampling in which it might be possible to miss subtle or minute structural changes.
The brain is a highly complex organ. The interaction of the regions of the brain, metabolism, and a host of external factors are far more complex than our current capability to predictably model neurotoxicity, even when using other vertebrate species. Although it is prudent to sample areas of the brain that are known to be affected by potential or known neurotoxicants, our predictive and modeling capabilities remain unable to fully anticipate where and how a neurotoxic reaction may occur. Perhaps the best case to be made for this unpredictability is an inherent acceptance of this fact by the pharmaceutical industry in their approach to drug discovery, which seeks compounds that differ in an advantageous way from those that are already known to be neurotoxic.
There are known candidate pharmaceuticals that are functionally effective, yet they unfortunately have known neurotoxic side effects. From a logical standpoint, if we assume compounds of the same class would be neurotoxic in the same way, there would be no need to test any more compounds in a class if any one was found to be neurotoxic. Rather, if we assume similar profiles, we could safely assume they are all neurotoxic in the same way. Of course, this supposition is not true, and much effort, money, and time are invested by industry (and rightly so) to explore compounds of a particular class that may be just as effective but lack the neurotoxic properties of other class members. MK-801 is an excellent example of this rule. Although it excels in its function as an N-methyl-D-aspartic acid (NMDA) receptor antagonist, MK-801 has an extensive profile for neurotoxicity (Wong et al. 1986). However, many other similar compounds are tested each year to find compounds with efficacy but not neurotoxicity. Thus, the neurotoxic assessment of such agents must be open-minded in considering the fact that the compound could be neurotoxic, but in a different manner than other structurally related compounds. Although these compounds may have similar neurotoxic (or non-neurotoxic) profiles, many exceptions exist, and it is not appropriate to assume the safety or neurotoxicity (or the location of effects) simply by extrapolation.
An illustrative example for this principle is a comparison of MPTP to 2′-NH2-MPTP. Although these two compounds are similar in structure and both cause neurotoxicity, the susceptible neural cell populations are different, thereby engendering distinct functional and structural changes (Figure 2). 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine damages the dopaminergic system, whereas 2′-NH2-MPTP attacks the serotonergic system. Note that the coronal levels (cross-sections) that would be used to sample the sensitive areas for one compound do not overlap with the levels needed to reveal the areas sensitive to the related agent. If a safety study were designed based only on the neurotoxic profile of one of these agents to assess the other one, not only would cell death not be found where expected for the second molecule, but also the coronal levels at which cell death did occur would not be available for analysis and detection.
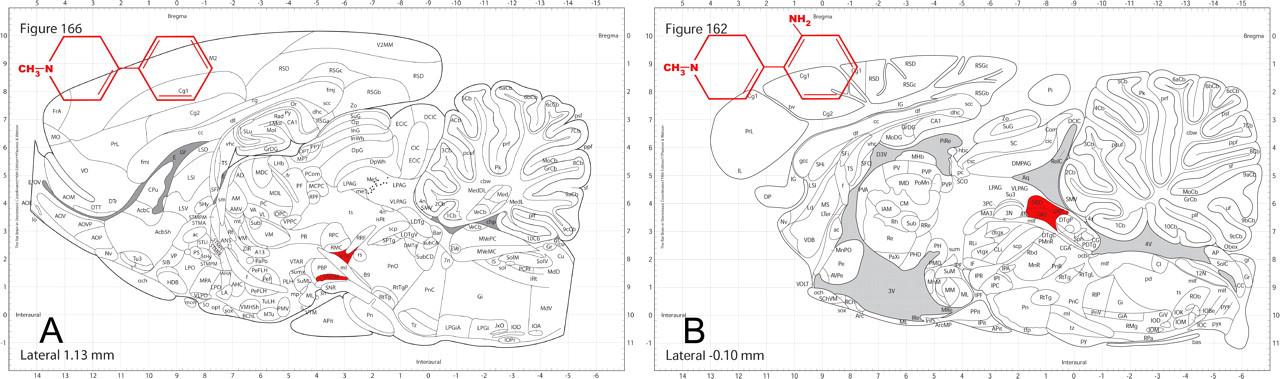
Structurally similar compounds do not necessarily have equivalent neurotoxicity profiles. (A) 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine destroys cells in the ventral tegmental area and pars compacta of the substantia nigra. (B) 2′-NH2-MPTP selectively destroys cells in the dorsal raphe. Thus, a targeted brain-sampling approach based on the neurotoxic damage produced by only one of these agents could miss neurotoxic changes induced by the related compound.
Another example illustrates the pitfall of only looking for neurotoxicity where it is expected. Figure 3 depicts the findings of two separate research groups that studied the neurotoxicity of D-amphetamine. In the first study, the researchers anticipated, looked for, and confirmed that D-amphetamine destroyed cells in the parietal cortex and somatosensory barrel field cortex (Belcher et al., 2005). A second study by the other group did not restrict the assessment to expected areas, so it not only confirmed the findings of the first group, but more importantly, it also found new sites of cell death in the frontal cortex, piriform cortex, hippocampus, caudate putamen, and ventral posterolateral nucleus of the thalamus (and also several other areas not shown in the figure, including the tenia tecta, septum, and other thalamic nuclei (Bowyer et al., 2004)). In this case, damage was found where expected, so neurotoxicity was at least recognized, although the full extent was not appreciated. In an opposite sense, if the assessment is restricted to only anticipated areas of concern and no lesions are found, a true assessment of neurotoxic risk cannot be compiled.
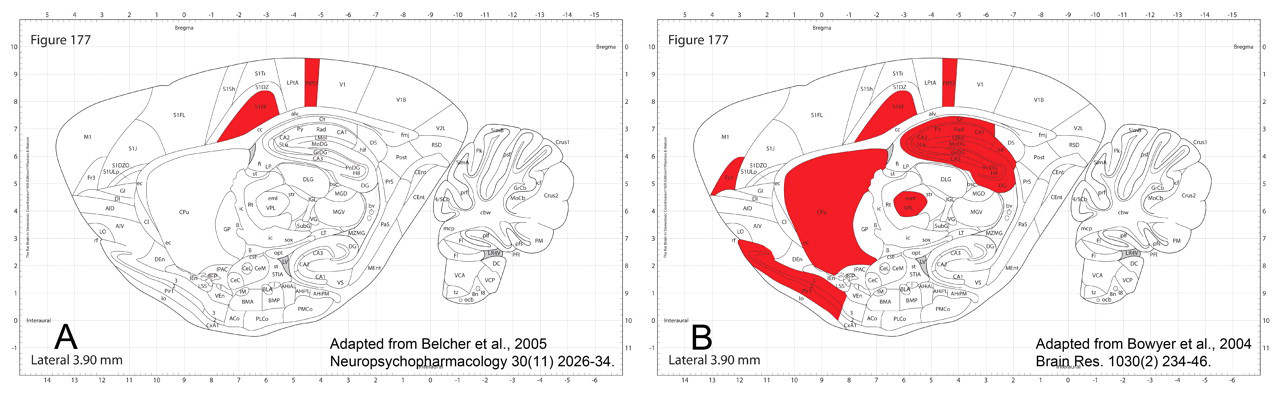
The sampling scheme dictates the distribution of cell death (red-shaded areas) attributed to D-amphetamine. (A) In one laboratory (Belcher et al, 2005), the brain was assessed only in areas expected to have neurotoxic lesions, and the apparent neurotoxicity signature is limited. (B) In another laboratory (Bowyer 2004), more comprehensive brain sampling revealed cell death in many more locations. The apparent absence of neurotoxicity (i.e., false negative results) can be reliably avoided by assessing more brain levels.
The final example of this principle relates to one of the most studied neurotoxic compounds, MK-801 (Figure 4). The first indication of neurotoxicity was the observation by John Olney of vacuoles in the posterior cingulate/retrosplenial cortex (Fix et al., 1993). As MK-801 was studied further, permanent damage was shown to occur in the form of cell death. The location of cell death coincided with the location of the early occurring vacuoles, but more significantly, cell death was found in other regions of the brain distant from the vacuolated sites.
Cell death induced by MK-801 is more broadly distributed than was predicted by the location of early neuronal vacuolation. (A) The earliest lesion in MK-801 neurotoxicity is acute neuronal vacuolation in the posterior cingulate/retrosplenial cortex occurring about four hours after exposure and peaking at six hours. (B, C, D) By two days after exposure and peaking at three to four days, MK-801–induced cell death occurs in the vacuolated regions but, significantly, it also occurs in many areas without vacuoles, including the tenia tecta, dentate gyrus, piriform cortex, amygdala, entorhinal cortex, and ventral CA1 and CA3 domains of the hippocampus.
Because of the neurotoxic profile of MK-801, the FDA routinely has requested safety studies to evaluate for the presence of the vacuoles (Olney’s lesions). As more was learned about the correlation of vacuoles with subsequent cell loss, markers of cell death have become the sole end point for these safety studies. In our experience with the FDA, the original regulatory requirement was restricted to vacuoles, but the approach evolved to encompass cell death assessment in both vulnerable areas and other regions. Specifically, in safety studies for NMDA receptor antagonists to be presented to the FDA, the investigator’s laboratory evaluates fifty (or more) evenly spaced coronal sections that sample the entire brain.
Principle #3
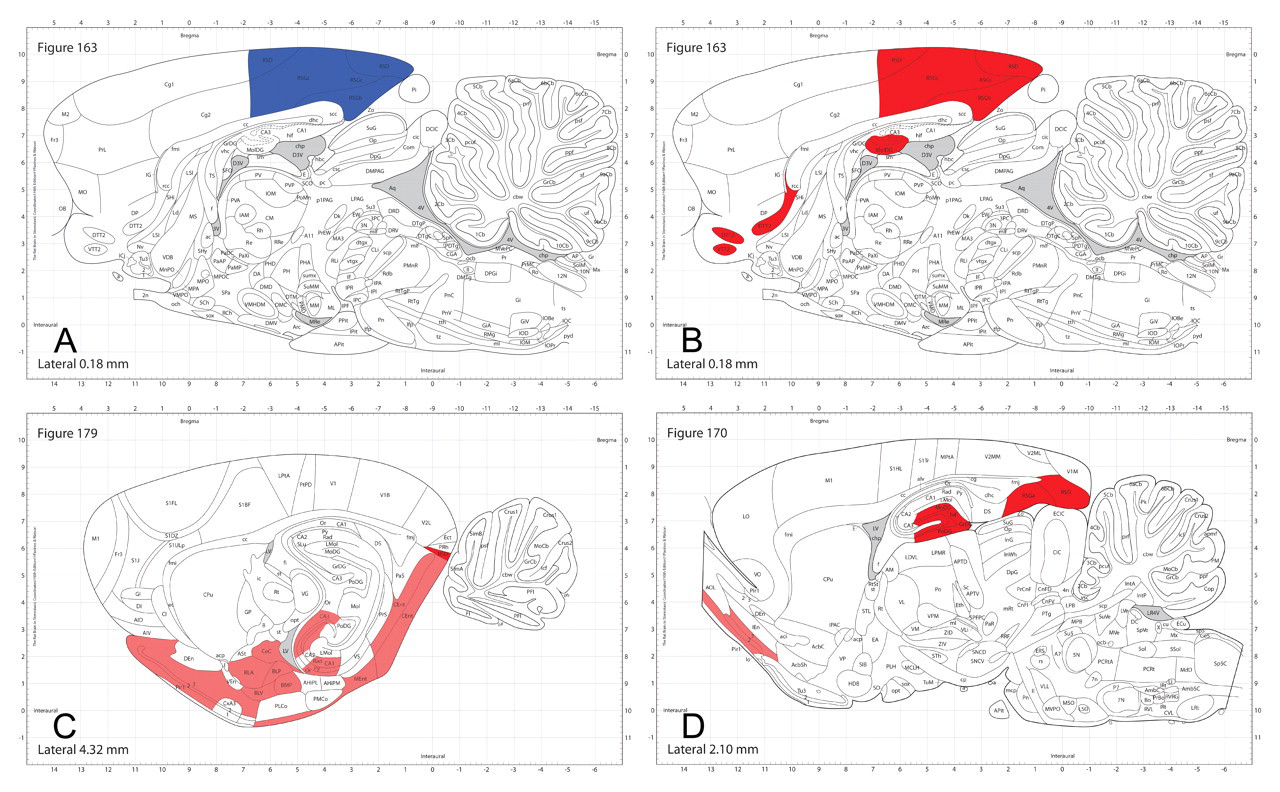
It is imperative to sample from representative minor divisions of the brain, not just from major divisions. Compounds do not necessarily have an impact on an entire “major” population, and different compounds may affect different subgroups of the same major population. For example, alcohol, domoic acid, and PCP have all been shown to destroy cells in the hippocampus. However, none destroys cells in all hippocampal regions. In fact, each of the three neurotoxicants affects unique, non-overlapping areas of this structure (Figure 5). When hippocampus is harvested in a conventional study, the level shown on the left of in Figure 5B is a typical (popular) choice as the single location for sampling. For these three compounds, it is significant to note that the impact of alcohol can be appreciated only by evaluating a more caudal and ventral level of the hippocampus (Figure 5C). The lessons of these examples are that (1) major divisions of the brain are not homogenous from a neurotoxicity standpoint and that (2) the coronal levels containing each subpopulation must be included in the sampling scheme if a complete assessment of neurotoxic risk is to be acquired.
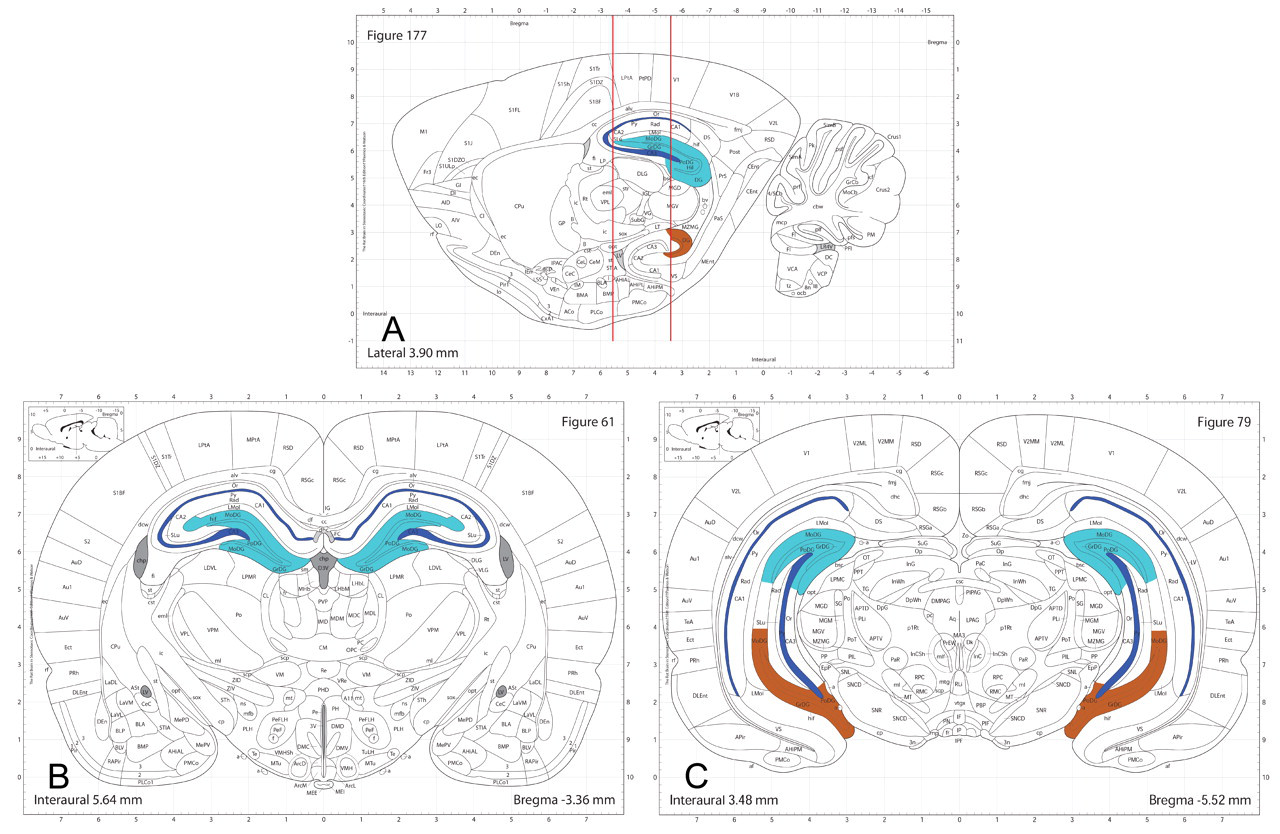
Different neurotoxicants affect distinct neural cell subpopulations in the hippocampus, a common site for neurotoxic lesions, indicating that adequate brain sampling cannot be attained using a single section through this structure. The vertical lines in the sagittal map (A) demonstrate two common coronal sections to evaluate the hippocampus; the level on the left is the most common level used in nonclinical safety evaluations. The hippocampal areas affected by the three known neurotoxicants are represented by unique colors: dark blue = domoic acid, which destroys cells in the pyramidal layer; light blue = phencyclidine (PCP), which destroys cells in the dorsal dentate gyrus; and brown = alcohol, which destroys cells in the ventral dentate formation. These diagrams also highlight that compounds are often toxic only to very discrete subdomains within a major structure rather than inducing a homogenous pattern of lesions across the entire region.
Application of Neurotoxicity Principles toward Study Design: Targeted Approach
A common strategy in using the targeted approach is to use a brain-slicing mold to produce specific levels for evaluation. Sampling brains at predefined linear coordinates in this way ensures relative (but not precise) consistency in the level being sampled. For discussion purposes, three levels that are often considered the standard for implementing this method in rats —taken through the optic chiasm, median eminence, and the cerebellum, corresponding with Paxinos Bregma coordinates of approximately −1, −3.5, and −11, respectively (Paxinos and Watson 2007)—will be considered for this discussion. Although these may not be the same levels or precise scheme used by any/all who still use a targeted scheme, the principles illustrated by the approach apply to this discussion. In Figure 6, a sagittal view of each brain has been adapted from Paxinos atlas images to shade the area(s) found to have experienced cell death by each neurotoxicant. Overlaid on each image are vertical lines that represent the Bregma coordinates near which samples would be taken in the targeted approach. The results are grouped into one of four categories, as shown by the various columns. The far left column includes compounds whose profiles should be easily detected if samples were taken at these three standard sections. The second column from the left shows the compounds that should also be detected by sampling at these levels, but since these compounds have a smaller profile, they would be somewhat less likely to be detected. The third column from the left demonstrates compounds that have very small profiles and/or the profile edge is very near to the sampling point; thus, the sampling scheme has the potential to miss the neurotoxic lesions. In the far right column are compounds for which the neurotoxic profile does not coincide with any of the levels in this standard sampling scheme. Using the described targeted approach outlined previously, these compounds would not be detected as being neurotoxic. All of these evaluations assume sampling will include all of the populations that exist at the identified levels. If populations within that level were selectively sampled, doing so would further reduce the chance of detection.
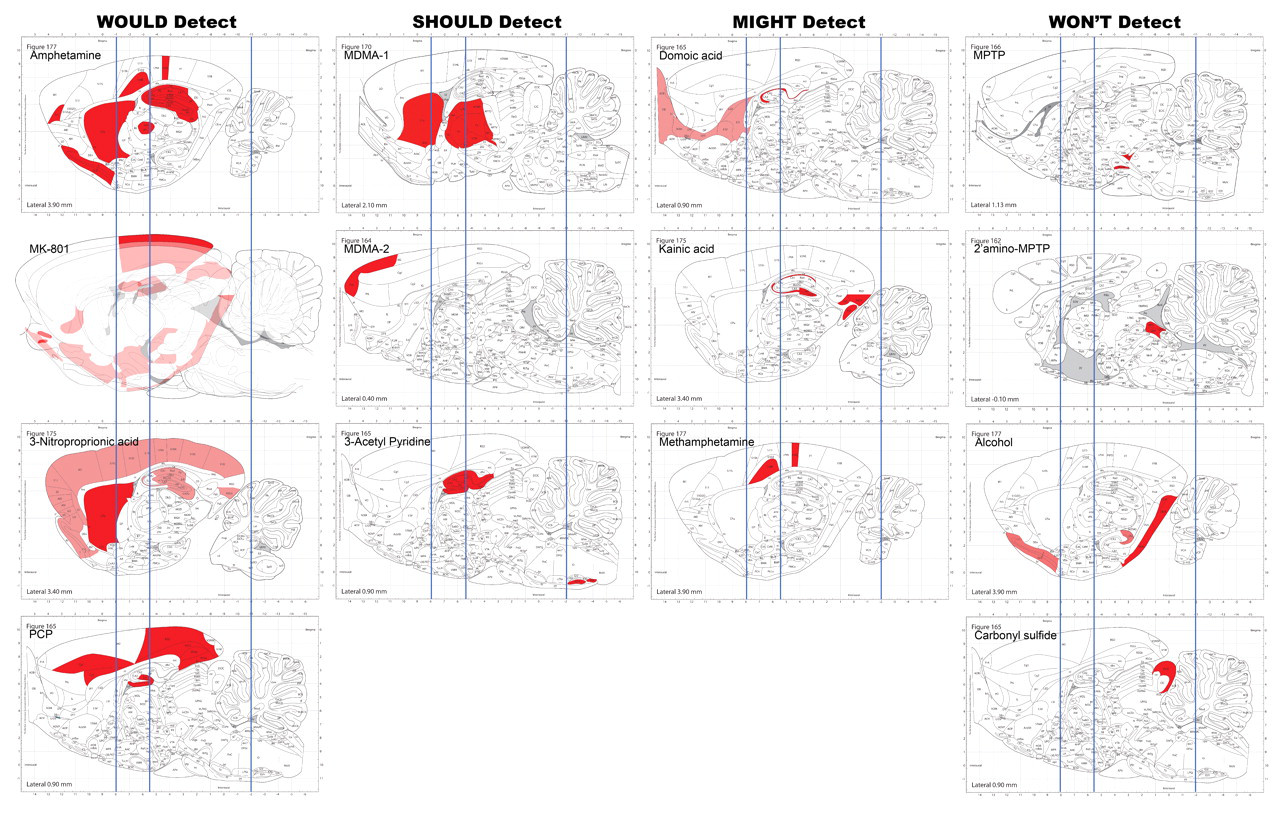
A conventional three-level sampling scheme for the rat brain detects less than 50% of known neurotoxicants. Vertical lines in each panel depict commonly used coronal levels for sampling brain regions that often exhibit toxicant-induced damage (e.g., cerebral cortex, hippocampus, and cerebellum/pons); lines that cross a red-shaded area would have successfully discerned neurotoxic lesions during a neuropathological examination. Compounds in the far left column would be easily detected, and those in the second column from the left should also be found by sampling at these three standard levels. In contrast, compounds that induce lesions in very small regions (third column from left) or that target regions at a distance from the standard levels (right column) are likely to be missed using targeted sampling schemes.
Of the thirteen compounds profiled (MDMA is shown twice at different levels), 30% (four of thirteen) would have missed an observation of neurotoxicity, and it is likely that another three would be at high risk for not being observed, making the success rate for detection using the three standard sections only 46%. The weakness of potentially missing significant neurotoxicity by inadequate sampling is highlighted by these examples. Although the cost and effort are minimized with this approach, this demonstrated risk of failed neurotoxicity recognition is not an acceptable outcome when seeking to minimize the neurotoxic risk to the human population.
Application of Principles toward Study Design: Comprehensive Strategy
The standard three-level approach as outlined above is obviously not adequate, so more sampling is clearly required to reliably detect many known neurotoxicants. The question then arises how to increase the sampling to an adequate level, ideally sampling all 600+ neural cell populations in the brain, without creating unnecessary increases in effort, time, or cost. One approach to a more comprehensive assessment is to systematically evaluate brain cross-sections (levels) taken at regular intervals from one end of the brain to the other. Defining the interval spacing between the levels then becomes the key variable in this sampling approach.
To begin this discussion, and continuing to focus on the rat as a model vertebrate brain, it is critical to understand how much the brain anatomy changes over a short interval. Using our sagittal view of rat brain once again in Figure 7 (A), a single cross-section represented by each vertical line yields a given coronal level. Any single level crosses a relatively small percentage of the 600+ brain cell populations, and the cell populations in the brain differ dramatically between levels that are separated by very short intervals. For example, Figure 7 (B–E) displays the structures present in coronal levels 1 to 4 from (A), each of which is separated from its neighbor by an interval of 1 mm. The shaded areas in each map represent populations that exist in that coronal level of the rat brain that do not occur in the prior level. Therefore, although 1 mm between levels (across a 21-mm-long adult rat brain) seems very close together, the illustration above indicates that there are extensive neuroanatomical variations across even very short differences in rat brains. Defining a sampling approach for histopathological safety assessments thus becomes a tradeoff between exhaustive sampling/laborious assessment and targeted sampling/easier examination. To sample and analyze every level (each section cut) throughout the brain would be 100% thorough, but it is impractical, expensive, and unnecessary. However, sampling levels at too great an interval can leave gaps that fail to assess some populations, and thus are inadequate. A compromise must be achieved that delivers reasonable safety assurance without imposing an excessive burden on both neurohistologists and neuropathologists. As discussed previously and as displayed in Figure 7, 1-mm sampling through a rat brain can be shown to leave broad gaps between samples, even though this scheme yields about twenty to twenty-three levels/sections.
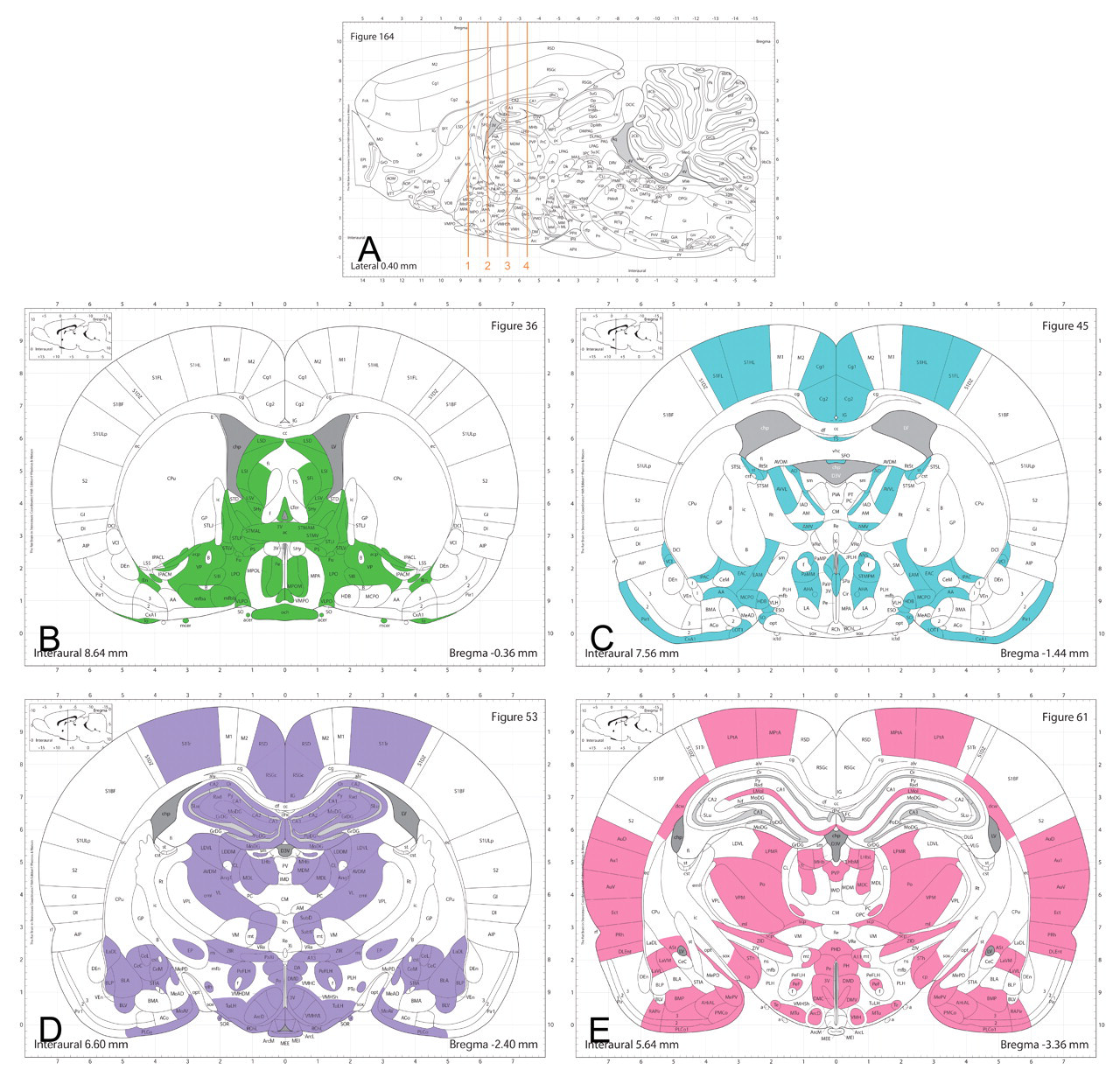
Profound neuroanatomical variations occur across very small distances in the rat brain. (A) The vertical lines depict the locations of coronal levels (shown in B, C, D, and E) that are 1 mm apart. The shaded areas represent the structures in that level that do not occur in the level 1 mm adjacent to it: (B) contains thirty-five structures not in level (C); (C) has fifty-five not in (B) and forty-five not in (D); (D) possesses sixty-two not in (C) and thirty-three not in (E); and (E) has forty-eight not evident in (D).
Doubling the sample rate to acquire a level at intervals of every 0.5 mm greatly improves the opportunity to sample all populations, but gaps can still occur. This approach typically delivers forty to forty-six levels/sections per rat brain. Doubling the sample size again yields an interval of 0.25 mm between levels, or eighty to ninety levels/sections per rat brain. This very thorough sampling rate is the one reflected in the original rat brain atlas by Paxinos and Watson (1982), and most neural cell populations will be sampled multiple times.
In practice, a 0.25-mm sampling interval is probably more rigorous than is necessary. As a meaningful benchmark, the most commonly used interval for basic research purposes in rat brain is 0.32 mm (Figure 8). This has proven to be a useful strategy for characterizing neurotoxic effects in the rat brain in industry and ensures adequate representation through most regions and neural cell subpopulations of the brain. This approach yields about sixty to sixty-five sections in a rat brain. This scheme is also the minimum sampling approach that has been approved by the FDA in the numerous Good Laboratory Practice–compliant regulatory studies in which one author (RCS) has participated during over a decade of applied neuroanatomy practice.
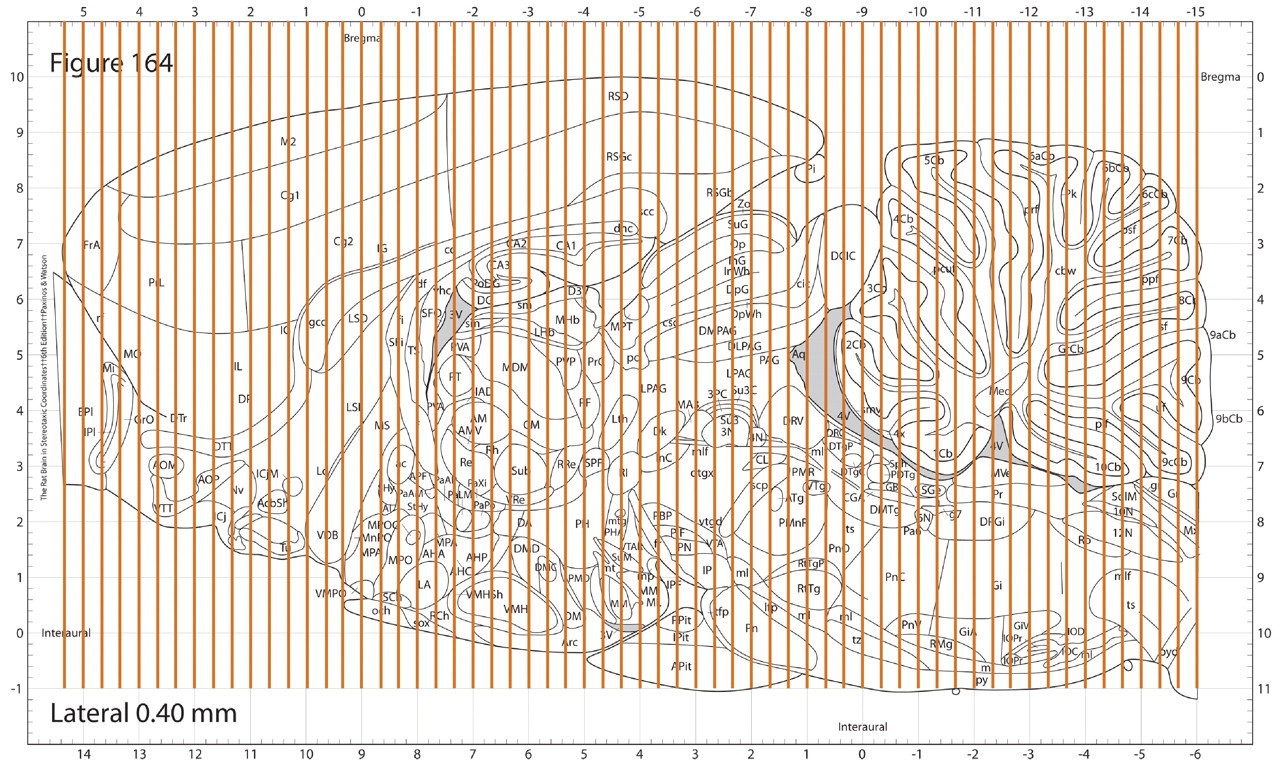
Depiction of the recommended sampling frequency for a systematic, comprehensive approach to sampling the rat brain. The vertical lines represent coronal levels spaced at a uniform interval of approximately 0.32 mm across the entire rat brain, which is a common sampling frequency for basic neurobiological research in this species. The gaps between levels are narrow enough that even the smallest of the more than 600 neural cell populations are unlikely to be missed.
In the distant past, the prospect of performing the neurohistology on sixty to sixty-five levels of each rat brain might have been considered unnecessary (hence the traditional approaches with less extensive sampling), and the effort to conduct such a study would have been considered to be prohibitive in time, cost, and effort. However, along with our increased understanding of the need for greater sampling, we have also made significant technologic advancements in assessment technology. Multiply embedding tissues into paraffin reduces the effort of sectioning and staining by the same factor as the number of tissues that can fit into the cassette (as many as eight rat brains). Even greater efficiencies are possible with larger-format embedding processes using gelatin-embedded sections and microtome slicing in which as many as forty rodent brains can be processed simultaneously. Once sectioned, a host of new specialty stains is now available that, unlike hematoxylin and eosin, stain only the feature (such as degenerating neurons) that the researcher needs to see. Using new staining technology can make the pathologic review much faster and more reliable than traditional methods. In short, the effort to process sixty sections versus three sections does not represent a linear increase in cost of 20×.
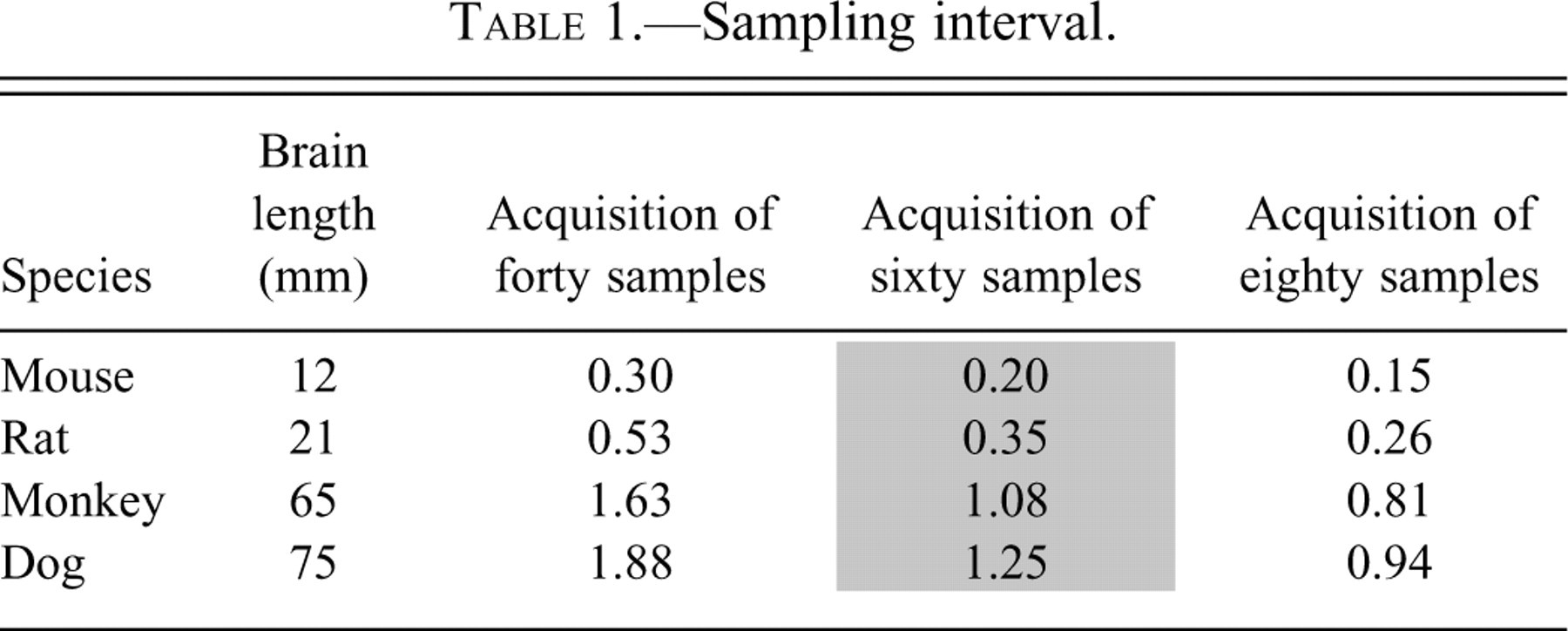
The rat brain has been the focus of this discussion, but certainly other animal species used for nonclinical safety testing need to receive a similarly comprehensive neuropathological evaluation. Conveniently, a rule of thumb has emerged by which the brain from any species can be sampled to produce comparable levels of representation (Table 1 ).
Sampling interval.
Timing and Methods of the Evaluation
Looking for neurotoxicity in the correct location is important, but equally critical is using the best tools (special stains) and looking at the correct time. Each of these topics is worthy of a manuscript in itself to discuss the evidence and justifications for recommendations. Although it is not the purpose of this manuscript to provide these details, it is important to introduce the concepts and a summary of guideline conclusions to optimize the utility of these recommendations for more comprehensive brain sampling.
Tools
Stains and antibodies used in histochemistry have been developed with the goal of revealing specific anatomical features. Therefore, the chosen stain should match the end point being evaluated. The minimum end point in a safety assessment should include the evaluation of cell death, as this outcome represents a marker of permanent damage to the brain. The disintegrative degeneration class of stains includes the Fluoro-Jade (Schmued and Hopkins 2000) and cupric silver (Switzer 2000) methods. These techniques are capable of revealing all four degenerating elements of a neuron, namely, the cell body, axons, axon terminals, and dendrites. These stains reveal a more comprehensive scope of cell death pathology than do hematoxylin and eosin and Nissl stains, and the results are more conspicuous and easier to interpret. Immunohistochemistry for microglia and astrocytes are standard tools to look for neurotoxicity end points such as inflammation and cellular reaction. Other, more specific tools may also be warranted depending on the nature of the compound being evaluated.
Timing
The timing of the necropsy following exposure to a compound is absolutely critical. Although damage can be permanent, the readily detectable pathology in the case of cell death (i.e., dead or dying cells and/or cell debris) persists for only approximately two or three days. To assess the brain before or after the period during which peak cell death occurs might lead to a lack of pathology and a potentially false negative result. It is also essential to probe for neurotoxicity during the course of the study, not just at one terminal time point. Empirically, the most common time course from the expression of cell death is from approximately two days until about five days following the first exposure, after which no additional cell death will usually be visible. Therefore, it is always imperative to conduct an acute assessment for neurotoxicity as well as subchronic and chronic evaluations. All three of the time frames should be seen as cumulative testing, not alternatives. Based on empirically derived data and the mechanics of cell death, disintegration, and clearance, the timing of evaluations (following the first dose) should typically be performed as follows: Acute: two and four days Subchronic: seven to ten days, sixteen to twenty days, twenty-five to thirty days Chronic: monthly from thirty to ninety days, quarterly for studies longer than ninety days.
A Practical Tiered Approach to Comprehensive Evaluation of the Brain
At a minimum, a compound should receive one comprehensive evaluation for neurotoxicity, as recommended here, in its development history prior to starting clinical trials. Some pharmaceutical companies may prefer to wait on this comprehensive evaluation until later in the development cycle, presumably to avoid the labor-intensive analysis for compounds that might fall out earlier during the development cycle.
An effective approach to tiered testing in the brain early in the development cycle uses the following strategy. Either before or after a “no observed adverse effect level” has been established in other test systems, the first tier of testing for neurotoxicity end points would be comprehensive sampling of the brain in high-dose animals (or the most effective dose). If the results were negative, the same comprehensive screening would still be required at lower repeated doses. If the results were positive for neurotoxicity, further testing at lower doses would be necessary to determine the no observed adverse effect level for neurotoxicity for that compound.
Conclusions
At the present time, the best technology available to us to predict cell death in human brains is the histologic evaluation of brains from animal models following exposure to the test article. In the future, we may have access to biomarkers, enhanced imaging modalities, predictive data modeling, or other tools that would allow this evaluation to occur in a noninvasive manner, but for now and the foreseeable future, the “gold standard” method to be used will remain neuropathologic assessment. The purpose of this article has been to evaluate design considerations that affect sampling strategy, and the findings reveal a classic risk/benefit tradeoff. There can be little debate that increased sampling of the brain decreases the potential that neurotoxicity would be missed, thus providing improved safety assurance. It can also be stated categorically that it requires less effort and cost to sample and evaluate fewer regions of the brain. The question thus becomes what level of sampling is most appropriate and prudent.
Using the approach the FDA takes to neurological safety testing for pharmaceutical candidates dictates that the best current scientific methods should be used. In the case of neurological sampling approaches, the optimal method would be the approach that has the greatest chance of detecting neurotoxicity and the least risk of missing a finding, performed with the least cost in terms of money, time, and effort. In our extensive experience, acquiring and evaluating fifty to sixty coronal levels of the brain is the minimum comprehensive sampling approach that provides the opportunity for nearly 100% detection of neurotoxicity with very little risk of a missed finding owing to inadequate sampling. As a minimum, a compound should receive one comprehensive evaluation for neurotoxicity during its development history, preferably prior to starting any clinical trials or other human exposure (if a potential environmental toxin) as a routine practice.