
Abstract
In situ hybridization (ISH) is used for the localization of specific nucleic acid sequences in cells or tissues by complementary binding of a nucleotide probe to a specific target nucleic acid sequence. In the last years, the specificity and sensitivity of ISH assays were improved by innovative techniques like synthetic nucleic acids and tandem oligonucleotide probes combined with signal amplification methods like branched DNA, hybridization chain reaction and tyramide signal amplification. These improvements increased the application spectrum for ISH on formalin-fixed paraffin-embedded tissues. ISH is a powerful tool to investigate DNA, mRNA transcripts, regulatory noncoding RNA, and therapeutic oligonucleotides. ISH can be used to obtain spatial information of a cell type, subcellular localization, or expression levels of targets. Since immunohistochemistry and ISH share similar workflows, their combination can address simultaneous transcriptomics and proteomics questions. The goal of this review paper is to revisit the current state of the scientific approaches in ISH and its application in drug research and development.
Keywords
This article is a product of a Special Interest Group of the European Society of Toxicologic Pathology (ESTP). The views expressed in this article are those of the authors and do not necessarily represent the policies, positions, or opinions of the ESTP. This article should not be construed to represent the policies, positions, or opinions of their respective organizations, employers, or regulatory agencies.
Introduction
In situ hybridization (ISH) is a tissue-based molecular technique for the detection and localization of specific nucleic acid sequences within cells or other matrices, which operates on a complementary binding of a nucleotide probe to a specific target sequence of DNA or RNA. 1 In the armamentarium of molecular experimental and toxicological pathology used in drug research and development, ISH has been traditionally less utilized than immunohistochemistry (IHC) on a routine basis, but lately it has gained increasing interest.2,3 This evolution is partly due to the lack of specific antibodies against a target of interest in a particular animal species, novel or rare targets, the development of nucleic acid-based therapeutics, and the easier accessibility of improved, ready-to-use ISH technologies.4,5
The first ever ISH protocol to visualize nucleic acids was described in 1969, and it was based on radiolabeled RNA probes to detect DNA sequences.6,7 In 1977, in situ applications used RNA probes marked with a fluorophore, called fluorescent ISH (FISH), to detect chromosomal targets.8,9 Few years later, chromogenic-based approaches were used with biotin and digoxigenin. 10 FISH for the detection of RNA was first shown in 1982, 11 and at the end of the last century, the ISH technology allowed the in situ tissue detection of single mRNA transcripts. 12
In the last few years, ISH technologies have become a powerful and versatile tool within research and diagnostics. ISH allows the detection of coding and noncoding RNA molecules (i.e., untranslated RNA regulating essential cell functions and adaptations) such as microRNA (miRNA/miR), as well as therapeutic oligonucleotides. Because it provides spatial information at the tissue level, ISH has become a good complementary tool to other RNA-based molecular techniques such as reverse transcription quantitative real-time PCR (RT-qPCR) or next-generation sequencing (NGS). 13 It has been also proven to be a useful complementary tool to IHC (i.e., to complement protein data and support IHC assay development) or even to replace it when no suitable antibodies are available to identify targets of interest.14,15
This review intends to provide a general overview of the ISH covering different facets of the technique including protocol-related aspects, types of probes and detection systems, as well as main applications in drug discovery and development. Multiplexing and ISH/IHC mixed techniques will be shortly alluded to, but not extensively reviewed.
Basic Technique Introduction
ISH is a technique used for the detection and localization of specific nucleic acid sequences within a tissue section. The target nucleic acid sequences can be endogenous nonmethylated DNA, methylated DNA, mRNA transcripts, noncoding RNAs (including small- or mid-sized RNA), or exogenous DNA or RNA from infectious agents or xenotransplanted cells with partially foreign nucleic acid sequences.16-21 Regardless of the final target, the underlying principle of ISH is the detection of nucleic acids by the specific hybridization of a complementary nucleic acid probe on which a reporter molecule is attached, that is, with a tagged probe.
With the increased use of ISH, different types of probes and detection methods have been developed for a variety of specific applications.1,22 Despite the different approaches available, the common following steps are performed: tissue preparation, hybridization, detection methods, visualization, and evaluation.
Tissue Preparation
Tissue preparation is a critical step for any ISH approach and consists of both tissue fixation and permeabilization. 1 Among various preanalytical factors that may affect ISH results, ischemia time, postmortem interval, fixative-to-tissue ratio, and fixation duration are critical, 23 since those influence the RNA integrity for the subsequent investigation (Figure 1). In the early days, ISH was mainly performed on frozen sections, 24 prepared by either snap-freezing or freezing after short (1-3 hours) fixation in 4% paraformaldehyde/0.1M PBS pH 7.4, followed by cryopreservation in 30% sucrose. While frozen sections provide several advantages (e.g., excellent nucleic acid preservation), there are also disadvantages (e.g., poor tissue morphology due to freezing artifacts, reactivation of RNases after tissue thawing with an increase in RNA degradation, storage considerations, and limited retrospective examination since freezing is normally not included in the standard routine fixation method). To overcome these caveats, a variety of chemical fixatives have been tested over the years,24,25 and today, 10% neutral buffered formalin (NBF), the standard fixative in pathology, is known to be the most suitable fixative for ISH when fresh–frozen tissue samples are not available.24,26 After tissue sampling, it is recommended to preserve the tissues (maximum thickness of 5 mm) in fixative as soon as possible to avoid postmortem degradation. Fixation time for 24 hours (±12 hours) at room temperature in 10% NBF at a 10:1 ratio of fixative to tissue (with or without postfixation in 70% ethanol depending on storage and shipping) has been demonstrated to provide optimal fixation. Shorter fixation periods might lead to insufficiently preserved tissue (under-fixation) and tissue over-digestion during the later permeabilization step with proteases, which leads to poor tissue morphology and RNA degradation. In contrast, longer fixation periods might require stronger proteases and/or retrieval treatments to avoid poorer tissue accessibility of the probes and lower signals. Some organs, like eyes and testes, might not be well preserved when using these fixation parameters and other fixatives like Davidson’s fixative are often used.27,28 Large animal brains are commonly fixed intact for a longer period. 29 If the fixation protocol deviates from the standard fixation parameters, additional validation steps might be needed (i.e., adjust pretreatment conditions). In the case of large organs, trimming prior to fixation or post sacrifice animal perfusion with formalin is also a way to use standard fixation parameters. Although decalcifying agents compromise the RNA quality, ISH in FFPE decalcified tissues is described.30,31
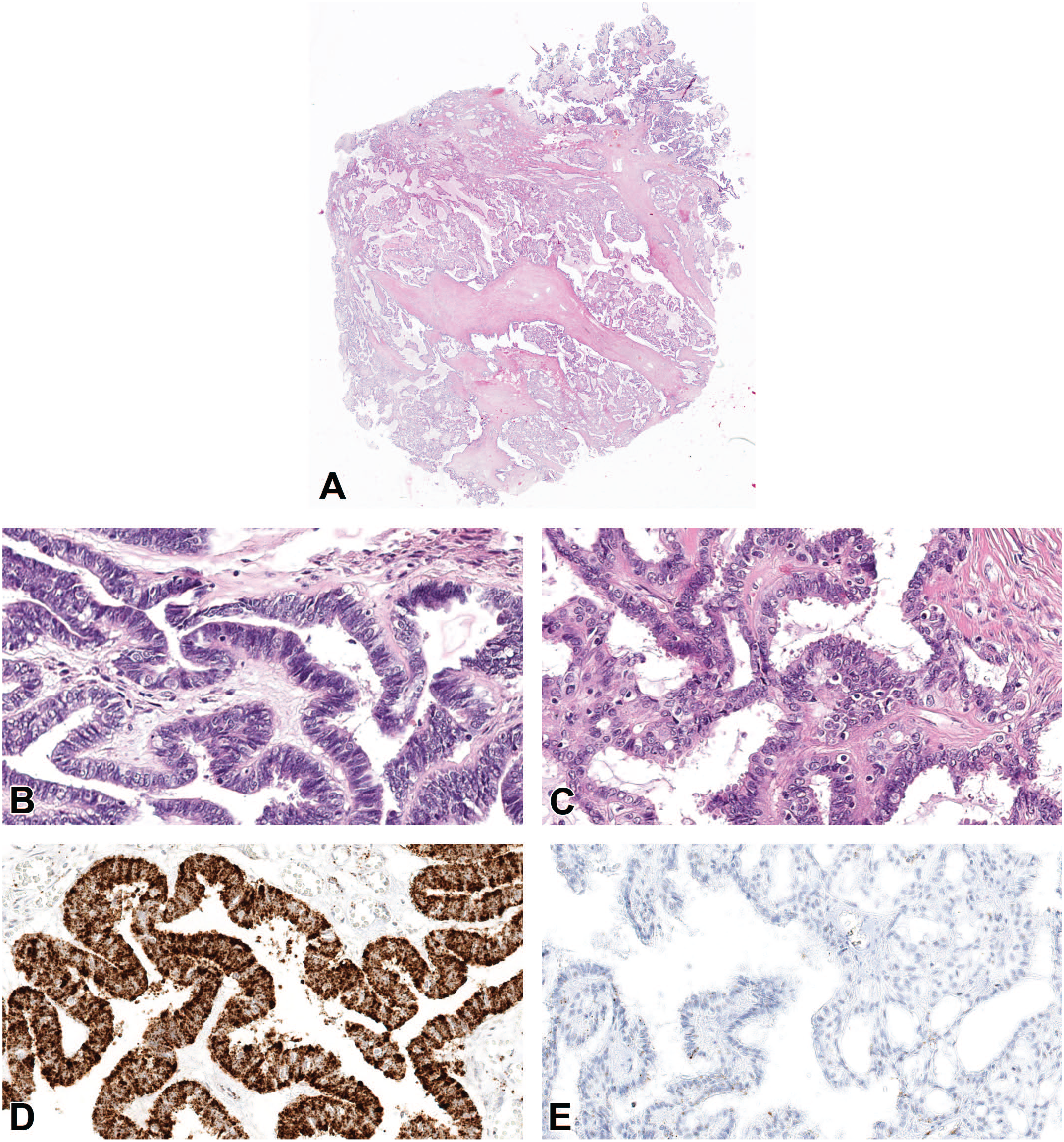
Preanalytical factors influence RNA integrity. Example of optimal or suboptimal fixed areas from a human carcinoma (A), different areas such as periphery (B) and central areas (C) look morphologically similar but in situ hybridization for human PPIB (peptidyl-prolyl cis-trans isomerase B) (housekeeping gene used as a positive control for RNA integrity) demonstrates higher positive staining in the optimal formalin-fixed periphery (D) compared to suboptimal formalin-fixed central areas (E).
The time and temperature at which the paraffin-embedded tissues are stored also influence the RNA integrity.32,33 Storage of paraffin blocks at room temperature for more than 5 years can greatly reduce the performance of ISH. 34 Therefore, storage at lower temperatures is recommended to preserve the RNA integrity in paraffin blocks for ISH.32,34 Also freshly cut slides are recommended, although vendor guidelines should be revised, for example, in RNAscope (Advanced Cell Diagnostics (ACD), Inc.) unstained tissues mounted in positively charged slides should be used within 3 months when kept at room temperature, or within 1 year when stored at −20°C or −80°C.34,35
In the first steps of the protocol, samples are permeabilized to allow proper penetration of hybridization reagents into the tissue and cells. 1 For this purpose, detergents (e.g., Tween-20, Triton X-100 and 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS)) at a concentration of 0.1% protease (e.g., proteinase K) treatments and heat-mediated pretreatments are used to improve tissue permeability and probe accessibility to its target.1,36 The intensity and duration of these pretreatments need to be adapted depending on the tissue and fixation duration. Too gentle or too strong pretreatments may affect the subsequent detection negatively (Figure 2). Traditionally, an RNase-free environment (solutions and surfaces) and RNase blockers (for endogenous RNases in tissues) were important to avoid RNA degradation by RNases, especially on fresh snap-frozen tissues. However, with the most recent commercially available ISH assays such as RNAscope, an RNase-free environment is not strictly required after tissue fixation since endogenous RNases are de-activated by NBF fixation.
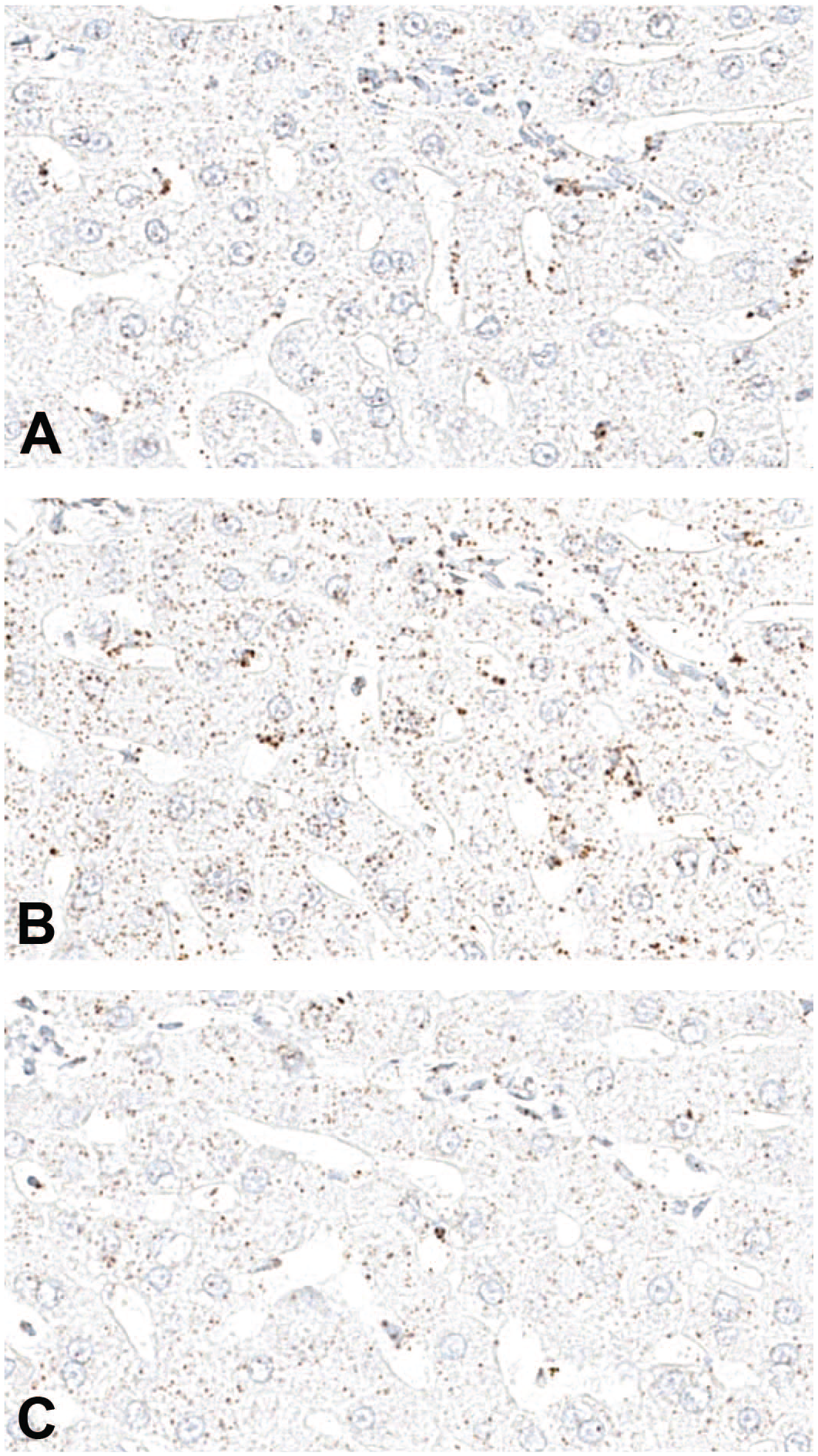
Optimization of pretreatment conditions for in situ hybridization. Example of differently pretreated liver sections from a cynomolgus macaque, where different pretreatment in 1X retrieval buffer at 98°C to 104°C for 20 (A), 30 (B), and 40 minutes (C). Note that 30-minute pretreatment gave the best signal for a housekeeping gene used as positive control-cynomolgus PPIB (peptidyl-prolyl cis-trans isomerase B), whereas under (20 minutes) or over treatment (40 minutes) was not optimal.
Hybridization
The next step after tissue/cell permeabilization is the probe hybridization with the target sequence. The stringency of this reaction is dependent on specific properties of the probe (e.g., length, GC content, solution molarity) and the level of complementarity between the probe and the target sequence.1,37 The above-mentioned physical properties will have a direct influence on hybridization parameters like probe concentration, hybridization temperature (usually between 55°C and 75°C), time of hybridization, and the concentration of monovalent cations to be added in the hybridization solution. 37 In addition, the concentration of specific reagents (e.g., formamide for lowering the melting temperature of probe–target hybrids) added into the hybridization solution has to be taken into account.1,37 The hybridization temperature should be a few degrees lower than the melting temperature, which can be estimated by the formulas presented in Supplementary Figure 1; however, other different formulas can be also found in the literature. 38 Optimal hybridization conditions have to be determined to maximize specific hybridization versus nonspecific binding (background). Furthermore, drying out by evaporation during the hybridization time must be avoided as this would reduce the reaction volume and increase the concentration of the used reagents.1,37 Finally, by tuning the washing stringency conditions during the post hybridization, the removal of nonspecific hybrids or the preservation of higher-intensity labeling can be achieved. 1 It is relevant mentioning that many of these protocol conditions are already standardized and validated in ready-to-use assays such as RNAScope.
Probes
The probes for ISH consist of short nucleic acid strands that may be composed of DNA or RNA and have an affinity for specific target sequences of nucleic acids. 1 Regardless of the type, the probes are composed of two elements: the nucleotide sequence complementary to a target sequence and a molecule that allows the direct (e.g., fluorophores or radioactive) or indirect (e.g., digoxigenin) detection and visualization.1,24,39,40
Initially, the self-made probes prevailed and the nucleic acids used for the probes were obtained by purifying the DNA of interest, which was then labeled by nick translation or random priming.24,41 The main disadvantage of this method included limited sensitivity and low specificity mainly due to cross hybridization between the sense and antisense complementary nucleic acid strands. 24 With the advent of molecular cloning, this method was largely replaced by the recombinant technology and the probes can either be custom-made and subsequently labeled or bought ready to use from various providers (e.g., ACD, OGT). Irrespective of the used approach, important criteria to consider for the probe design and selection are their sensitivity and specificity, the tissue penetration ability and the stability of probe–target hybrids. 37 To maintain the balance between sensitivity, specificity, and physical properties, the composition of the probes and sequence lengths are crucial. 1 Knowing the exact nucleotide sequence of the DNA or RNA target will allow designing of precise complementary probes. When more than 5% of base pairs are not complementary, the probe will bind loosely to the target sequence increasing the risk of being washed away during the post hybridization washing. 1 Self-made probe design is typically performed using open-source software (e.g., ProbeDealer, OligoMiner), which allows the customization of different parameters according to the experimental needs, for example, the GC content influences the melting temperature (Supplementary Figure 1).
The probes can be composed of natural or modified nucleic acid elements, both come with advantages and disadvantages (Table 1). Among natural nucleic acid probes, DNA probes provide a high sensitivity; however, they do not hybridize as strongly to target mRNA molecules compared to RNA probes, so formamide should not be used in the post hybridization washes in this application. 1 In the case of double-stranded DNA probes a predenaturation step is necessary before hybridization, which might introduce technical challenges. 42 RNA probes can be produced as antisense or sense probes which could help to identify sense and antisense strands of viral DNA/RNA or to be used as a correct probe (antisense strand) and negative control (sense strand) probe. 43
Overview of different probe types
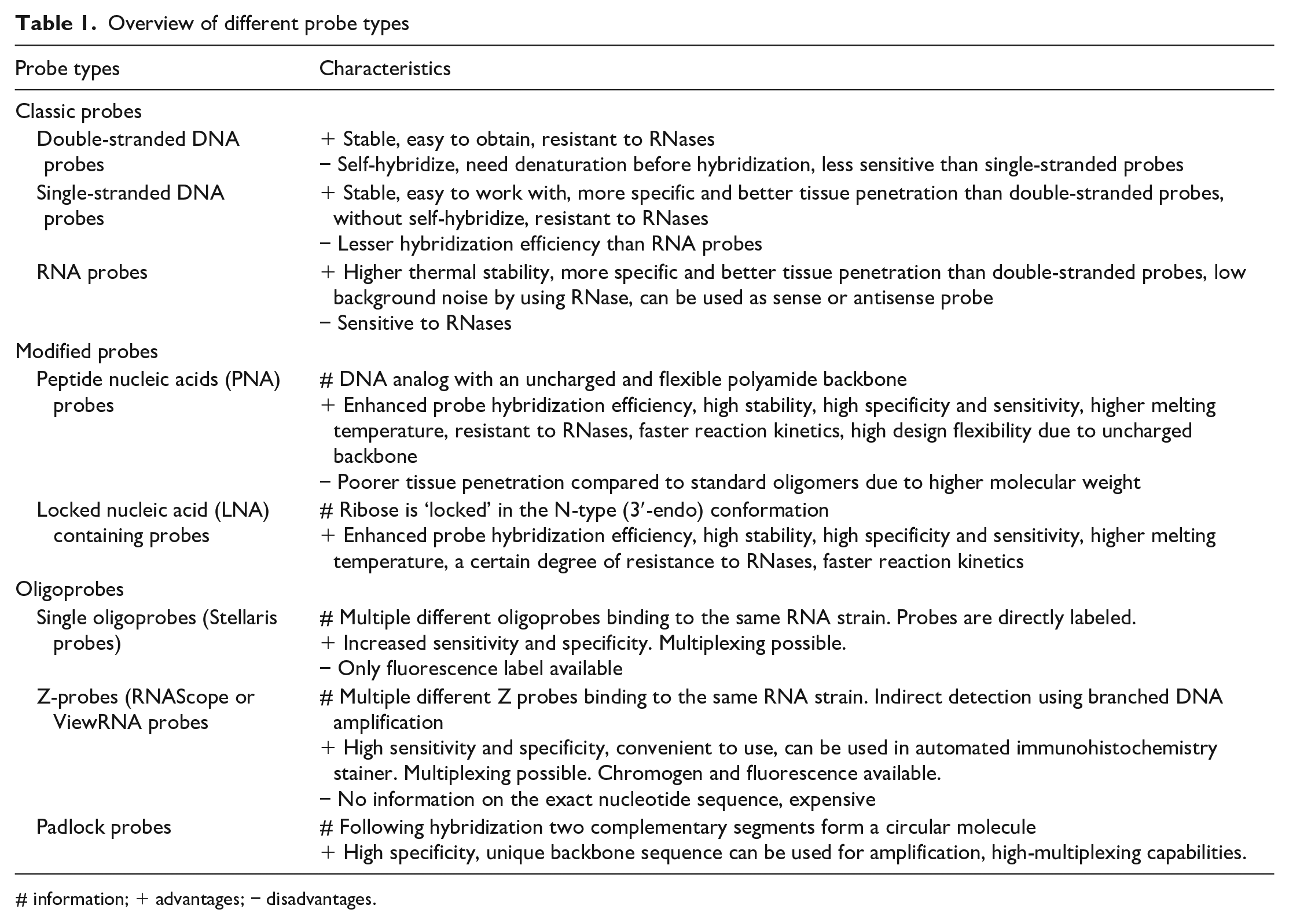
# information; + advantages; − disadvantages.
Modified nucleic acid probes contain modifications in their backbone structure giving resistance to extra- and intracellular nucleases without affecting the binding affinity of the probe–target sequence tandem. 44 The most commonly used modified nucleic acid probes are the peptide nucleic acids (PNAs) (Figure 3) and the locked nucleic acids (LNAs). PNAs are modified DNA analogs in which the phosphodiester backbone is replaced by repetitive units of N-(2-aminoethyl) glycine to which the purine and pyrimidine bases are attached via a methyl carbonyl linker. 45 Due to their uncharged and flexible polyamide backbone, PNAs hybridize with complementary DNAs or RNAs with high affinity and specificity (Table 1). On the other hand, LNAs have an O-methyl group, which bridges 2′ and 4′ carbons of the ribose ring covalently and effectively “locks” the ribose in the N-type (3′-endo) conformation. 46 This conformation enhances base stacking and phosphate backbone preorganization. Therefore, LNA-containing probes have an improved affinity for complementary DNA or RNA sequences resulting in a higher melting temperature (Table 1). This structure can be attributed to the increased stability against enzymatic degradation such as exo- and endonucleases. 47 In LNA probes, DNA and RNA bases can be positioned within a sequence wherever needed. By that, the properties of a probe can be altered and probes can be designed to detect nucleic acids with single-nucleotide variants. 48
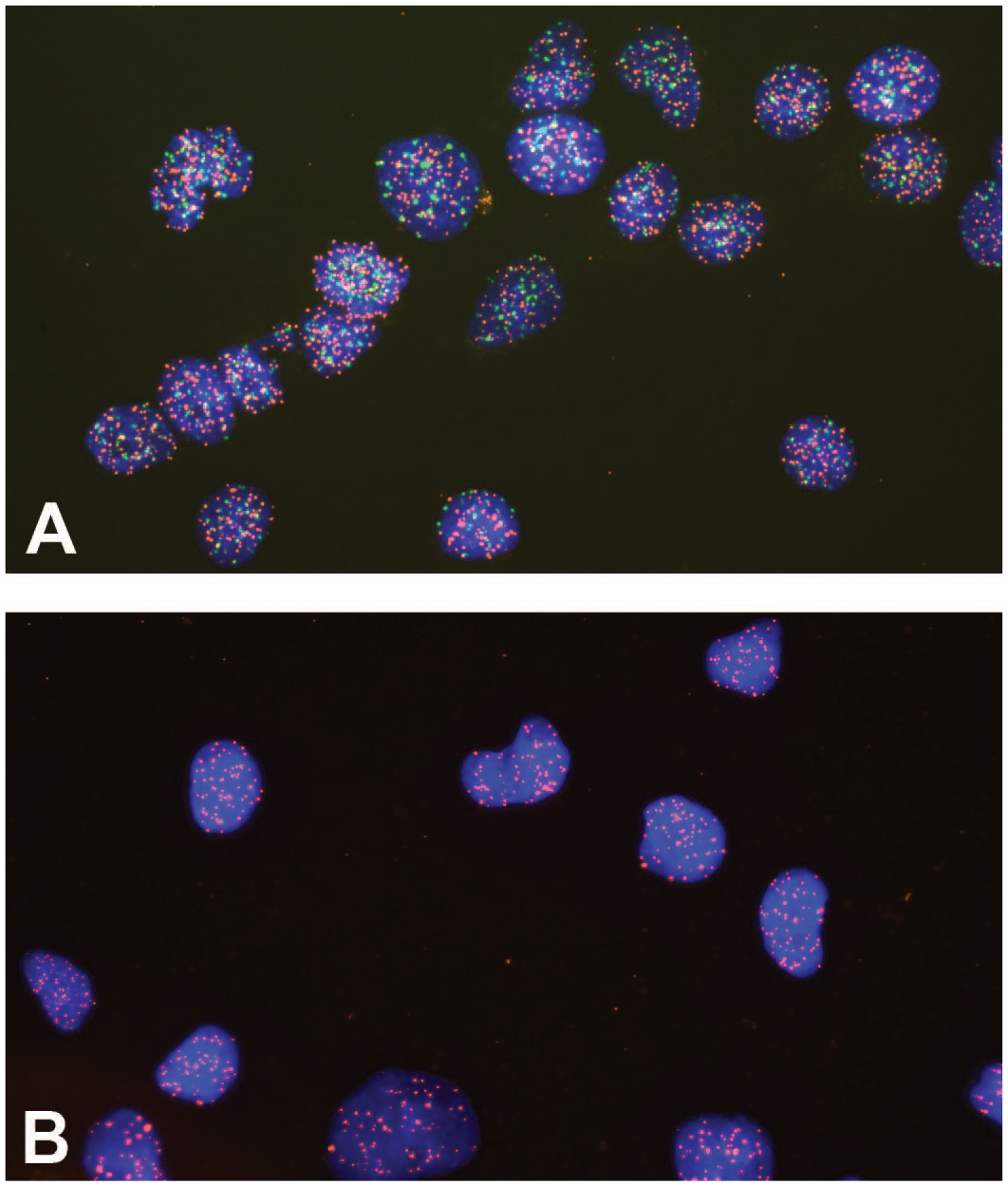
Example of FISH using peptide nucleic acid probes to discriminate between two genetically close species. FISH using probes composed of peptide nucleic acid (PNA) is used to discriminate between human and monkey cells. Human cells are double positive (A), whereas on the monkey cells, only one probe yielded a positive signal (B).
Not only is the probe composition influencing the hybridization performance but also the length. 1 A probe length of 50 to 300 nucleotides is preferred, although probes of up to 1500 might be used.1,40,42 In general, longer probes have higher specificity (and good sensitivity owing to numerous covalently attached reporter molecules) but are more difficult to synthesize. 40 Designs of shorter sequences of 20 to 50 nucleotides (oligonucleotides) have been introduced in recent years. Since RNA decay occurs by fragmentation or shorting from the end, 49 the shorter oligonucleotide probes have better accessibility to their target, are more suitable for partially degraded mRNA and may also be used to discriminate between different nucleic acid isoforms.40,50 One of the great advantages of oligonucleotides is the ability to target a single transcript by using multiple probes, a method called single-molecule FISH (smFISH).12,51 SmFISH developed over time and today this technique is well implemented and commercialized (Stellaris probes). 52 This single-molecule approach has significantly improved the sensitivity and specificity. However, at least 20–30 probes (each probe being 18-20 nucleotides long) need to bind the transcript to receive a signal. Therefore, a minimum length of target RNA sequence is required. 53 Another ISH technology that is based on the so-called “Z probe” has emerged in the last years, which also allows the detection of single transcripts. 54 Z probes have two binding sites, one complementary to the target mRNA molecule (18-25 base pairs long) and another one complementary to the preamplifier for the signal amplification, described in the following section. Only those probes that bind contiguously in tandem making a complementary binding site for the preamplifier will emit a signal. 54 Approximately 20 double Z probes are designed for a single RNA transcript. This technology is commercialized, as RNAscope (ACD, Inc.) and ViewRNA (ThermoFisher Scientific) and the detection system is not limited to fluorophores but also includes chromogenic substrates. Single molecule-based techniques are continuously improving to detect shorter RNA targets between 17 and 50 nucleotides (i.e., miRNA, siRNA or splice variants), 20 or nascent RNA molecules by targeting intron sites. 55
Another type of probe is the so-called padlock probe, which is composed of two terminal arms complementary to the target sequence that are connected by a DNA linker sequence. 56 The two probe arms bind side by side the target sequence and are subsequently brought together via enzymatic ligation (e.g., T4 DNA/RNA ligase) to form a circularized DNA that serves as a template for rolling circle amplification technologies (RCA) (described in the following section).57,58 Only probes that have a successful dual binding of the arms and enzymatic ligation to form the circular template will produce a signal, hence adding high sensitivity and specificity. 59 Padlock probes are capable of detecting very short sequences or even point mutations and are consequently often used in miRNA detection studies, genotyping studies or for the detection of viral pathogens with high genetic variations.60-65 Padlock probes are also used in spatial transcriptomic technologies, based on the application of multiple padlock probes containing different barcode sequences in the DNA linker. 66
Using ISH with commercial probes has certain advantages. The time needed to establish these techniques is normally a month or less. Furthermore, once the technique is established, it can be used with different probes with only minimal adaptions related to tissue particularities and preparation. However, the main drawback is that companies use proprietary probes; hence, the specific sequences of the probes are not public. This makes alignment with the sequence used in other techniques like RT-qPCR challenging. Furthermore, commercial ISH techniques are more expensive compared to traditional self-made probe-based ISH techniques.
Detection Methods
To detect where the probe has bound within the cells or tissue, the probe needs to be labeled before hybridization (one-step protocol/direct labeling) or following hybridization (two-step protocol/indirect labeling). In directly labeled probes, the fluorophore is incorporated by using either random priming, nick translation, or Taq DNA polymerase. On the contrary, in indirect labeling protocols, primary probes contain an additional sequence site for the hybridization or a hapten for the hybridization of secondary labeled probes or antibodies, respectively.67-69 The presence of the label should not interfere with the hybridization reaction. Three basic approaches to visualize the DNA or RNA target sequences in situ can be used: radioactive ISH, chromogenic ISH (CISH) and fluorescent ISH (FISH). Each detection method has inherent characteristics that make them tailored for different applications. The field is constantly evolving, and many different signal amplification modalities and combinations are described in the literature. In this article, we present a general overview of the commonly used methods (Figure 4).
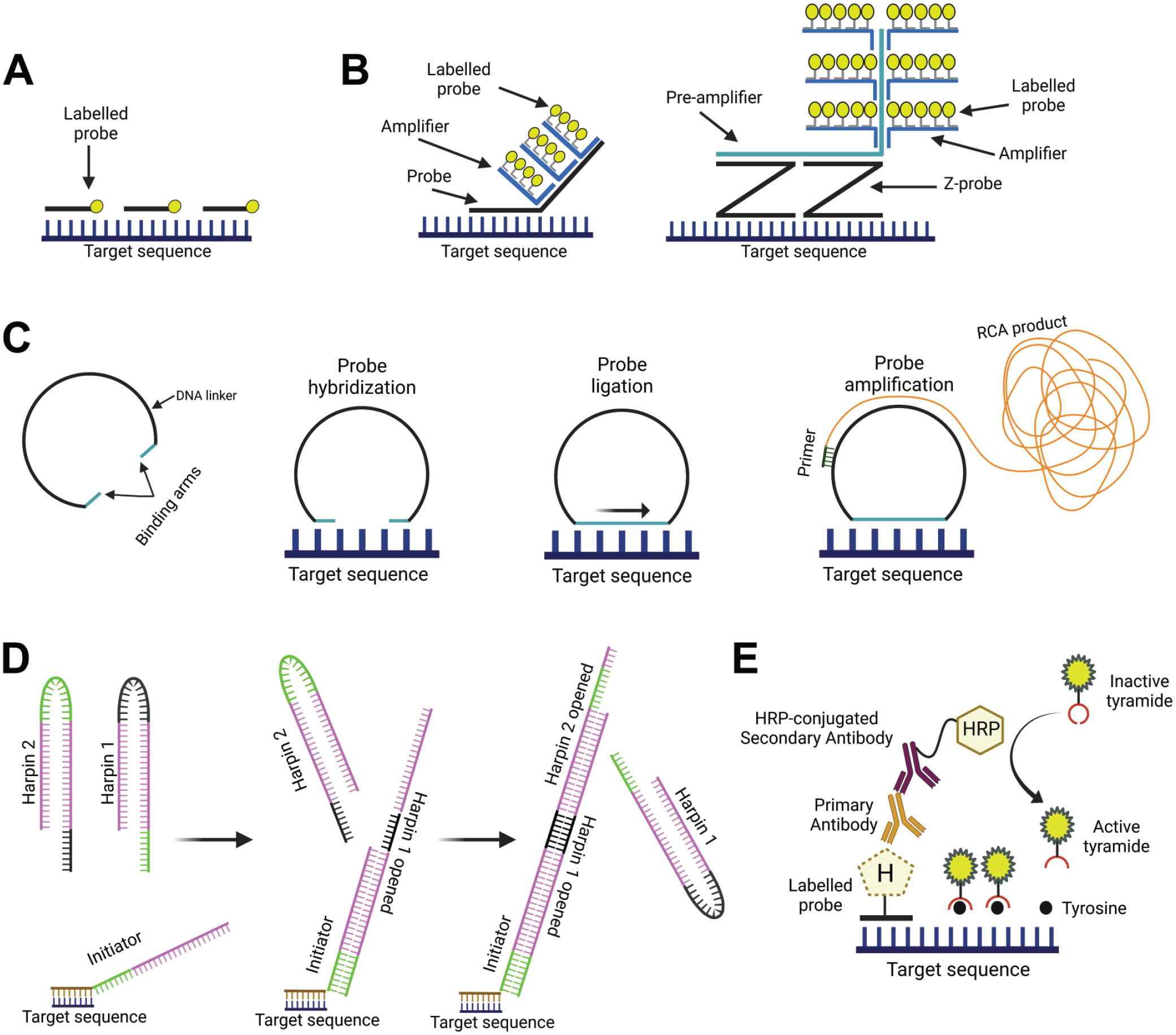
Different amplification methods for detection. (A) Example of directly labeled fluorescent oligoprobes that bind directly to the target sequence, in this case no amplification method is applied; (B) two similar amplification methods, the Sequential tethered and intertwined oligodeoxynucleotides complexes FISH (FISH-STICs) method consists of a two-step sequential method that uses an amplifier probe binding the target probe followed a labeled probe binding the amplifier probe, whereas the branched DNA method is a three-step amplification method that uses a pre-amplifier probe that binds the target probe followed by an amplifier and a labeled probe. DNA; (C) Rolling circle amplification technique uses a circular padlock probe as a template to produce long single-stranded tandem repeat sequences complementary to the template (so-called “RCA product”), the RCA product is subsequently targeted by fluorescently labeled probes; (D) Hybridization Chain Reaction technique uses self-assembling hairpin-structured nucleotide strands for elongation, three types of nucleotides are used: an initiation and two different harpin-nucleotide probes; (E) The Tyramide Signal Amplification method uses peroxidase-labeled antibody to activate tyramine substrates which bind and deposit to nearby tyrosine residues. FISH, fluorescent in situ hybridization; RCA, rolling circle amplification.
Radioactive approaches
In the past, ISH, was typically performed using radioactive probes, including 3H, 35S, or 32P. Radiolabeled approaches do not need signal amplification providing a linear relationship between signal and transcript quantity that allows high sensitivity combined with low background. Because of the technical and safety caveats related to the release of ionizing radiation, this approach could never be scaled up and has been gradually replaced by nonradioactive approaches.70,71
CISH and FISH
The nonradioactive probes are used in CISH, which relies on enzymatic reaction to detect colored substrates, and FISH, which is based on fluorochrome-based labels. For instance, the commercial Stellaris is an example of directly labeled fluorescent oligoprobes (generally 30-48 bp) that allows the detection of individual mRNA molecules without signal amplification.52,72 The RNAscope and ViewRNA are examples of assays relying on a two-step indirect labeling of the oligoprobes and allow CISH or FISH modalities.
An important limitation of directly labeled probes is the low-intensity signal when a low amount of probe is bound to a specific site; for instance, for FISH, it might be challenging to discern background autofluorescence from a real signal. 51 The low-intensity signal could be compensated by using a higher amount of oligoprobes, which in turn increases the signal-to-background ratio. 73 Several techniques based on a two-step methodology have been developed for CISH and FISH to increase the detection limits of the probe signals and can increase signals by 10 to 100 times. 74
Sequential tethered and intertwined oligodeoxynucleotides complexes (FISH-STICs) and branched DNA (bDNA) (Figure 4B) methods consist of successive hybridization cycles using (pre-) amplifiers and labeled probes. 68 The FISH-STICs method can be applied in fluorescence protocols and consists of a two-step sequential method that uses an amplifier binding the target probe and a detection probe binding the amplifier probe. 75 On the other hand, bDNA is a three-step amplification method that uses a preamplifier probe that binds the target probe followed by an amplifier and a labeled probe. 76 In both methods the (pre-)amplifier probes carry multiple binding sites for the subsequent binding of an amplifier (i.e., bDNA) which in turn carries additional binding sites for the labeled probe. The bDNA amplification method is currently the most widely used in CISH and FISH commercial technologies such as RNAscope and ViewRNA. The output consists of signaling dots that represent RNA molecules which can be visualized via bright field or fluorescent microscopy using chromogenic or fluorescent assays, respectively. The degree of amplification through this structure branching can be predetermined by the design of oligonucleotides, and the sensitivity further increased by expanding the number of probes per target when feasible. Therefore, bDNA was initially applied for quantitative diagnostics of viral infections, for example, human immunodeficiency virus and hepatitis C virus on solid-phase microwell plates and later for large-scale transcriptomics by smFISH.74,77,78
The Rolling Circle Amplification (RCA) method uses a circular padlock probe as a template (described in the probe section) to produce long single-stranded tandem repeat sequences complementary to the template (RCA product) (Figure 4C). 58 As described in the probe section, the two binding arms of the padlock probe bind the target sequence followed by enzymatic ligation to produce a circular DNA template that will be used by one or multiple primers during the amplification phase to produce the RCA product.57,64 The RCA product can be described as “DNA-balls” of 0.5 to 1 µm diameter which remain coiled adjacent to the padlock probe. 63 There are different strategies to visualize the RCA product such as direct labeling (i.e., incorporation of conjugated dNTPs), using DNA binding dyes like SYBR green, and hybridization of labeled probes among others. 57 RNA detection with padlock probes is more challenging than DNA, 61 and often the protocol relies on reverse transcription reactions before padlock probe hybridization. 79 Therefore, different strategies have been developed to improve sensitivity and specificity like the RollFISH amplification method which uses a smFISH probe as a docking sequence for a padlock probe. 80 More recently, RCA has been used in spatial transcriptomic technologies (i.e., Xentium in situ technology by 10× Genomics) in which a barcoding sequence contained in the padlock probe is detected in the RCA product. A detailed description of spatial transcriptomic technologies goes beyond the scope of this article, and we refer the reader to relevant literature. 81
Hybridization chain reaction (HCR) is an enzyme-free method that consists of elongation of probes by self-assembly of hairpin-structured nucleotide strands. 82 The reaction contains an initiator and two sets of metastable hairpin probes (Figure 4D). The reaction starts when the initiator binds the overhanging toehold sequence of the first hairpin. This initiator-hairpin binding triggers the opening of the hairpin and the exposure of the other side for the toehold for the binding and subsequent opening of the second hairpin probe. This chain reaction can continue until one of the probe sets is fully consumed. The length of toehold, stem, and loop of the hairpin probes and stringent hybridization conditions have been optimized for specific multiplex detection of mRNAs in tissues with high signal-to-background ratios.83,84 Recently developed split-initiator probes and binary probes further improved the specificity to visualize multiple targets in whole-mount chicken embryos and to distinguish single-nucleotide variations.85,86
The Tyramide Signal Amplification (TSA) method is based on the peroxidase-mediated oxidation of tyramine-labeled substrates. Labeled inactive-tyramine substrates become activated when exposed to peroxidase enzymes (e.g., horseradish peroxidase), this activation results in covalent binding and accumulation of tyramine to the nearby tyrosine residues (Figure 4E). 87 Detection of biotin (or digoxigenin)-labeled DNA probes using peroxidase-conjugated streptavidin (or antidigoxigenin antibody) and tyramine-conjugated fluorophores in cultured cells has shown that this method can also be used for FISH signal amplification. 88 Hybridization probes labeled with various haptens, peroxidase-conjugated antibodies, and various fluorophores conjugated with tyramine offer a broad range of combinatorial capabilities and application potential. However, the low penetration efficiency of the large peroxidase-conjugated antibody molecule may cause a concentration gradient in thick tissue samples resulting in different signal intensities depending on the distance from the surface. 89
Visualization and Evaluation
The ISH glass slides can be examined directly on a microscope, or using whole slide images (WSI) which are digitized glass slides that are visualized on a computer screen, or both. 90 CISH requires a bright-field microscope or WSI, whereas FISH can be detected by fluorescence-based techniques, such as epifluorescence, confocal microscopy, or fluorescent WSI. FISH has the advantage of visualizing and co-localizing multiple targets in the same sample, but limited tissue morphology evaluation. 91 Although it can be achieved by matching a serial section with a standard morphological staining such as hematoxylin-eosin (HE). Tissue slides initially stained with FISH can also be stained in a secondary step with HE, IHC, or CISH and subsequently aligned (Figure 5). As a rule, single DNA or a single copy of mRNA molecules are detected as pinpoint dots. 54 Therefore, adequate image quality and focus are crucial to see sharply demarcated signals.
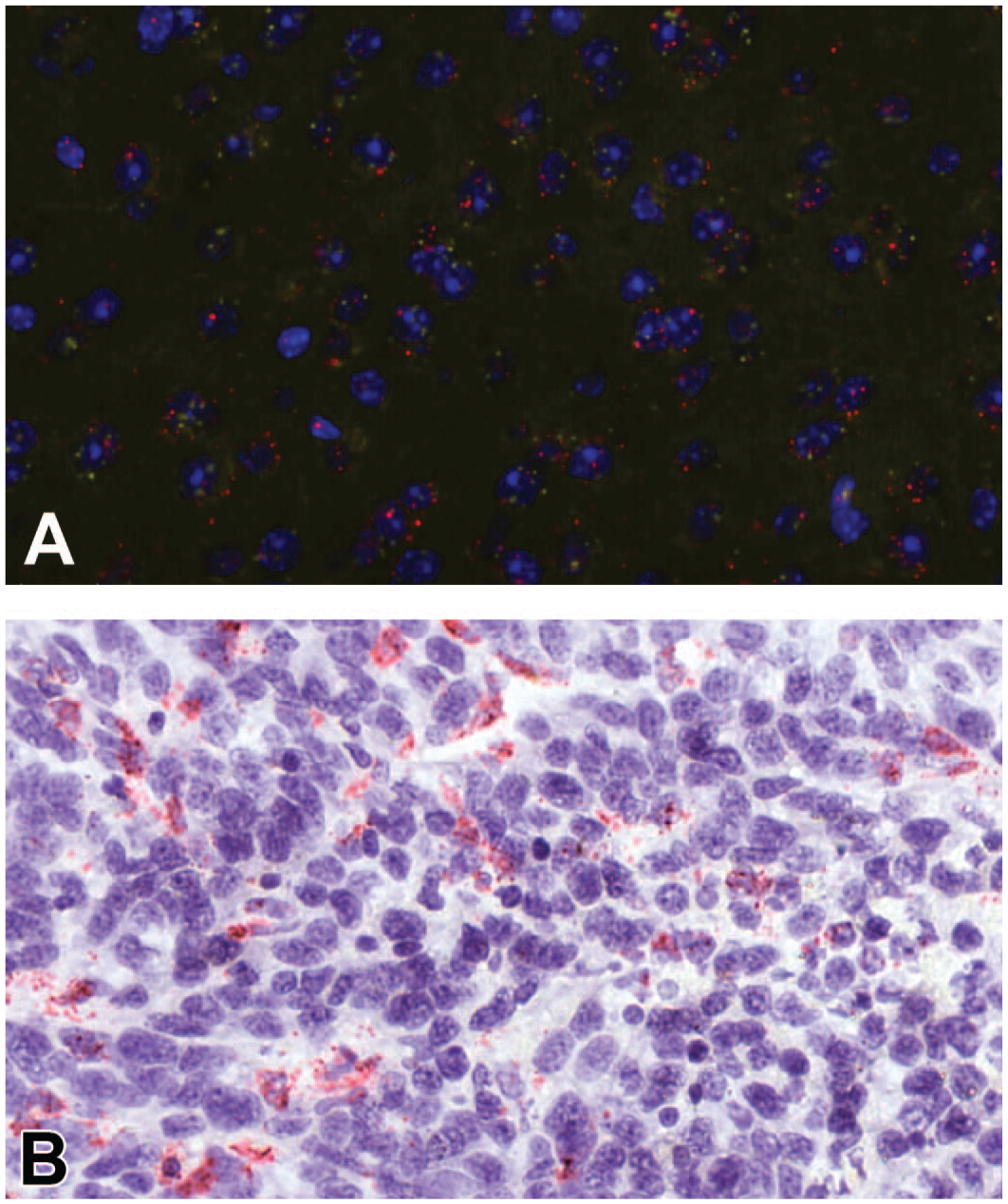
Sequential FISH-CISH staining. Example of sequential ISH staining of the same slide. Tissue was initially stained with FISH using housekeeping gene probes as a positive control to check the RNA integrity (A) followed by CISH staining of the target gene (B). FISH, fluorescent ISH; CISH, chromogenic ISH; ISH, in situ hybridization.
In CISH, the main chromogen is peroxidase- or alkaline phosphatase-based.
Several aspects could affect the outcome of the ISH analysis, for example, the identification of false positives, the background noise, the visualization limit based on the dot size, the proximity of dots to the nucleus, and the challenging identification of cytoplasmic filaments or axons and dendrites in the case of neurons (Supplementary Figure 2). Clusters of dots may result from the overlap of signals coming from multiple molecules such as multiple mRNA molecules or could be related to the number of probes bound to the target molecule. 51 Another aspect to consider in CISH is the counterstain intensity for adequate cell and nuclear detection, as in some instances may mask weak but relevant signals. For FISH, autofluorescence of certain structures should be considered and excluded (e.g., collagen, hemoglobin, and lipofuscin), including the intrinsic fluorescence different from the true signal but resulting from overfixation or overdigestion of the sections. 91 Semiquantitative or quantitative readouts of dots signaling can be obtained. Semiquantitative scoring of ISH-images is done via visual assessment, and ACD, Inc. company provides a guidance example related to RNAscope, as orientation, which might need adaption depending on the tissue, the sublocalization in the organ, and the range of staining (Figure 6). Analyzing digital images gives the possibility to use specialized software that allows more precise and reproducible quantification of the positive signal. However, a review by a pathologist is still recommended to confirm the results obtained by image analysis. 92 Artificial intelligence algorithms are evolving at incredible speed, which allows the extraction of a large amount of information from a tissue section and reduces the human bias that comes from visual assessment. 93 Deep learning artificial intelligence (AI) solutions and services for image analysis requiring expensive licenses are nowadays available and provide predesigned algorithms (e.g., Indica Labs’ HALO, Visiopharm’s Oncotopix Discovery, Aiforia Platform, and more). Some other open-source free software (e.g., ImageJ Fiji, CellProfiler, QuPath, Icy, and more) are also available. Cell and dot count on selected regions of interest is a feature available from all major free image analyzers (or even whole slides for ImageJ Fiji and QuPath).
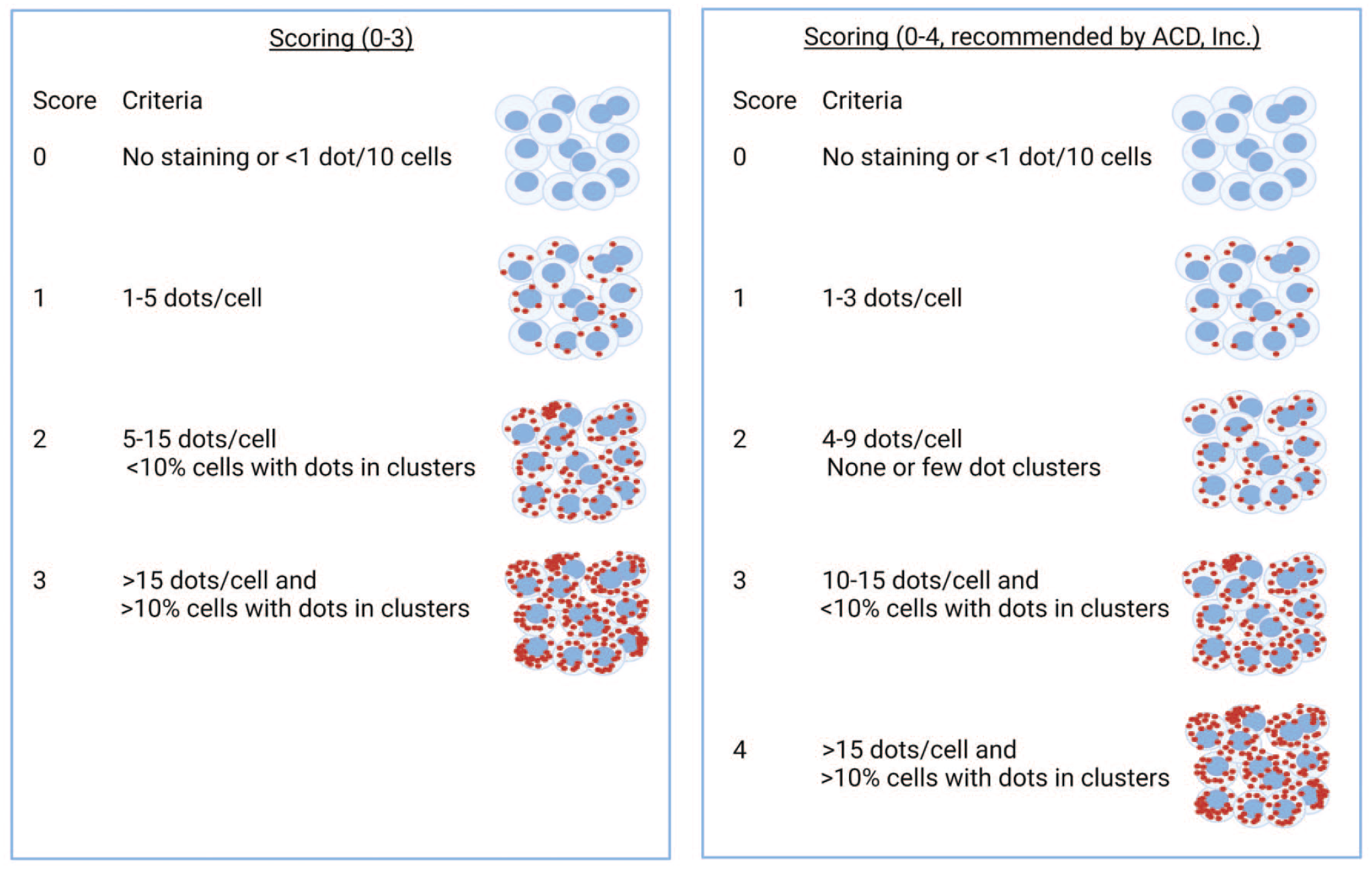
Example of semiquantitative scoring criteria. ACD, advanced cell diagnostics.
Quantification can be planned as the total number of positive cells, the total number of dots, or the average number of dots per cell. One of the major challenges of ISH analysis is the separation of single and multiple probe dots, achievable exclusively by sophisticated dot-determining algorithms and high-quality image acquisition systems. This type of automatic tool requires manual tuning of several parameters characterizing the images destined for analysis, specific for different experimental conditions. Sometimes, the cluster of signals cannot be separated into individual dots. In such a case, the area of the signal can best serve as a quantitative output. 94
Workflows to quantify ISH staining have been developed. 95 If probe quantification should be done on individual cells, the initial step is to identify cells by detecting the nucleus, to segment them by using the nuclei as seeds (Supplementary Figure 3). 95 Cell separation can also be performed by using, for example, IHC markers to stain cell membranes. 95 The ISH stained spots can be subsequently detected by, for example, thresholding and attributed to the nucleus or cytoplasm of the individual cell if required. If a heterogeneous expression of the ISH target occurs, semi quantitative scoring can be used to calculate an H-score.96-98 The H-score can also help to show different ISH staining in different cell types and/or different subpopulations by showing the percent of cells expressing the target at different levels. 97
Several aspects could affect the outcome of the ISH analysis, for example, the identification of false positives, the background noise, the visualization limit based on the dot size, the proximity of dots to the nucleus, and the challenging identification of cytoplasmic filaments or axons and dendrites in the case of neurons (Supplementary Figure 2).
Multiplexing
Although single-plex ISH is widely used in pathology, this technique is not sufficient to evaluate cellular interactions and/or spatial context of multiple markers in tissue sections. Techniques such as flow cytometry or other omics technologies (e.g., bulk and single-cell RNAseq) are helpful to investigate multiple analytes in a tissue, but they do not provide spatial context. 99 Multiplex molecular localization methods allow the visualization of multiple analytes and their spatial relationships in a single tissue section and maximize the utilization of valuable tissues (Figure 7). Innovation and adoption of multiplexing methods have recently progressed at extraordinary rates, 100 which occur not only at the chemistry level but also in the development of new imaging and computational techniques. 101 It is not the scope of this article to provide a detailed description of multiplexing approaches but only a general overview of the different possibilities of ISH-based technologies.
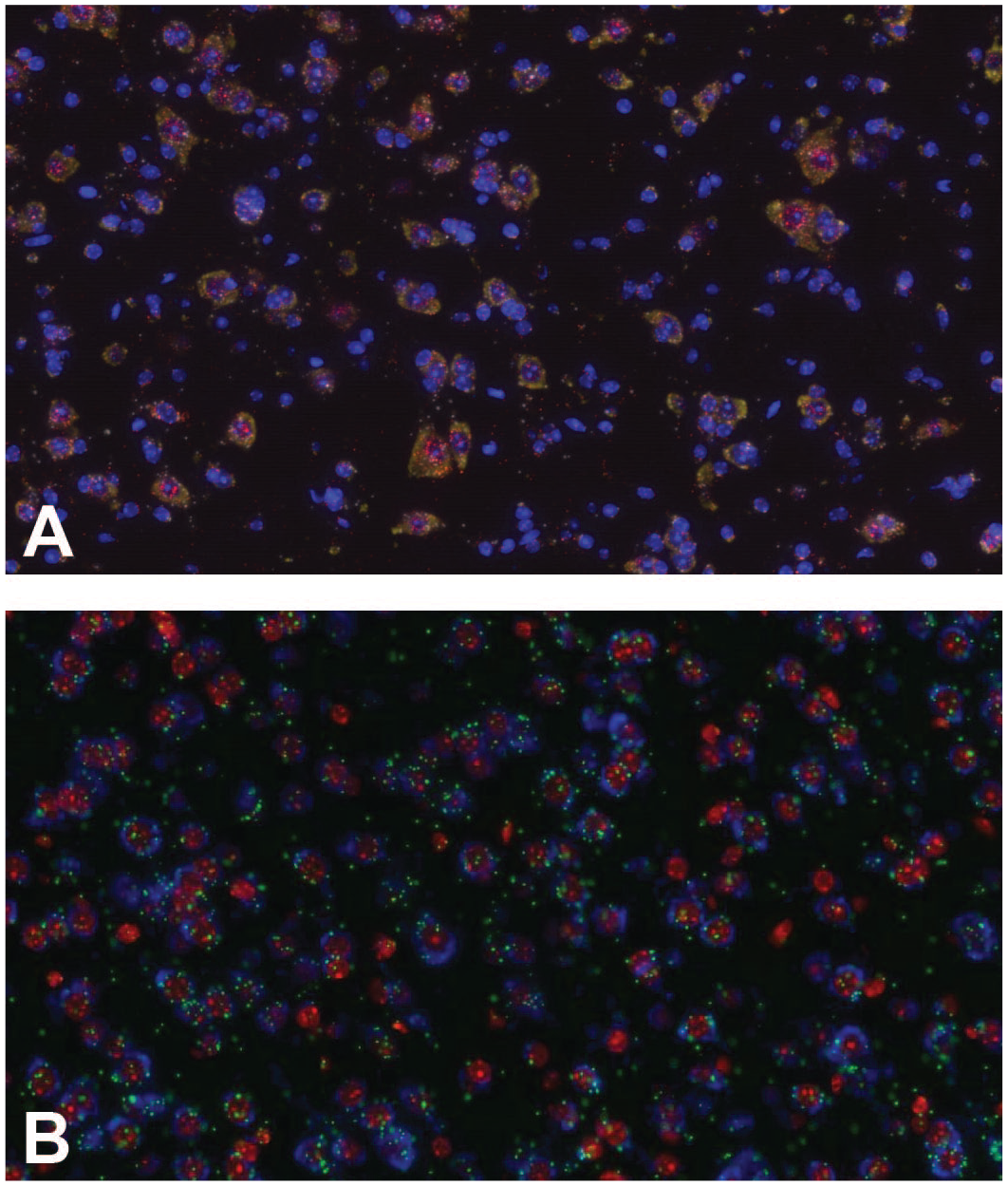
Multiplexed in situ hybridization staining with three different markers. Examples of multiplexed fluorescent in situ hybridization using three different markers on murine brain. Three different housekeeping genes as positive controls, murine peptidyl-prolyl cis-trans isomerase B (PPIB), murine RNA polymerase II subunit A (POLR2A) and murine ubiquitin C (UBC) (A); three different target genes detected by the multiplexed staining (B).
The evaluation of more than one target simultaneously in a tissue can be achieved either in CISH or FISH technologies. As already mentioned, CISH allows better morphological assessment than FISH. However, chromogenic approaches based on precipitates are often limited to the simultaneous identification of two targets within the same cell. 102 Therefore, over the years multiplex ISH protocols have been developed on FISH-based technologies, 8 and nowadays, many different fluorescent-based protocols with a wide range of labeling strategies have been developed as well as sophisticated imaging techniques to capture a wide range of color spectra. 103 The main limitation of multiplex FISH technologies has always been the number of targets due to a limited number of fluorophores and overlap in the spectral range. 79 To overcome these limitations, protocols have incorporated sequential rounds of hybridization and imaging to expand the multiplex capabilities; this is the case, for example, of ACD, Inc., which can detect up to 48 targets. However, smFISH has become in many aspects the foundation of high multiplexing ISH-based technologies and allows the identification of a higher number of targets simultaneously. 12 SmFISH-based technologies have evolved to target mRNA molecules with a combination of probes carrying different fluorophores, hence giving specific spatial barcoding (combination of fluorophores in a specific order) or spectral barcoding (combined spectral barcoding independent from the order). 104 Spatial barcoding has been also combined with sequential rounds of hybridization, imaging, and stripping to create a unique barcode that allows the identification of up to 10,000 genes.99,105-107
Controls
A great emphasis should be placed on the inclusion of appropriate controls as an integral part of any experimental procedure. When performing an ISH experiment, one should be confident that the probe is selectively binding to the intended target. On the one hand, failure to hybridize should be the consequence of the lack of target expression in the tissue and not a technical failure preventing a successful hybridization reaction. On the other hand, hybridization should be intended binding to the correct target and not an unintended binding to other not targeted molecules. With appropriate controls, these questions can be answered with a high degree of confidence. Several positive and negative controls should be used to assess nucleic acid integrity (tissue quality) and the hybridization reaction sensitivity and specificity.
Positive Control Probe: directed against a target uniformly expressed in a specimen of interest (i.e., housekeeping gene). This probe is used for tissue quality assessment (i.e., good DNA and RNA preservation). This probe should be similar to the test probe in base pair composition, type of nucleic acid, labeling, and final concentration. Also, the expression level should be similar to the target to optimize pretreatment and hybridization conditions. 108 When controls are used to verify the correct function of the applied experimental protocol, ubiquitously expressed genes (housekeeping genes) such as actin and tubulin can be used and the spatially discrete and predictable staining pattern should be assessed.1,109 The most appropriate control has to be chosen for each tissue.
Negative Control Probe: directed against a target absent in the specimen of interest; this probe is used to monitor the test sample for nonspecific reactions with probe or detection reagents. Similar to the positive control probe, it should equal the test probe in the conditions mentioned above. The hybridization binding specificity can be verified using probes targeting nucleic acid species from a different animal species (no signal expected) or the opposite sense probe in parallel with its correct sense counterpart (no signal vs specific signal approach). However, when both sense and antisense probes are used it should be ensured that the gene of interest is not transcribed from both sense and anti-sense DNA strands.1,110-113 Results of the ISH cannot be interpreted unequivocally if there is a signal with the negative control probe. Pretreatment of the samples with RNase or DNase will also help to ensure that the probe binds exclusively RNA or DNA (depending on the probe) as no signal should be expected in treated samples. 1
Control Specimens: Control materials should be derived from the same specimen type recommended for use with the assay to control the analytic performance of the probe. In the case of FFPE specimens, cell lines with a known quantity of target expression could be chosen to mimic tissue sections. Three kinds of control specimen may be useful: positive controls expressing the target, a mixture of cells from positive and negative controls, and negative controls not expressing the target. The positive sample control must be included with each experiment and the expected staining pattern (where known) should be confirmed. Positive controls could help for example to identify run-to-run differences related to technical abnormalities. The control sections should have the same process pre-analytical and experimental conditions of the test samples.
Regulatory (ISH Assay Validation and Level of Compliance)
Good Laboratory Practices (GLPs) are a series of federal regulations passed in the United States by the FDA under 21 CFR Part 58 to ensure the validity, integrity, and reliability of data submitted for regulatory evaluation and approval (U.S. Food and Drug Administration’s Good Laboratory Practice for Nonclinical Laboratory Studies, Title 21, Vol. 1, Part 58.) 114 and in the European Union by several directives of the European Commission (e.g., Directive 2009/9/EC, Directive 2004/10/EC), 115 by the OECD Series on Principles of Good Laboratory Practice (GLP) and Compliance Monitoring, and by national regulations of the Member States.
Since a detailed description of the individual requirements for a GLP-compliant ISH study would exceed the scope of this publication, only the main aspects of GLP-compliant ISH studies are mentioned here. In brief, a study director and study personnel with appropriate experience/training should be assigned by the management. In addition, a standard operating procedure for ISH should be established. The publication of Yoon and Andersen 116 can serve as guidance. A report should be prepared at the end of the study, and all study records should be archived and retained according to applicable regulations. Responsibility for the validity of the ISH assay is by the laboratory performing the experimental work (test site or test facility). Upon study completion, a statement of GLP compliance must be issued and signed by the principal investigator and the responsible scientist.
Besides assays that require a complete validation for GLP studies (i.e., lot release assays, excipient testing, etc.), experiments used for GLP animal toxicity or related safety studies are expected to be validated and it should be demonstrated that the methods are accurate, safe, and reproducible for the intended use. 117 The major steps for ISH assay validation are listed in Figure 8.
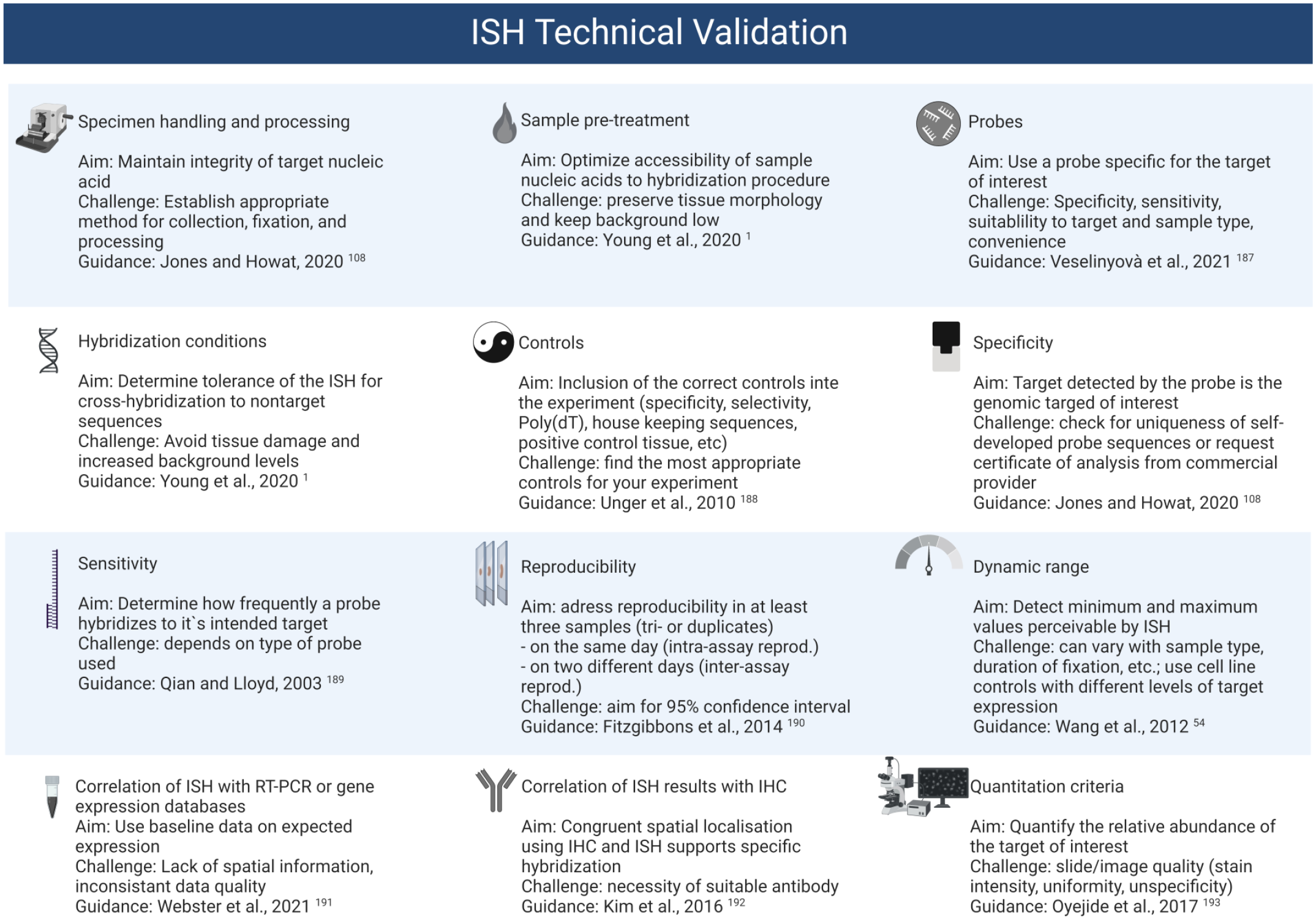
Overview of ISH validation requirements. ISH, in situ hybridization; RT-PCR, real-time polymerase chain reaction; IHC, immunohistochemistry.
Complementary sequences to nucleic-acid-based test items can be used to evaluate biodistribution/DMPK/ADME-T. Although such studies are seldom GLP-validated, as they often are performed on excised tissues from diseased human patients,118,119 there are, to our opinion, no obstacles to do so, especially since many controls are included in such assays. However, this is seldom performed in practice, due to the rarity of those assays in toxicology studies. Validating ISH assays for common applications, for example, for key cytokines, could be the initial step in specialized laboratories.
Currently, there are no regulatory requirements to test complementary sequence specificity in situ. Therefore, there are no nucleic acid therapeutic screening assays for specificity in situ with ISH approaches. 120 Consequently, in contrast to an antibody or complex protein therapeutics with IHC, ISH assays are not used, to the best of our knowledge, as test article/test item binding to on-target or off-target sequences in target cells in humans. But due to the unique and complex nature of the gene therapy medicinal products (GTMPs), which are in large majority AAV viral vectors, ASO, LNP, and Gal-NAc, it is unlikely that these assays would be executed with nonmodified GTMP in the foreseeable future. 121 In the situation of modified mRNA therapeutics, the therapeutic of which are often translation-related, the need for such studies is not warranted. 122 In this perspective, an ISH assay, confirming no modified mRNA had entered the nucleus might be highly advisable. However, published work on such investigations are not found.
For mRNA-based gene editing, it is very unlikely that the specificity of the target sequence edition will be performed by assays like ISH, but rather by other experimental and computational tests. 123 To conclude, an equivalent for in situ tissue cross-reactivity of therapeutic monoclonal antibodies currently being carried out by IHC and another in vitro approach, is very unlikely to be implemented for nucleic acid therapeutics, thereby limiting the scope of GLP regulation for GTMP and therapeutic mRNA. But this could change, given the robustness of the novel ISH platforms.
Applications of ISH in Investigative and Toxicologic Pathology
Considering the recent technological advancements, ISH technology is widely used across the process of drug discovery and development as a complementary tool to IHC to better characterize drug pharmacological effects and on- and off-target safety risks. In the following paragraphs, common applications and examples of ISH in drug R&D are presented (Figure 9).
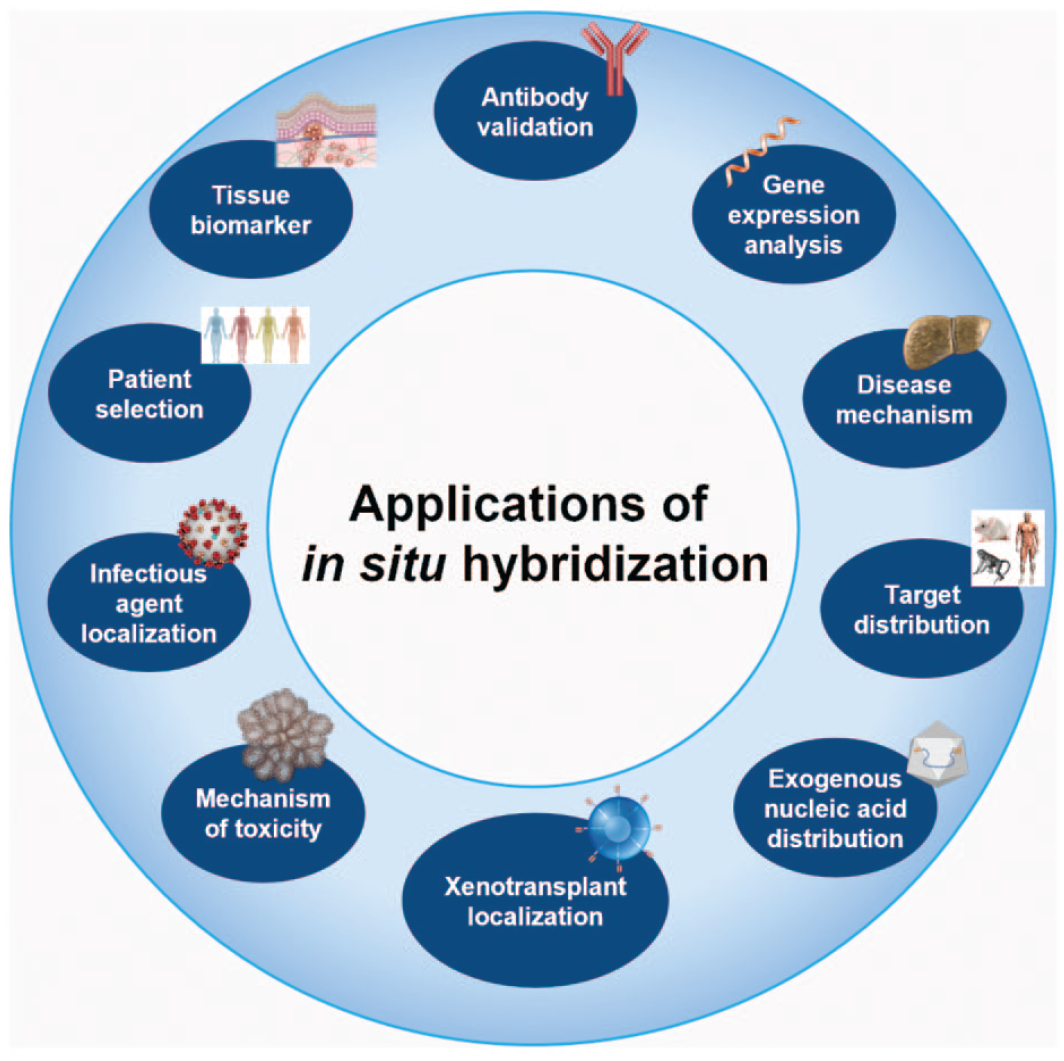
Overview of possible application of in situ hybridization in investigative and toxicologic pathology in drug research and development.
Validation and Adds-on to Biomarker Reference Antibodies to Pharmacological Targets
In drug discovery and development, the validation of large immunogenic or haptenic pharmacological targets in tissues is often performed by flow cytometry (FC), and other omics technologies (e.g., RNAseq) across species. However, as already mentioned elsewhere in this article, these technologies do not offer spatial information. As such, IHC has been widely used to provide the spatial context of the protein target. IHC often employs polyclonal antibodies with potential cross-reactivity across species. To evaluate the performance of polyclonal antibodies cross-reactivity, species-specific monoclonal antibodies (mAbs) are commonly used. When species-specific mAbs are not readily available, a more rapid and convenient approach would be ISH, where target- and species-specific probes can be easily synthesized. 124 In addition to enabling specificity, ISH can be used to assess the sensitivity of the antibodies. ISH can be used in a more systematic way and more upfront during IHC optimization to avoid false negative results. 118 Compared to IHC, a wider use of ISH to survey transcripts of pharmacological targets would offer a thorough target distribution and validation in normal and diseased human and model animal tissues.
Validation of Bulk/Single Cell Gene Expression Analysis
Single or multiplex ISH and IHC are often used to validate the results from bulk and single-cell RNAseq analysis.125-129 For instance, the molecular classification of foveal retinal cells versus nonfoveal retinal cells was reported. 126 Here, single-cell RNAseq results were validated by ISH for some of the targets demonstrating that ISH can replace IHC if specific antibodies are not readily available.
Elucidation of Disease Mechanisms
Defining spatial localization of targets in specific cell types in tissues is shown to provide mechanistic insights into physiological and pathological processes.130-133 There are several published examples illuminating the significance of ISH in establishing disease pathogenesis and we highlight a few here.134-143 For example, FISH was used to localize Ifnar1 and Ifnar2 in mouse dorsal root ganglia (DRG) sensory neurons supporting the idea that activation of these receptors could modulate nociceptive signaling during viral infection. 144 In addition, downregulated expression of neuronal genes, such as TRPV1, SLC17A7, and PRDM12 was demonstrated in L4 DRGs of patients further providing new insights into intraganglionic pathology and mechanisms causing neuropathic pain in patients with type 2 diabetes mellitus. 145 Cheng 146 showed that glucagon is an important regulator of metabolic zonation in the liver. ISH revealed the effects of glucagon on the expression of periportal and perivenous genes in specific layers of liver zonation. In all these studies, ISH was used to precisely localize the targets as specific antibodies were not available underpinning the advantages and utility of the ISH assays.
Characterization of Animal Models
Animal models have played an important role in many medical advances that saved or enhanced many human lives and therefore need thorough characterization. IHC and ISH contributed significantly to animal model characterization for validation of pathogenesis and target pathways to be intervened pharmacologically.147-149 Similarly, in vaccine development, ISH can be very useful to monitor viral pathogenesis in animal models. Using ISH, key data were provided to show that the cynomolgus monkey was as good as, if not better than the rhesus monkey, to model SARS-Cov-2 infection and disease. 150
Comparative Target Distribution/Validation in Human and Preclinical Species
Comparative target tissue distribution studies with molecular localization studies in human and preclinical pharmacology and toxicological species add value to efficacy and safety studies. Characterization of the animal model and the human disease for precise localization of target is critical before initiating therapeutic interventions in animal models. Similarly, adequate understanding of the target distribution is important to predict the relevance and importance of the toxicological findings for humans. For example, multiplex ISH was used to elucidate the distribution of several unique and classic neuronal mRNAs in peptidergic (CGRP-expressing) and nonpeptidergic (P2X3R-expressing) nociceptor subpopulations in mouse and human DRGs highlighting the significance of comparative distribution between preclinical models and humans to ascertain translational potential and therapeutic efficacy. 151 ISH has been extensively used in comparative target localization studies, particularly for novel targets.118,124,152,153 For instance, to determine relevant toxicology species for assessing the safety of cadherin6 (CDH6) targeting antibody–drug conjugates, the expression of CDH6 across normal rat, cynomolgus monkeys, and human tissues were studied. 124 ISH-based localization studies were found to be overall comparable target distribution across species and enabled toxicity testing in rat and cynomolgus monkeys.
Exogenous Nucleic Acid (Oligonucleotide, siRNA, Viral Vector) Distribution
Together with quantitative PCR, ISH is now a routine technique to investigate the biodistribution of therapeutic nucleic acid in tissues and to whether the intended protein molecule is modulated.154-156 For instance, a decoy oligonucleotide was used to neutralize the over-transcription of the transcription factor AP-1 in Marfan disease, and ISH was used to demonstrate the persistence in tissues of this decoy oligonucleotide, which was effective in alleviating/neutralizing the Marfan’s disease phenotype in diseased aorta from a mouse model, the fibrillin-1 hypomorphic mice. 157
Localization of Xenotransplant Cells/Tissues
ISH techniques are instrumental in the localization of xenotransplant cells in homologous/heterologous species. For instance, RNAscope ISH (to detect the CAR mRNA’s 3′-untranslated region) was used to show anti-EGFRvIII CAR-T cell infiltration in glioblastoma tumor sections before and after intravenous infusion. 158 Moreover, anti-EGFRvIII CAR-T cell infiltration into tumor cells was shown to correlate with EGFRvIII downregulation in tumor cells. ISH was employed to show anti-ROR1 and anti-BCMA CAR-T cell distribution and tumor infiltration in xenograft tumor models. 159 Furthermore, ISH has been used quite frequently in target engagement and other mechanistic studies by characterizing human versus mouse-specific target localization in patient-derived xenografts (PDX).160-162
Investigation of Toxicity in Preclinical Species and its Potential Translation in Patients
Investigations of mechanisms of toxicity using either or both ISH and IHC is not a new concept, 163 yet the widespread adoption of such approaches is yet to come. A PubMed simple search of “toxicity or ISH or mechanisms” yielded only 536 articles, many of which deal with cancer-associated toxicity, environmental biotoxicity (algae), or virus distribution and associated toxicity. Notable studies in model organisms include ISH in whole zebrafish (Danio rerio) adults or embryos,164-167 fish gill to study environmental toxicants, 168 tritium-labeled probes in toxicant-induced tumors in mouse models, 169 NMDA-induced toxicity in the eyes of mice, and atropine-induced toxicity in the rat testis.170,171
Precise Localization of Infectious Agents
The widespread adaptation of commercially available ISH techniques (e.g., RNAscope) has opened many avenues to better understand tissue reservoirs,172,173 pathogenesis, viral loads, and tissue distribution in both deceased humans and animal models of viral infections.150,173 Particularly, localization and quantification of viral nucleic acids in tissues and bodily fluids and their clearance mechanisms in animal models may inform to therapeutic design in human clinical trials.172-174
Patient Selection/Stratification in Clinical Trials
In general, modern RNA ISH techniques are specific and sensitive that have been increasingly utilized in clinical studies, particularly in patient selection and/or stratification.175-178 FGFR mRNA ISH was used to select patients with advanced cancers for treatment with FGFR inhibitor.175,179 Similarly, DKK1 mRNA ISH was used to screen patients with gastric and gastroesophageal adenocarcinoma in clinical trials.176,177
Tissue-Based Biomarker
ISH-based tissue-based biomarkers have increasingly been used in understanding basic biology, disease diagnosis, and treatment response in both preclinical and clinical studies.94,180-184 For instance, RNAscope ISH with quantitative image analysis algorithm was used to study changes in mRNA levels of targets in mouse brain and liver sections in response to biotherapeutics. 94 For breast cancer diagnosis, FISH is widely used for the determination of HER2 status, especially to resolve indeterminate IHC scores.185,186 α-Fetoprotein mRNA ISH was found to be a highly specific marker of hepatocellular carcinoma. 183
Conclusion
This article provides a review of the technical basis, main features, and applications of in situ hybridization in the context of drug research and development. It reflects the discussions and opinions of the Pathology 2.0 Molecular Pathology Special Interest Group from the European Society of Toxicologic Pathology. The members of this Special Interest Group are experienced pathologists from a broad range of pharma and academic research.
Overall, ISH techniques have been developed in the 1970s but have recently gained in specificity and accessibility. ISH could be used for a wide range of applications in drug research and development. The ISH technology is evolving at a high speed not only at the level of the molecular technology platforms (i.e., probes and detection methods) but also in visualization techniques. Higher plex, higher resolution, and better image analysis and data evaluation have placed ISH as an indispensable tool in the field of pathology. A good understanding of the basic principles behind this molecular technique is essential for pathologists to prepare and execute the studies.
Supplemental Material
sj-docx-1-spp-10.1177_01926233231178282 – Supplemental material for European Society of Toxicologic Pathology (Pathology 2.0 Molecular Pathology Special Interest Group): Review of In Situ Hybridization Techniques for Drug Research and Development
Supplemental material, sj-docx-1-spp-10.1177_01926233231178282 for European Society of Toxicologic Pathology (Pathology 2.0 Molecular Pathology Special Interest Group): Review of In Situ Hybridization Techniques for Drug Research and Development by Josep M. Monné Rodríguez, Anna-Lena Frisk, Robert Kreutzer, Thomas Lemarchand, Stephane Lezmi, Chandrassegar Saravanan, Birgit Stierstorfer, Céline Thuilliez, Enrico Vezzali, Grazyna Wieczorek, Seong-Wook Yun and Dirk Schaudien in Toxicologic Pathology
Footnotes
Acknowledgements
Figures 4, 6, and 8, and Supplementary Figure 1 are created with BioRender.com. Figure 3 is provided by Stella Reamon-Buettner, Fraunhofer Institute for Toxicology and Experimental Medicine, Germany. Supplementary Figure 3A and ![]() is provided by Lorenzo Limana, Ipsen Innovation, Saclay site, France.
is provided by Lorenzo Limana, Ipsen Innovation, Saclay site, France.
The studies were all conducted under the respective Animal Welfare Act. All nonclinical studies conducted in the US study were complied with the Animal Welfare Act, the “Guide for the Care and Use of Laboratory Animals” and the applicable approved IACUC protocol for this species. All animal care and experimental procedures were nationally approved in the respective European country by the responsible authorities according to International and National law and policies (EU Directive 2010/63/EU and the respective National Animal Welfare Acts in the respective European Countries).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.