
Abstract
Nonalcoholic fatty liver disease (NAFLD) is a common chronic liver disease and its influence on drug-induced liver injury (DILI) is not fully understood. We investigated whether NAFLD can influence acetaminophen (APAP [N-acetyl-p-aminophenol])-induced hepatotoxicity in a diet-induced obese (DIO) mouse model of NAFLD. The male C57BL/6NTac DIO mice, fed a high-fat diet for more than 12 weeks, developed obesity, hyperinsulinemia, impaired glucose tolerance, and hepatomegaly with hepatic steatosis, similar to human NAFLD. In the acute toxicity study after a single dose of APAP (150 mg/kg), compared with control lean mice, the DIO mice had decreased serum transaminase levels and less severe hepatocellular injury. The DIO mice also had altered expression of genes related to APAP metabolism. Chronic APAP exposure for 26 weeks did not predispose the DIO mice with NAFLD to more severe hepatotoxicity compared with the lean mice. These results suggested that the C57BL/6NTac DIO mouse model appears to be more tolerant to APAP-induced hepatotoxicity than lean mice, potentially related to altered xenobiotic metabolizing capacity in the fatty liver. Further mechanistic studies with APAP and other drugs in NAFLD animal models are necessary to investigate the mechanism of altered susceptibility to intrinsic DILI in some human NAFLD patients.
Keywords
Introduction
In humans, nonalcoholic fatty liver disease (NAFLD) is the most common liver disease, with a global prevalence of 25%.1,2 It is commonly associated with metabolic comorbidities, such as obesity, diabetes mellitus, and dyslipidemia. NAFLD is clinically defined as the presence of fatty liver in the absence of other causes of chronic liver diseases or excess alcohol consumption. Recently, a new definition of metabolic-associated fatty liver disease (MAFLD) was proposed for better reflection of its risk factors3,4; the criteria are based on the presence of fatty liver disease with the co-occurrence of any one of the following three conditions: overweight/obesity, type 2 diabetes mellitus, or evidence of metabolic dysregulation. NAFLD is pathologically classified into nonalcoholic fatty liver (NAFL or simple steatosis) and nonalcoholic steatohepatitis (NASH).5,6 NAFL is defined by the presence of steatosis in > 5% of hepatocytes without evidence of hepatocellular injury in the form of hepatocellular ballooning, while NASH is characterized by hepatic steatosis, lobular inflammation, and hepatocellular ballooning with variable degree of fibrosis. NASH can progress to liver fibrosis, cirrhosis, and eventually hepatocellular carcinoma. As most (> 80%) of NAFLD patients have hepatic steatosis without progression to NASH, it should be clarified whether hepatic steatosis itself can alter hepatic function, such as xenobiotic metabolism. To date, no animal model recapitulates the full spectrum of NAFLD, 7 and most NASH-related studies are focused on the molecular mechanisms and therapeutic target of NASH progression.
Drug-induced liver injury (DILI) is a form of adverse drug reactions in the liver, which is classified into intrinsic (direct, predictable, dose-dependent, and reproducible in animal models) and idiosyncratic (indirect, unpredictable, not dose-dependent, and poorly reproduced in animal models) types based on the presumed mechanism of action.8,9 DILI is the leading cause of acute liver failure in the United States and some European countries, and the most common reason for regulatory actions after drug approval. Recently, there has been an increasing recognition that NAFLD patients may be more susceptible to DILI.10 -15 In addition, NAFLD patients usually have other comorbidities, such as obesity, diabetes mellitus, dyslipidemia, hypertension, and cardiovascular diseases, and are frequently subjected to polypharmacy than healthy individuals. Thus, NAFLD patients are exposed to a higher risk for DILI via both endogenous and exogenous factors. Some drugs (e.g., acetaminophen [APAP]) can induce severe acute liver failure in patients with NAFLD, as NAFLD is associated with altered xenobiotic metabolizing enzymes, resulting in increased generation of toxic metabolites and/or impaired detoxification pathways.10,11,16 In contrast, hepatotoxicity induced by several drugs (e.g., amiodarone and statins) does not seem to be more frequent in patients with NAFLD. 11 To date, the risk of NAFLD for intrinsic or idiosyncratic DILI is poorly understood and the underlying mechanisms contributing to these altered risk profiles are largely unknown.
APAP is a representative example of drugs that induce intrinsic (dose-dependent and predictable) DILI and well known to induce severe DILI after an acute overdose.8,9 Large retrospective studies have suggested that patients with NAFLD have a 4- to 7-fold higher prevalence of acute liver injury after APAP overdose compared with those without NAFLD.17,18 Besides, recent studies suggested that therapeutic dose of APAP can induce mild to moderate liver injury in morbidly obese patients, and even fulminant hepatitis in a few patients with predisposing factor, such as fatty liver and obesity. 10 Interestingly, some clinical studies showed that obese patients are less or similarly susceptible to APAP-induced acute liver injury compared with nonobese patients.19,20 To specify the cause of this variability in the susceptibility to APAP-induced liver injury, animal models replicating the condition of human fatty liver are required.
The results of animal studies on the influence of NAFLD condition on APAP toxicity are variable between studies.10,11,16 For instance, APAP hepatotoxicity is greater in db/db (leptin receptor deficient) obese mice but is similar in ob/ob (leptin deficient) obese mice compared with their respective wild type lean controls. 21 Possible factors influencing the susceptibility to APAP-induced hepatotoxicity include preexisting hepatic CYP2E1 induction, decreased hepatic glutathione (GSH) stores, and mitochondrial dysfunction10,11; however, the overall mechanism is still not completely understood. The purpose of this study is to clarify whether APAP-induced liver injury is altered in a mouse model of NAFLD caused by diet-induced obesity (DIO) and to identify molecular mechanisms underlying the altered susceptibility to APAP-induced hepatotoxicity.
Materials and Methods
The phenotype of the DIO mouse was characterized in the first two experiments described below. The acute and chronic APAP toxicity in lean and DIO mice were evaluated in Experiments 3 and 4, respectively. Experimental designs of these 4 studies are presented graphically in Figure 1.
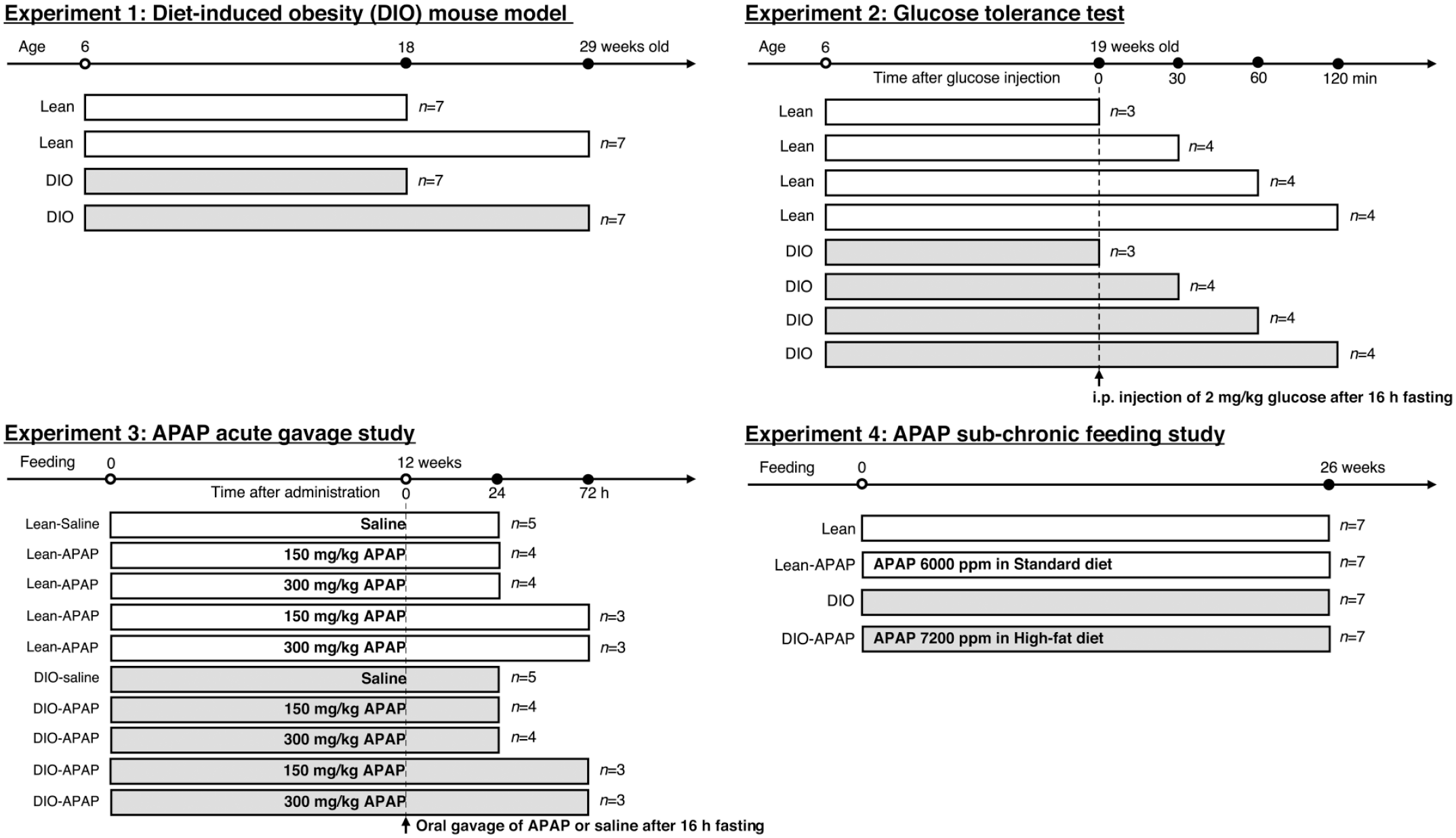
A graphical summary of the experimental designs of the 4 studies conducted in this project are presented. Lean and DIO C57BL/6NTac mice sourced from Taconic Biosciences were fed either a standard diet (10 kcal% fat; D12450Ji) or a high-fat diet (60 kcal% fat; D12492), respectively. The acetaminophen (APAP) dosage in each of the treatment groups is indicated in the corresponding bars. The timeline of each of the treatments is indicated at the top of each figure and the sample size is indicated on the right side of each bar. In Experiment 4, the DIO mice received a higher dosage of APAP after accounting for the differences in the amount of feed consumption between the high-fat diet versus the standard diet. The endpoints in Experiment 1 included clinical chemistry and histopathology; Experiment 2 included analyses of glucose, insulin, and leptin; Experiment 3 included clinical chemistry, histopathology, and RT-PCR (for APAP metabolizing enzymes); Experiment 4 included clinical chemistry, and histopathology. APAP indicates N-acetyl-p-aminophenol; DIO, diet-induced obesity.
All experiments conducted in this study are reported in accordance with the ARRIVE guidelines (https://arriveguidelines.org). All mice were housed in individual cages and were provided food and water ad libitum, except during fasting treatment before APAP or glucose administration. Based on previous studies, a sample size of 6 animals per group (7 with 25% extra) was considered to be necessary for this study as the DIO phenotype may not equally be present in all the mice and the hepatotoxicity (serum transaminase levels) induced by APAP is known to have some variation. 22 Male mice were selected to be used in this study as female mice are well known to develop less hepatic necrosis than male mice after APAP exposure. 22 Glucose tolerance study was done with minimal sample size (3 or 4) as it contains multiple time points. In APAP acute gavage study, the sample size of 7 was split into 3 or 4 to accommodate the 2 time points (24 and 72 hours) needed to evaluate the temporal changes of the liver injury. One mouse fed the standard diet without APAP group was excluded from the experiment at Week 2, because of an abnormal circling behavior and decreased body weight in the APAP chronic feeding study. All mice sampled were included in the analyses described below without any exclusion. Allocation of the mice to lean and DIO groups had been conducted by the supplier (Taconic Biosciences) before the mice were introduced into the animal facility of the National Institute of Environmental Health Services (NIEHS). Mice were allocated to control and APAP treatment groups without randomization since there were no significant differences in the body weight range. Two of the authors (TI and RAC) were aware of the group allocation at the different stages of the experiments. All experiments were performed after 1- to 2-week acclimatization periods.
DIO Mouse Model
Six-week-old male C57BL/6NTac mice (Taconic Biosciences, Rensselaer, NY) were fed a high-fat diet (60% fat, derived from lard and soybean oil; D12492, Research Diets, New Brunswick, NJ) alone are hereafter referred to as the DIO mice or a standard diet (10% fat, derived from lard and soybean oil; D12450Ji, Research Diets) alone from here onwards are referred to as the lean mice until 18 and 29 weeks of age. Mice were then euthanized by CO2 inhalation and were necropsied. Serum was collected for clinical chemistry and tissues were collected for histopathological examination. The liver was weighed at necropsy and the organ weights relative to body weights were calculated. For glucose tolerance test, DIO and lean mice at 19 weeks of age were injected intraperitoneally with D-glucose (Sigma-Aldrich, St Louis, MO) at the dose of 2 g/kg after 16-hour fasting, and euthanized by CO2 inhalation at 0 (control), 30, 60, and 120 minutes. Serum was collected for analyses of glucose, insulin, and leptin as previously described. 23 All experiments and methods in this study were performed in accordance with relevant guidelines and regulations and were approved by the Animal Care and Use Committee at National Institute of Environmental Health Science (code number: 2018-0002).
APAP Acute Gavage Study
Six-week-old male C57BL/6NTac mice were fed the high-fat diet (D12492) diet alone (DIO mice) or the standard diet (D12450Ji) alone (lean mice) until 18 weeks of age. Mice were then gavaged with a single dose of APAP (at 7:30-9:00 am; 150 or 300 mg/kg; A7085, Sigma-Aldrich) or physiological saline after 16-hour fasting (refeeding 3 hours after gavage following fasting at 6:00 pm the day before necropsy). The APAP doses, 150 and 300 mg/kg, were determined based on published scientific literature.22,24 They were sampled at 24 and 72 hours after gavage. Necropsy, histopathology, clinical chemistry, and real-time polymerase chain reaction (RT-PCR) (for APAP-metabolizing enzymes) were performed to clarify whether susceptibility to APAP-induced acute liver injury is altered in DIO mice.
APAP Chronic Feeding Study
DIO mice were fed a high-fat (D12492-based) diet containing 7200 ppm of APAP or the D12492 diet while lean mice were fed a standard (D12450Ji-based) diet containing 6000 ppm of APAP or the D12450Ji diet for 26 weeks. The concentration (6000 ppm) of APAP in the D12450Ji-based diet was determined according to the previous NTP 13-week study, in which no hepatotoxicity was observed in male B6C3F1/N mice fed a diet containing 6200 ppm of APAP. 25 There was no effect on the palatability in mice with APAP feeding at the doses below 6200 ppm in the NTP 13-week sub-chronic study. To keep the same APAP ingestion levels per mouse, the concentration of APAP in the high-fat (D12492-based) diet was adjusted to 7200 ppm on the basis of the difference in food consumption between lean (3.6 g/day) and DIO (3.0 g/day) mice. The calculated ingestion levels are equal between the two mice groups: 3.6 g/day × 6000 ppm = 21.6 mg/day in lean mice, and 3.0 g/day × 7200 ppm = 21.6 mg/day in DIO mice. Human equivalent dose (HED) can be calculated from the mouse dose: 21.6 mg/0.02 kg /12.3 = 87.8 mg/kg. 26 The HED is 1.3-fold higher than the upper limit of total daily dose of APAP in human medicine (4000 mg/60 kg = 66.7 mg/kg) 27 and 2.5- to 5-fold lower than lethal dose range in humans (13,000 to 25,000 mg/60 kg = 217-417 mg/kg). 28 Necropsy, histopathology, and clinical chemistry were performed to clarify whether fatty liver can alter susceptibility to hepatotoxicity induced by persistent, chronic ingestion of APAP.
Histopathology
Liver tissues from the left lateral, right medial, and caudate lobes were fixed in 10% neutral buffered formalin within 24 hours, routinely processed, embedded in paraffin, sectioned at 5 μm in thickness, and stained with hematoxylin and eosin (H&E). For histopathological evaluation of fatty liver in the present DIO mouse model, a simple scoring system for rodent NAFLD models was selected. 29 Macrovesicular and microvesicular steatoses, and hepatocellular hypertrophy were individually evaluated: score 0, < 5%; 1, 5% to 33%; 2, 33% to 66%; 3, > 66% of hepatocytes. Hepatocellular hypertrophy in this scoring system is defined as a simple increase in the size of the cytoplasm irrespective of accumulation of lipids and is distinct from the hepatocellular lesion induced by certain chemical compounds like phenobarbital. Inflammation was evaluated by counting the number of inflammatory foci in the hepatic parenchyma, defined as a cluster of ≥ 5 inflammatory cells, per 100× field (average of 5 different 100× fields): score 0, < 0.5; 1, 0.5 to 1.0; 2, 1.0 to 2.0; 3, > 2.0 foci. For histological evaluation of APAP acute gavage study, degeneration/necrosis of hepatocytes was evaluated with the following scores: score 0 (absent); 1 (mild), degeneration of hepatocytes in some part of zone 3; 2 (moderate), necrosis of hepatocytes in some part of zone 3 or extensive degeneration of zone-3 hepatocytes; and 3 (marked), extensive coagulation necrosis of zone-3 hepatocytes.
Clinical Chemistry
Serum was isolated by centrifugation at 3000 rpm for 10 minutes at 4°C within 45 minutes of blood collection. Alanine aminotransferase (ALT), aspartate aminotransferase (AST), total bilirubin, and alkaline phosphatase (ALP) were measured using Beckman Coulter AU480 chemistry analyzer and reagents obtained from the manufacturer (Beckman Coulter, Inc., Brea, CA).
Reverse transcription polymerase chain reaction
Liver tissues from the left lateral lobe of lean and DIO mice with administration of saline and 150 mg/kg APAP were immediately frozen by liquid nitrogen and stored at −80°C before use. Total RNA was extracted with miRNeasy Mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. One microgram of total RNA was reverse-transcribed to cDNA with iScript cDNA Synthesis kit (BioRad, Hercules, CA). RT-PCR was performed with Taqman Gene Expression Assays using QuantStudio 3 system according to the manufacturer’s instructions. The following Taqman probes/primers (Applied Biosystems, Foster City, CA) were used: Cyp2e1 (Assay ID: Mm00491127_m1), Cyp2d22 (Assay ID: Mm00530542_m1; an orthologue of human CYP2D6), 30 Ugt1a1 (Assay ID: Mm02603337_m1), Ugt1a9 (Assay ID: Mm03809914_sH), Gstp1 (Assay ID: Mm04213618_gH), Sult1a1 (Assay ID: Mm00467072_m1), Abcc4 (Assay ID: Mm01226381_m1), Abcg2 (Assay ID: Mm00496364_m1), and 18S rRNA (Assay ID: Hs99999901_s1). The selected genes were reported to be involved in the metabolism of APAP in the liver at therapeutic and toxic doses. 31 Data were quantified by the ΔΔCt method and are expressed as fold change to lean-saline mice; 18rRNA was used as a reference gene.
Statistics
Data were expressed as mean ± standard deviation. Statistical analyses were performed by Tukey’s or Sidak’s multiple comparison using Prism software ver. 9 (GraphPad, San Diego, CA). Pearson’s correlation coefficient was used for correlation analysis.
Results
Phenotypes of the DIO Mouse Model
The DIO mice had significantly (P < .05) increased calorie intake, increased body weight, and increased absolute liver weight at 18 weeks of age compared with the lean mice (Table 1). The DIO mice had increased body weights and increased absolute and relative liver weights at 29 weeks; the increased liver weights were more significant (P < .05) in the DIO mice at 29 weeks than at 18 weeks. Grossly, the DIO mice had obesity with increased subcutaneous and visceral fat and hepatic discoloration (paler with macroscopically distinct lobules) compared with lean mice (Figure 2A and D). Microscopically, hepatocytes in the centrilobular region (Zone 3) were enlarged with predominantly microvesicular steatosis in the DIO mice at 18 weeks of age; a few hepatocytes with macrovesicular steatosis were present in the midzonal region (Zone 2) (Table 2 and Figure 2B and E). The microvesicular steatosis became more extensive with greater admixture of macrovesicular steatosis in the DIO mice at 29 weeks of age (Figure 2C and F). Mild inflammatory cell infiltrate within the steatotic areas (mainly Zone 3) consisted of mostly macrophages and fewer neutrophils and fibrosis was absent in the liver of DIO mice based on H&E stains, and the inflammatory cell infiltration distributed randomly tended to increase slightly in the DIO compared with that of lean mice. No special stains or immunohistochemistry (IHC) analysis were conducted to confirm the lack of fibrosis in the study. Steatosis was absent in lean mice at 18 weeks of age, while macrovesicular steatosis was present mildly (in 10%-20% of hepatocytes) and minimally (in < 5% of hepatocytes) in one and four of the seven lean mice at 29 weeks of age, respectively. Microvesicular steatosis was absent in lean mice at both 18 and 29 weeks of age.
Food consumption and necropsy data of C57BL/6NTac lean and DIO mice.
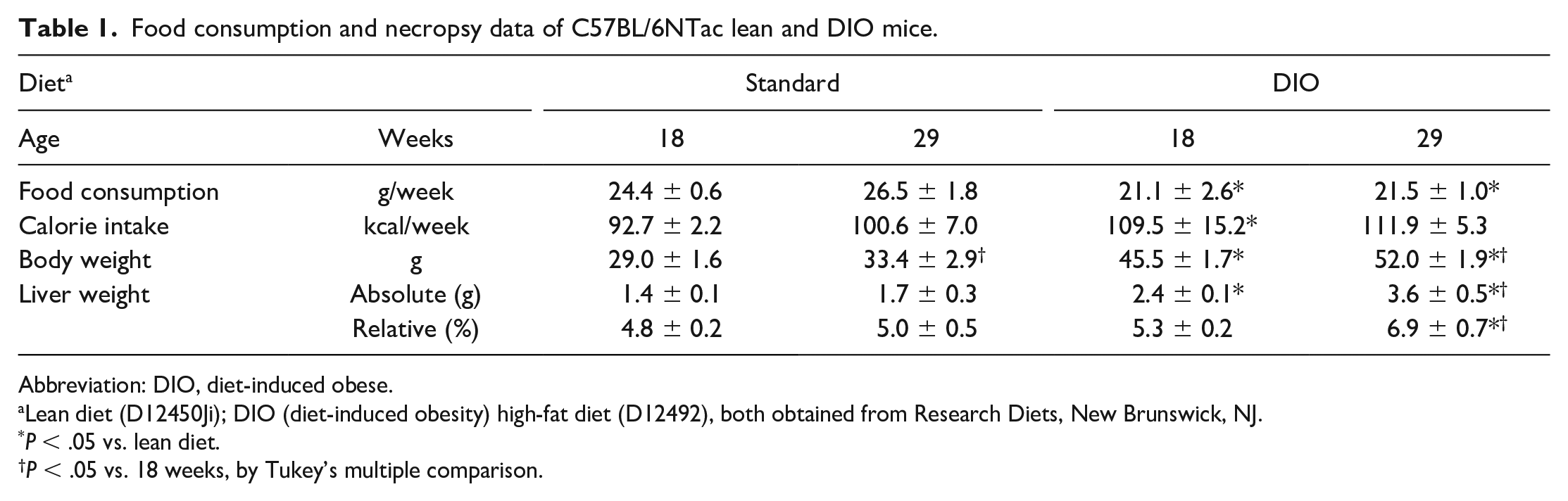
Abbreviation: DIO, diet-induced obese.
Lean diet (D12450Ji); DIO (diet-induced obesity) high-fat diet (D12492), both obtained from Research Diets, New Brunswick, NJ.
P < .05 vs. lean diet.
P < .05 vs. 18 weeks, by Tukey’s multiple comparison.
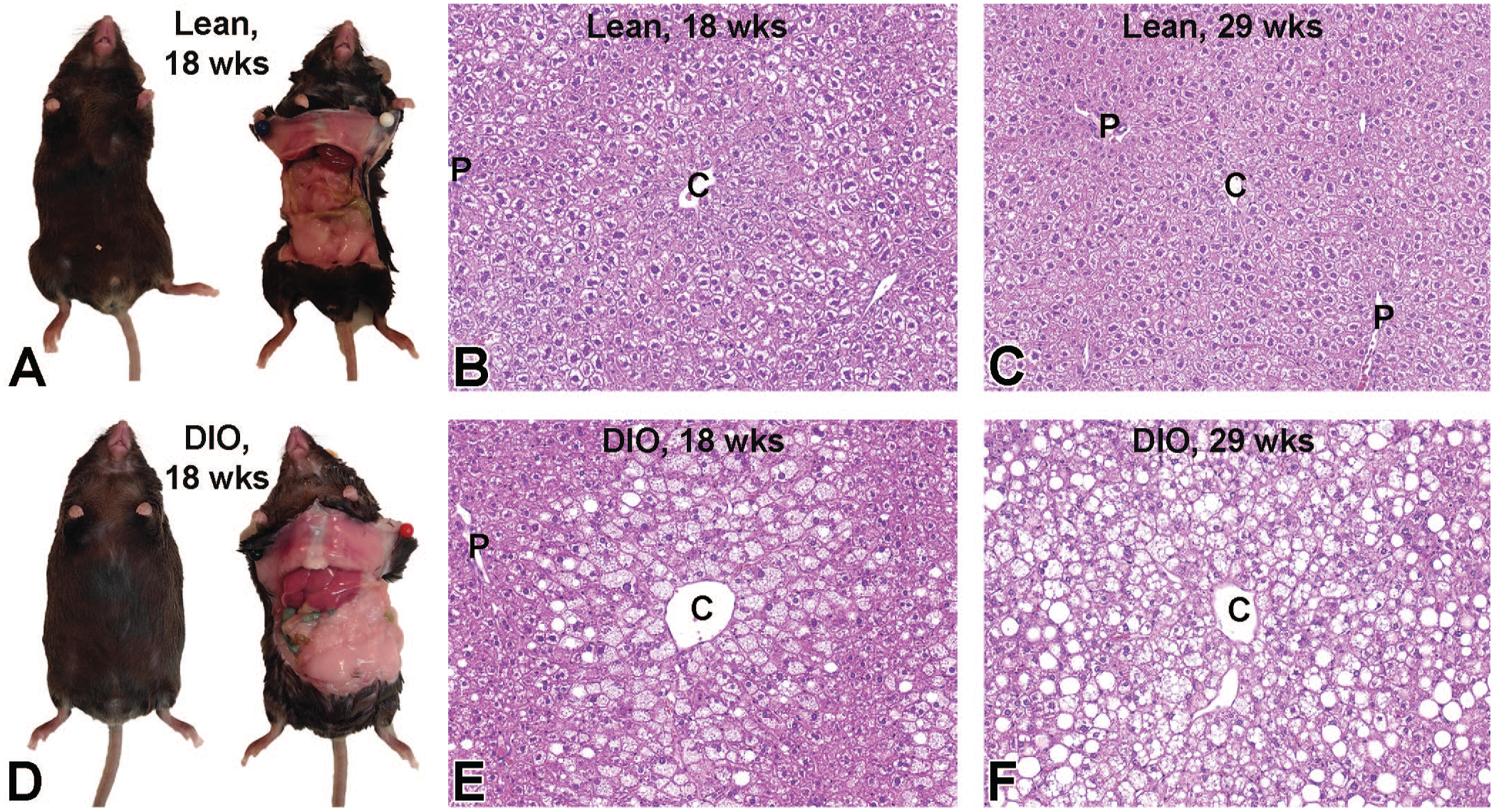
Gross appearance of lean (A) and C57BL/6NTac DIO mice (D) at 18 weeks at necropsy. The DIO mouse shows obesity with an increased amount of subcutaneous fat and hepatomegaly with pale discoloration. Histopathology of the liver in lean (B and C) and DIO (E and F) mice at 18 (B and E) and 29 weeks (C and F). At 18 weeks, hepatocytes in the centrilobular region (Zone 3) are swollen with predominantly microvesicular steatosis in DIO mice; a few hepatocytes with macrovesicular steatosis are present in the midzonal region (Zone 2). At 29 weeks, the microvesicular steatosis is more extensive with more macrovesicular steatosis admixed in DIO mice. DIO indicates diet-induced obese.
Histological scoring of the liver from C57BL/6NTac lean and DIO mice.
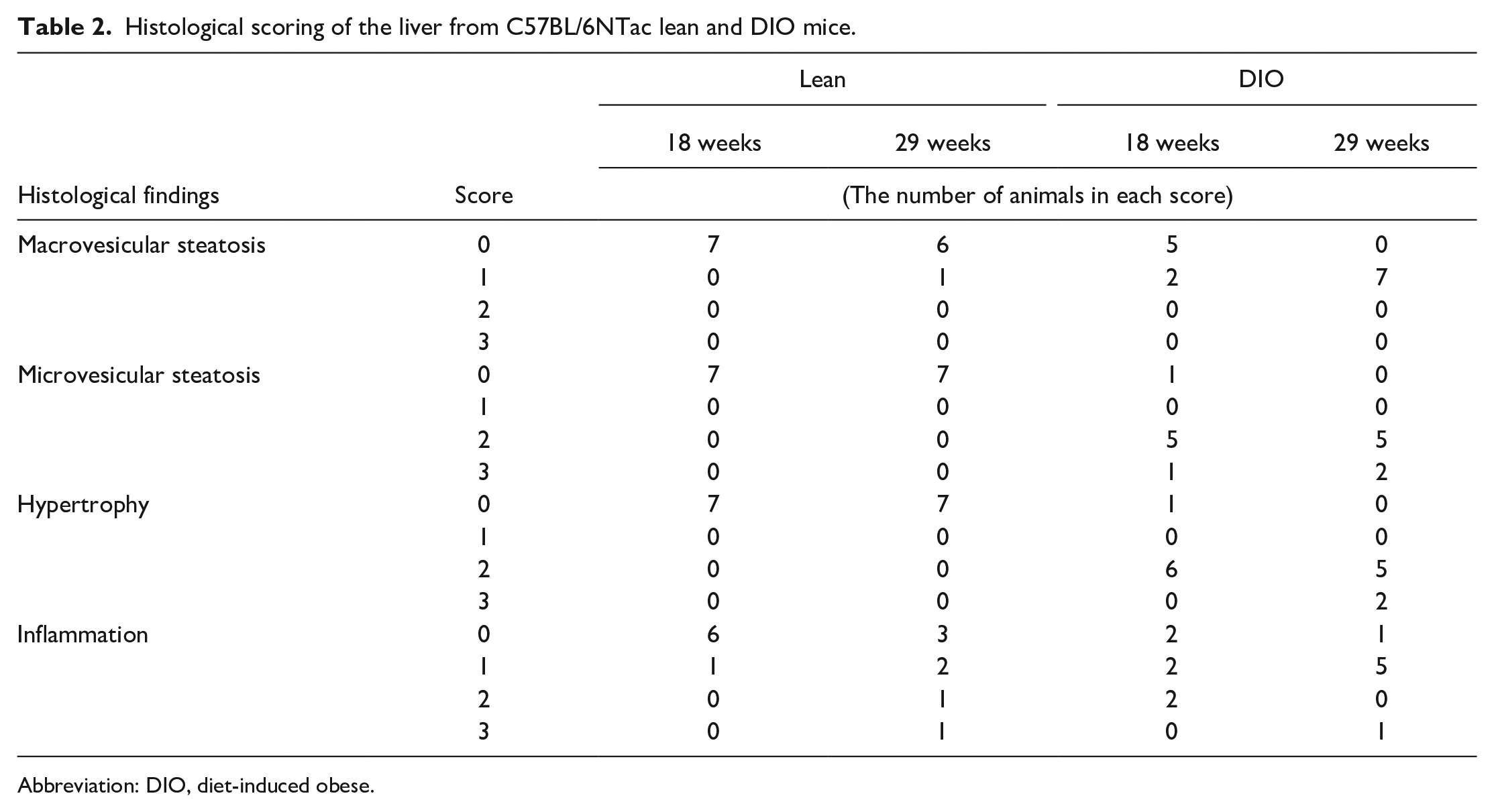
Abbreviation: DIO, diet-induced obese.
Glucose tolerance test revealed that serum glucose levels were persistently higher in the DIO mice than in lean mice at 60 and 120 minutes after glucose injection (Figure 3A), suggesting an impaired glucose tolerance. Serum levels of insulin and leptin were also significantly (P < .05) higher in the DIO compared with the lean mice, irrespective of glucose injection (Figure 3B and C). These results suggest that the present DIO mouse model develops hepatic steatosis in association with glucose intolerance, hyperinsulinemia, and hyperleptinemia, similar to NAFLD in humans.
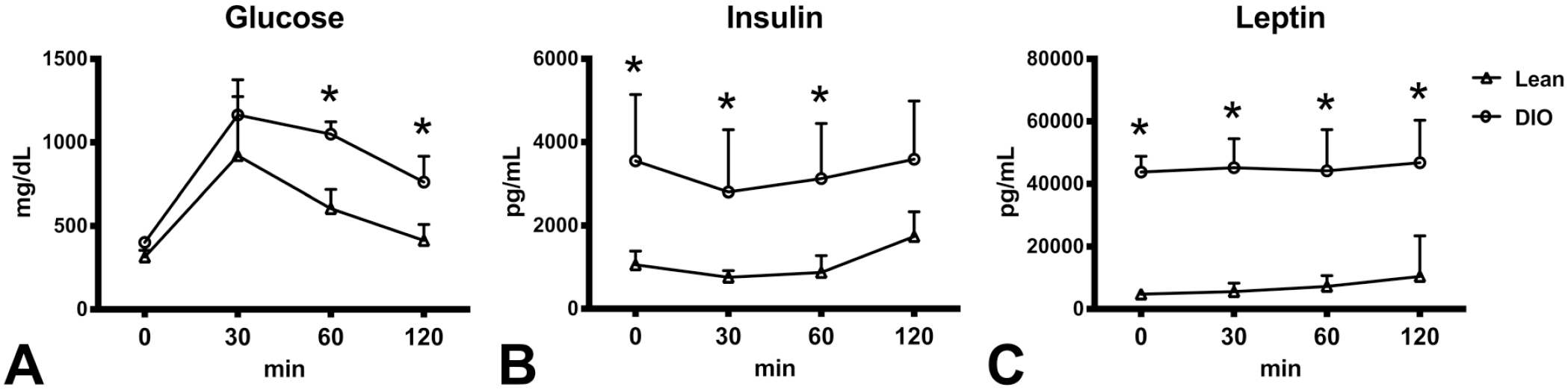
Temporal changes of serum glucose (A), insulin (B), and leptin (C) in lean and DIO C57BL/6NTac mice with glucose tolerance test. Glucose levels are higher in DIO mice at 60 and 120 minutes after glucose injection (2 mg/kg, i.p.) than in lean mice. Insulin and leptin levels are higher before and after the glucose injection in DIO mice than in lean mice. DIO indicates diet-induced obese.
APAP Acute Gavage Study
Compared with the DIO mice, the lean mice had a significant (P < .05) elevation of serum transaminase levels (AST and ALT) with increased absolute and relative liver weight at 24 hours after APAP gavage at 150 mg/kg; interestingly, the transaminase levels were significantly (P < .05) higher at 150 mg/kg than at 300 mg/kg dose (Table 3). In the lean mice, compared with the 24-hour time point, the transaminase levels at 72 hours decreased significantly (P < .05) in the 150 mg/kg group but the decreases at 300 mg/kg dose were not significant. Compared with the saline controls, the DIO mice had a significant (P < .05) elevation of serum transaminase levels at 24 hours after APAP gavage at 150 and 300 mg/kg; the transaminase levels were significantly (P < .05) lower in the DIO than in lean mice at 150 mg/kg dose. The transaminase levels at 150 mg/kg dose decreased significantly (P < .05) at 72 hours, compared with those at 24 hours in the DIO mice. Total bilirubin significantly (P < .05) increased at 24 hours after APAP gavage in lean mice at 150 mg/kg and in the DIO mice at 300 mg/kg when compared with their respective saline controls and then decreased significantly (P < .05) at 72 hours in both lean and DIO mice compared with the 24-hour time point. ALP increased at 24 hours in the DIO mice at 300 mg/kg compared with the respective saline group. On average ALP is also higher (but without statistical significance) in lean mice at 150 and 300 mg/kg than lean saline group suggesting that acute APAP overdose may induce damage to biliary system, but the degree of the damage is very mild.
Necropsy and clinical pathology data of acute APAP gavage study in lean and DIO C57BL/6NTac mice.
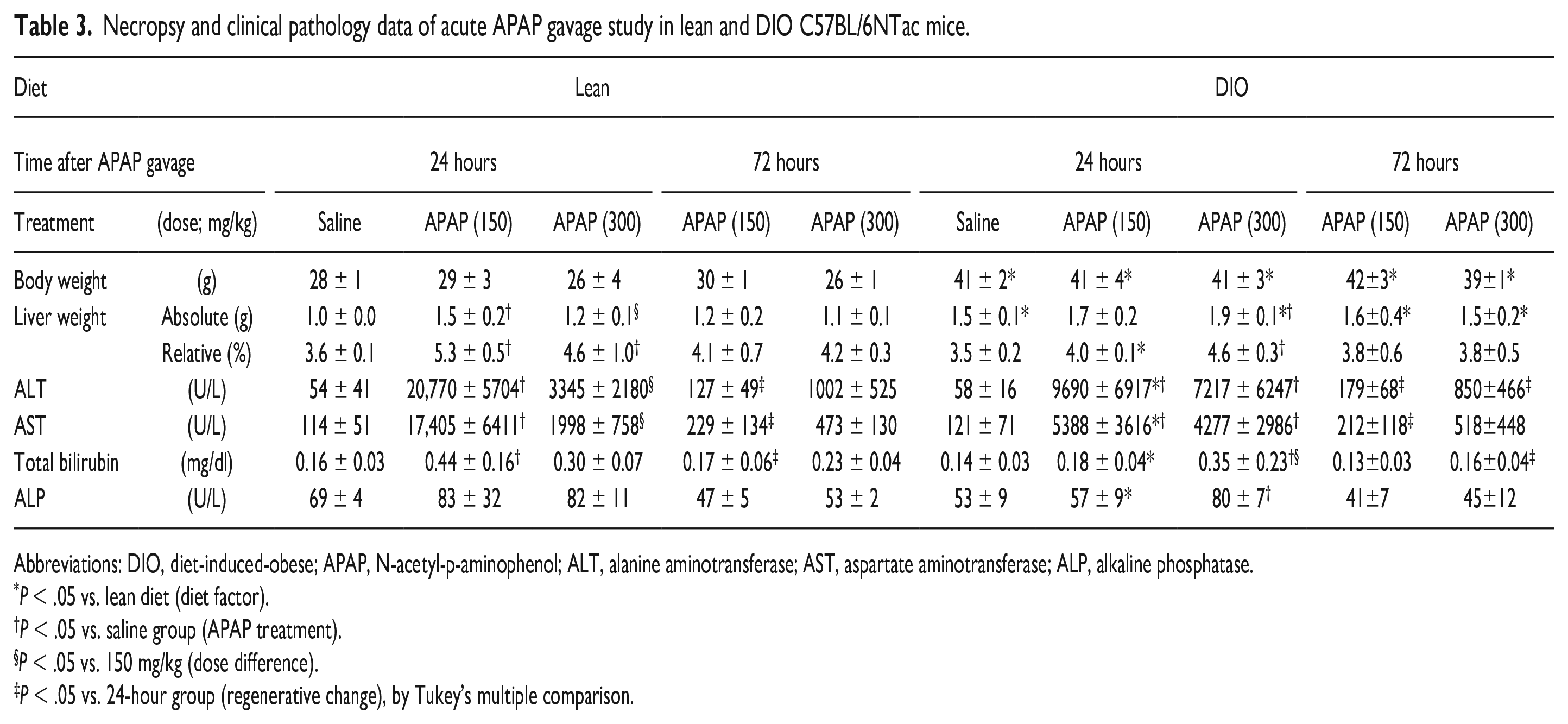
Abbreviations: DIO, diet-induced-obese; APAP, N-acetyl-p-aminophenol; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALP, alkaline phosphatase.
P < .05 vs. lean diet (diet factor).
P < .05 vs. saline group (APAP treatment).
P < .05 vs. 150 mg/kg (dose difference).
P < .05 vs. 24-hour group (regenerative change), by Tukey’s multiple comparison.
Representative histopathology images as well as the results of histopathological scoring of hepatocellular degeneration/necrosis are shown in Figure 4A-F and Table 4, respectively. There was a severe, centrilobular (Zone 3) coagulative necrosis with moderate infiltration of mononuclear cells and neutrophils in the liver of lean mice with APAP gavage at 150 mg/kg (Figure 4B) and surprisingly the severity of necrosis was lower in lean mice at 300 mg/kg dose (Figure 4C). In the DIO mice, degeneration and coagulative necrosis of hepatocytes with mild to moderate inflammatory infiltrates was found in the centrilobular region at 24 hours after APAP gavage at 150 and 300 mg/kg (Figure 4E-F), but was less severe than in lean mice at 150 mg/kg dose (Figure 4B). There was no apparent difference in morphology between 150 and 300 mg/kg doses in the DIO mice at 24 hours but the severity was slightly lower at 150 mg/kg compared with 300 mg/kg. At 72 hours, lean and DIO mice at 150 and 300 mg/kg doses had mild hepatocellular degeneration/necrosis admixed with mild to moderate inflammatory infiltrates in the centrilobular region (Figure 5A, B, D, and E); massive necrosis extending multiple hepatic lobules were focally present in all groups but was always less than at 24 hours (Figure 5C and F). Based on the increased severity of transaminase levels and increased hepatic injury in the lean mice compared with DIO mice, it appears that susceptibility to APAP-induced hepatotoxicity is altered in the DIO mice. The extent of hepatic steatosis (both microvesicular and macrovesicular types) tended to decrease at 72 hours after APAP gavage. The degree of histologic lesions was similar between the hepatic lobes examined.
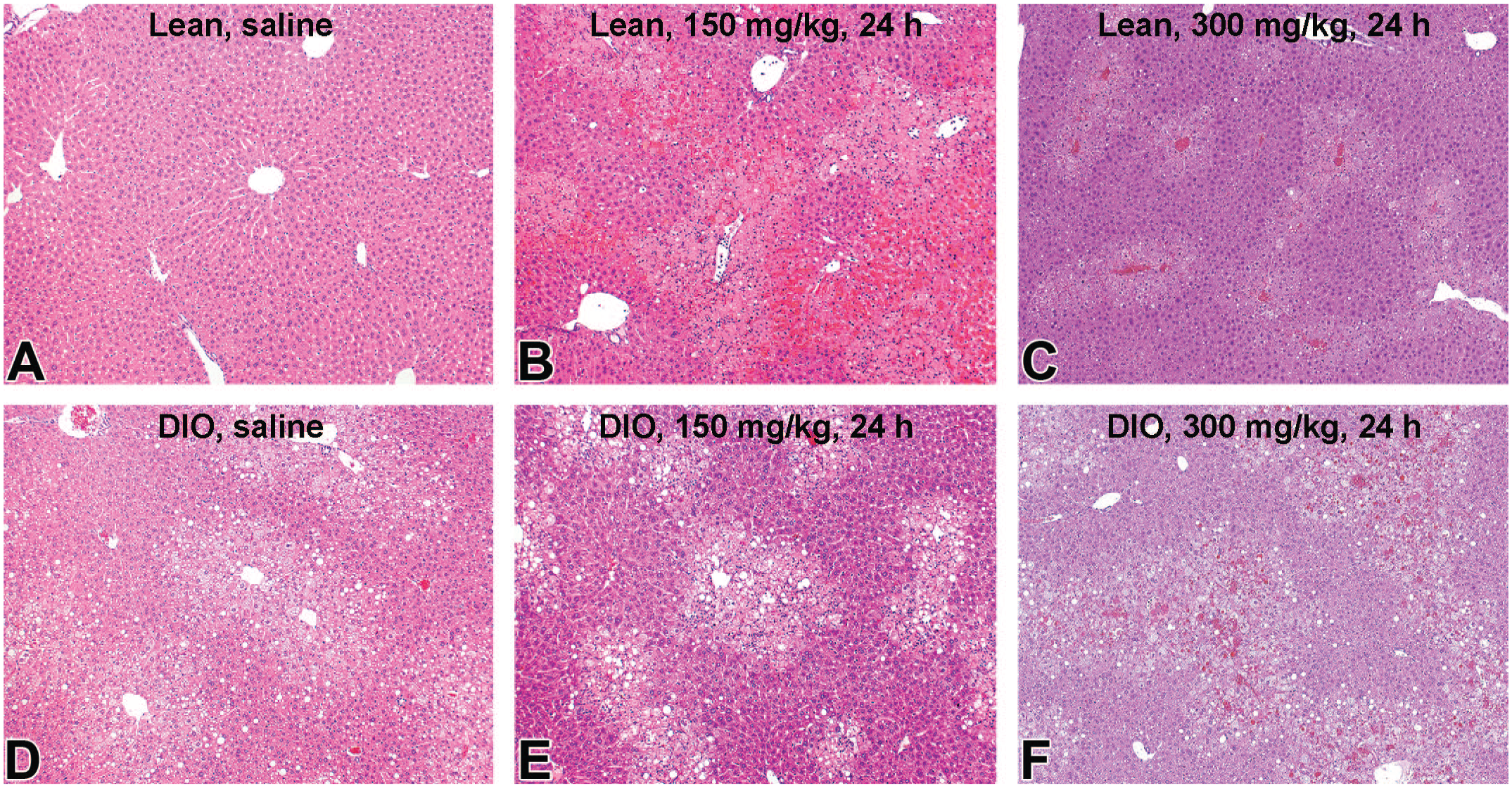
Histopathology of the liver in lean (A-C) and DIO (D-F) C57BL/6NTac mice at 24 hours after oral administration of saline (A and D), and acetaminophen (APAP) at the dose of 150 (B and E) and 300 (C and F) mg/kg. Centrilobular necrosis is most prominent in lean mice at 150 mg/kg APAP dose (B); it is less extensive in DIO mice at the same dosage I. The necrosis is also less extensive in lean mice at 300 mg/kg (C) than at 150 mg/kg dosage (B). APAP indicates N-acetyl-p-aminophenol; DIO, diet-induced obesity.
Histological scoring of the liver from C57BL/6NTac lean and DIO mice with acute acetaminophen (APAP) exposure at 24 hours.
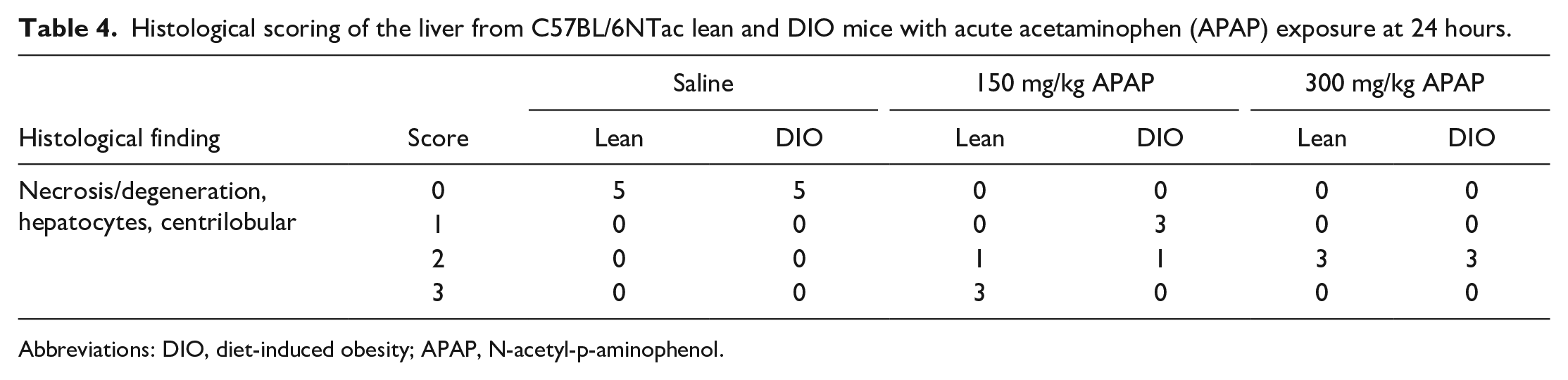
Abbreviations: DIO, diet-induced obesity; APAP, N-acetyl-p-aminophenol.
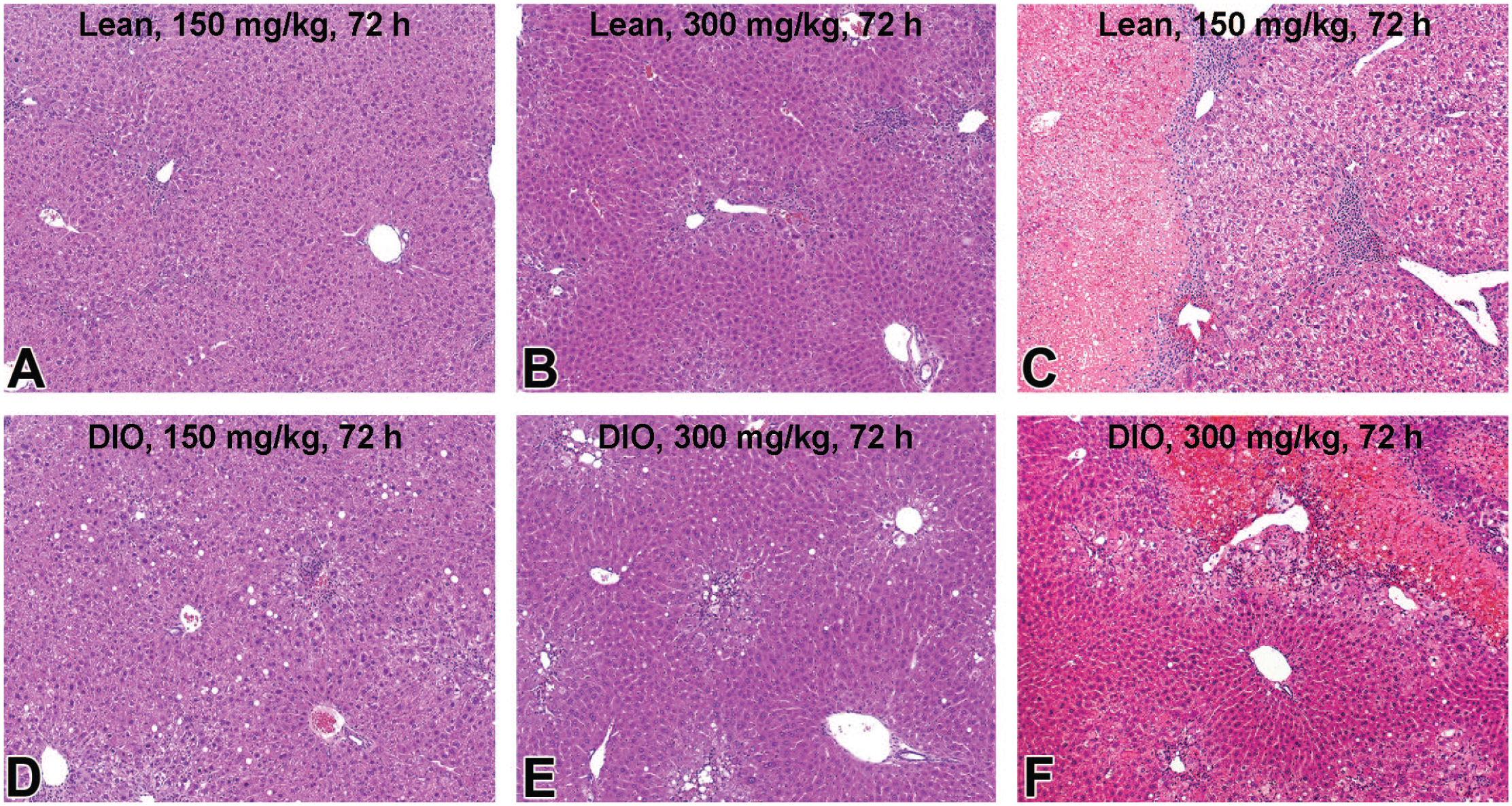
Histopathology of the liver in lean (A-C) and DIO (D-F) C57BL/6NTac mice at 72 hours after oral administration of APAP at the dose of 150 (A and D) and 300 (B and E) mg/kg. Massive necrosis, extending multiple lobules with hemorrhage, is focally present in both leI(C) and DIO (F) mice at 72 hours after APAP dosing. APAP indicates N-acetyl-p-aminophenol; DIO, diet-induced obesity.
To investigate whether fatty liver can alter xenobiotic metabolizing enzyme levels and whether such changes in enzyme levels can contribute to the altered susceptibility to APAP-induced hepatotoxicity, gene expression of APAP-metabolizing enzymes was analyzed. Hepatic expression of Ugt1a9 and Sult1a1 decreased in the DIO mice compared with lean mice, regardless of administration of 150 mg/kg APAP (Figure 6D and F). Expression of Abcc4 increased following administration of APAP in both lean and DIO mice; the levels were lower in the DIO than in lean mice (Figure 6G). Expression of Cyp2d22 was lower in the DIO mice than in lean mice (Figure 6B). Expression of Cyp2e1, Cyp2d22, and Ugt1a1 decreased in lean mice and tended to decrease in the DIO mice following administration of APAP (Figure 6A-C). Expression of Cyp2e1 tended to decrease in the untreated DIO mice compared with untreated lean mice. Expression of Gstp1 and Abcg2 did not differ significantly between groups (Figure 6E and H). These results suggest that expression of APAP-metabolizing enzymes, particularly Ugt1a9, Sult1a1, Abcc4, and Cyp2d22, is altered in the fatty liver of DIO mice. Correlation analysis revealed that expression of Abcc4 was positively correlated with ALT (R = 0.8560, P < .0001), AST (R = 0.7979, P < .0001), and histological score of hepatocellular degeneration/necrosis (R = 0.9190, P < .0001) while expression of Cyp2d22 was negatively correlated with ALT (R = −0.6119, P = .0070), AST (R = −0.5594, P = .0158), and histological score (R = −0.6895, P = .0015). Expression of Ugt1a9 and Sult1a1 was not correlated with serum transaminase levels or histology.
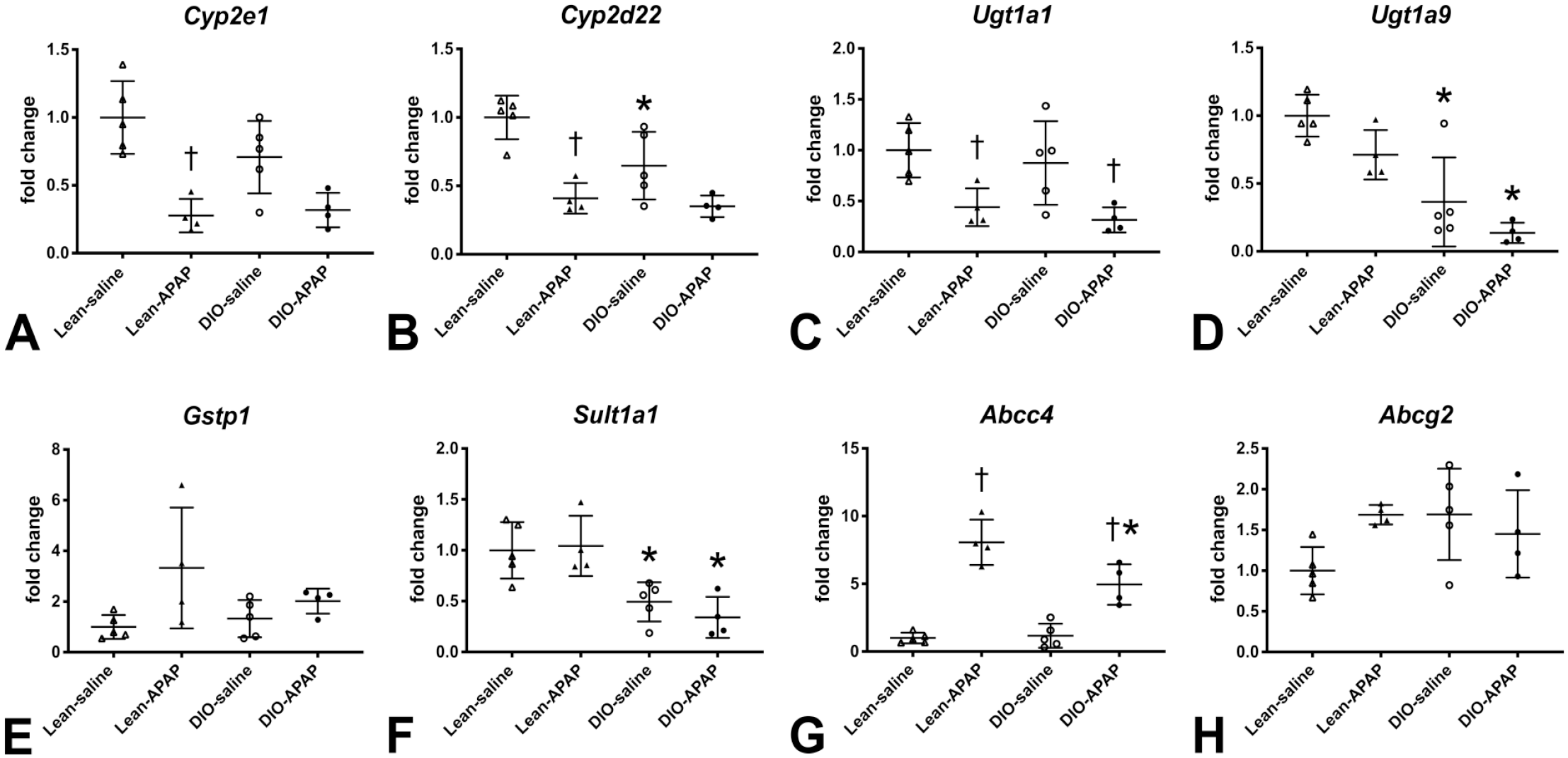
Hepatic expression of APAP-metabolizing enzymes Cyp2e1 (A), Cyp2d22 (B), Ugt1a1 (C), Ugt1a9 (D), Ip1 (E), Sult1a1 (F), Abcc4 (G), and Abcg2 (H) in lean and DIO C57BL/6NTac mice with or without APAP dosing. APAP indicates N-acetyl-p-aminophenol; DIO, diet-induced obesity.
APAP Chronic Feeding Study
Chronic (26 weeks) exposure to APAP in feed did not change body weight, liver weight, or serum hepatic enzymes in lean or the DIO mice suggesting that the diet-induced NAFLD condition does not predispose to hepatic injury at a dose well tolerated in lean mice (Table 5). As seen in the DIO phenotyping study (Table 1), the DIO mice without APAP exposure had increased body weight and increased absolute liver weight compared with lean mice (Table 5). The DIO mice with APAP exposure had an increased body weight compared with lean mice with APAP; interestingly, the liver weight was similar between the two groups. Scoring of hepatic steatoses is shown in Table 6. Macrovesicular and microvesicular steatosis was more extensive in lean mice after 26-week feeding of standard diet (51 weeks of age; Figure 7A) than in young lean mice (18 and 29 weeks of age; Figure 2B and C), suggesting that the C57BL/6NTac mice spontaneously develop hepatic steatosis with age. Chronic APAP exposure reduced microvesicular steatosis in the lean mice (Figure 7B); the degree of macrovesicular steatosis was similar between lean mice with and without APAP exposure. Microvesicular steatosis was more extensive in the DIO mice (Figure 7C) than lean mice (Figure 7A), while macrovesicular steatosis was less extensive in the DIO than lean mice, suggesting that the high-fat diet feeding facilitates microvesicular and suppresses macrovesicular steatosis in this mouse strain. Chronic APAP exposure reduced microvesicular steatosis in the DIO mice (Figure 7D), as in lean mice. These results suggest that the Chronic feeding of APAP in this study is non-toxic to the C57BL/6NTac mouse strain and can reduce microvesicular steatosis (fatty liver) induced by the high-fat diet feeding.
Necropsy and clinical pathology data of 26-week APAP sub-chronic feeding study in lean and DIo C57BL/6NTac mice.
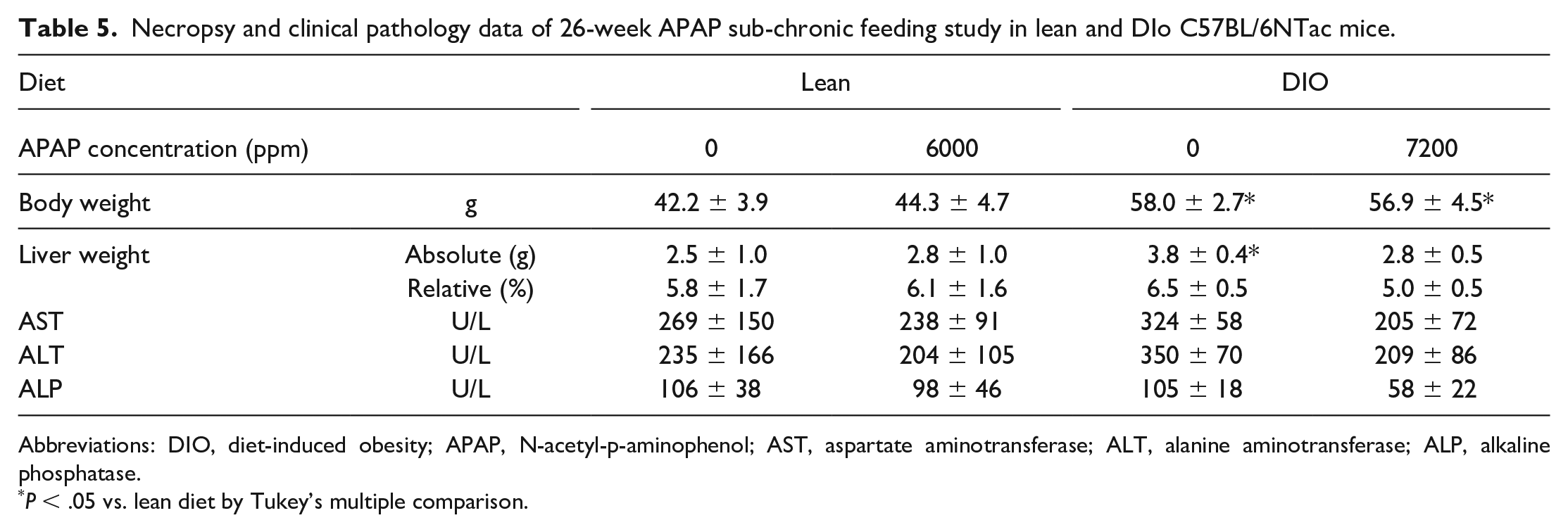
Abbreviations: DIO, diet-induced obesity; APAP, N-acetyl-p-aminophenol; AST, aspartate aminotransferase; ALT, alanine aminotransferase; ALP, alkaline phosphatase.
P < .05 vs. lean diet by Tukey’s multiple comparison.
Scoring of hepatic steatosis in the 26-week APAP sub-chronic feeding study.

Abbreviations: APAP, N-acetyl-p-aminophenol; DIO, diet-induced obesity.
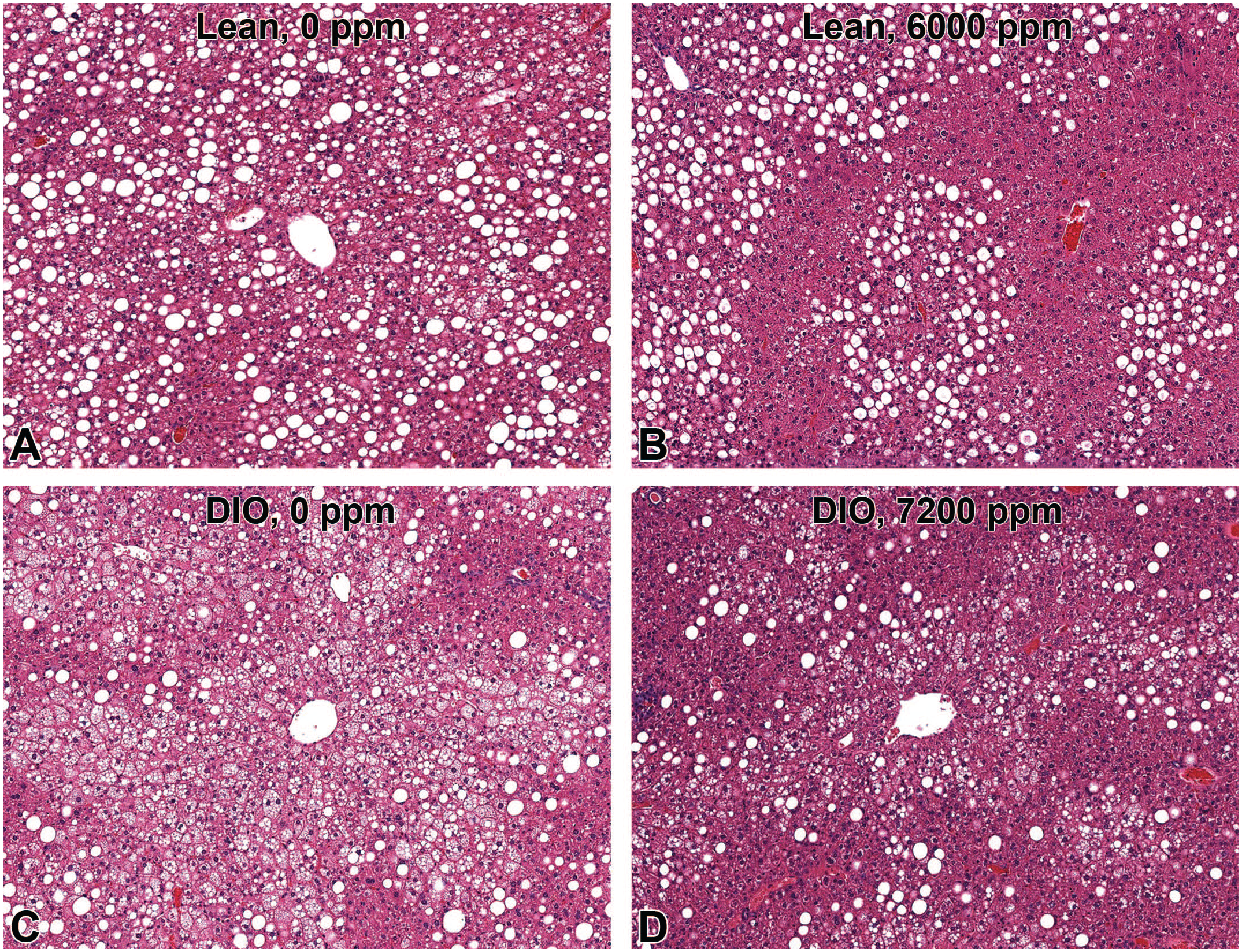
Histopathology of the liver in lean (A and B) and DIO (C and D) C57BL/6NTac mice with or without 26-week chronic exposure of APAP. Microvesicular steatosis is less prominent in lean and DIO mice with APAP (B and D) than without APAP (A and C). APAP indicates N-acetyl-p-aminophenol; DIO, diet-induced obesity.
Discussion
The C57BL/6NTac DIO mice have an increased body weight (obesity), hyperinsulinemia, hyperleptinemia, impaired glucose tolerance, and hepatomegaly with hepatic steatosis; these phenotypes are similar to those in human NAFLD. However, there is an important difference in liver histology between the DIO mice and human NAFLD patients, that is, the hepatic steatosis is mainly microvesicular in the DIO mice while it is predominantly macrovesicular in human NAFLD induced by diet, but in both cases, it is centered mainly on Zone 3 and extending into Zone 2, and even diffuse in very severe cases. In addition, features, such as Mallory-Denk bodies were absent in the DIO mouse.5,32 Hepatic microvesicular steatosis is patchy and never diffuse in human NAFLD. Diffuse microvesicular steatosis can indicate severe mitochondrial injury seen in a variety of diseases, such as alcoholic liver disease, drug-induced hepatotoxicity (e.g., tetracycline, valproic acid), and acute fatty liver of pregnancy in humans. A large retrospective study showed that microvesicular steatosis is present in 10% of liver biopsies from NAFLD patients and is associated with more advanced histology of NAFLD. 33
On the other hand, the pattern of hepatic steatosis is inconsistent among rodent models of NAFLD: mainly macrovesicular in choline-deficient, and choline-deficient amino acid-supplemented diet models,34,35 mixed microvesicular and macrovesicular in a variety of high-fat diet feeding models,36,37 and more microvesicular in rodents fed a diet with high cholesterol content.35,37 Despite such great inconsistency, the histological pattern of hepatic steatosis has been disregarded in rodent models until recently. 7 Animal models truly relevant to human NAFLD are required to develop fatty liver mainly consisting of macrovesicular steatosis in the centrilobular region, associated with metabolic disorders, such as obesity, insulin resistance/hyperglycemia, and dyslipidemia.7,38 A recent study demonstrated that feeding of “Western diet” (high-fat plus high-fructose corn syrup) to an inbred isogenic strain of C57Bl6/J and S129S1/svlmJ mice (DIAMOND mice) can replicate the key metabolic, pathological, and transcriptomic features of human NASH: obesity, insulin resistance, hypertriglyceridemia, and progression of liver diseases from NAFL (with extensive macrovesicular and microvesicular steatosis, Mallory-Denk bodies) to NASH, liver fibrosis, and hepatocellular carcinoma. 39 The present C57BL/6NTac-DIO mouse model recapitulates most features of human NAFLD. It is anticipated that the inclusion of high fructose corn syrup along with the high-fat diet (Western diet) feeding regimen in the DIO C57BL/6NTac strain could replicate additional features of human NAFLD in this mouse model.
Based on the lower hepatic injury (liver enzymes, histological scores) in DIO mice compared with lean mice after acute APAP exposure, we conclude that steatosis in the C57BL/6NTac-DIO mouse model does not increase susceptibility to APAP-induced acute liver injury, rather partly reduces liver injury in the DIO mice. These results are inconsistent with some human clinical data that suggest an increased risk of APAP-induced liver injury in NAFLD patients.10,11,13,16 -18 On the other hand, other clinical studies showed less or similar susceptibility to APAP-induced acute liver injury in obese patients compared with nonobese patients.19,20 Similarly, the influence of NAFLD condition on APAP toxicity are variable between models in rodent studies.10,11,16 It is well known that occurrence of APAP-induced hepatotoxicity in rodent models depends on a subtle balance between bioactivation and detoxification pathways.10,11,16,22 At a therapeutic dose, APAP is mostly converted in the liver to pharmacologically inactive glucuronide and sulfate conjugates by UDP-glucuronosyl transferases (UGTs) and sulphotransferases (SULTs), respectively. 31 These inactive metabolites are released into blood by ABCC4 (ATP Binding Cassette subfamily C Member 4) and ABCC3 transporters while they are excreted into bile by ABCG2 and ABCC2 transporters. A minor fraction (5%-10%) is oxidized to a reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI) by P450 enzymes, such as CYP2E1 and CYP2D6. NAPQI is detoxified by glutathione-S-transferases (GSTs) in healthy condition. At a highly toxic dose, both SULT and UGT pathways are saturated, and higher proportion of APAP is oxidized to NAPQI. Excess NAPQI eventually depletes hepatic GSH stores and starts to form protein adducts, resulting in the development of liver injury. In the present DIO mouse model, down-regulation of some APAP-metabolizing enzyme genes (particularly Cyp2d22 [an orthologue of human CYP2D6] and Cyp2e1) may lead to decreased production of the toxic metabolite NAPQI in the liver and thus decreased susceptibility to APAP hepatotoxicity. Interestingly, hepatic expression of Abcc4 and Cyp2d22 showed a positive and negative correlation with hepatocellular injury following administration of APAP, respectively. However, it remains unknown how the decreased induction of Abcc4 gene following APAP administration and the decreased expression of Cyp2d22 gene without APAP administration contribute to the altered susceptibility to hepatotoxicity in the DIO mice. Further toxicokinetic analysis of APAP metabolites, combined with analyses on activity of multiple APAP-metabolizing enzymes, detoxification pathway (e.g., GSH stores), and mitochondrial function (e.g., ATP levels) in the liver at an earlier time point (e.g., hours after APAP exposure), would clarify how APAP-metabolizing pathways are altered in the DIO mice.
Acute liver injury at 24 hours after APAP exposure did not show a dose-response relationship in this study, where serum transaminase levels and degree of necrosis were lower at 300 mg/kg than at 150 mg/kg dosing in lean C57BL/6NTac mice. At 72 hours, the liver injury was higher at 300 mg/kg than at 150 mg/kg in lean mice, suggesting that the peak of APAP-induced acute hepatotoxicity may be delayed (later than 24 hours) at 300 mg/kg or that the hepatotoxicity may develop more slowly at 300 mg/kg than at 150 mg/kg. It is considered that acute liver injury after an overdose of APAP appears in a dose-dependent manner in mice but the severity may differ between mouse strains. 22 Further toxicokinetic analysis of APAP metabolites is required to investigate the mechanism underlying this discrepancy of the dose-related changes in this background strain.
In contrast to our hypothesis, DIO mice with NAFLD did not have greater susceptibility to hepatotoxicity after chronic APAP exposure for 26 weeks. This hypothesis is partly based on studies reporting that NAFLD patients generally suffer from co-morbid conditions related to obesity and are susceptible to hepatotoxicity.10 -15 Similar results were obtained from a recent 13-week sub-chronic study of APAP at a maximum dose of 8000 ppm in Zucker fatty rats (Leprfa/Leprfa), showing that the Zucker fatty rats may be less susceptible to APAP-induced hepatotoxicity than their lean counterparts. 40 Decreased immunoreactivity to CYP2E1, compared with a standard rat strain (F344), is considered to contribute at least partly to the decreased APAP-induced hepatotoxicity in the Zucker fatty rats. In the present study, chronic APAP exposure to the lean C57BL/6NTac mice also did not induce any hepatic lesions; similar results were observed in lean B6C3F1 mice exposed to APAP at a dose of 6200 ppm for 13 weeks. 25 Further study with hepatotoxic dose (> 12,500 ppm) of APAP is needed to elucidate the influence of fatty liver on hepatotoxicity to chronic APAP exposure.
Interestingly, chronic APAP exposure decreases fatty liver with decreased microvesicular steatosis and liver weight. Sub-chronic (13 weeks) APAP exposure to the Zucker fatty rats did not modify the histologic steatosis, liver weight, serum triglyceride, or serum transaminases, suggesting that the effect of sub-chronic APAP exposure on fatty liver may differ between experimental models. Further transcriptomic analysis focusing on hepatic lipid metabolism could clarify the molecular mechanism underlying the decreased fatty liver by APAP exposure in this C57BL/6NTac DIO model.
This study suggests, among other possibilities, that the markedly increased lipid vacuoles in fatty livers might have contributed to reduced xenobiotic metabolizing capability in the DIO mice and as a result the toxic metabolites are produced to a lesser extent. In addition, alterations in mitochondrial metabolism, immune system, oxidative stress response and other detoxifying mechanisms may have contributed to the differential susceptibility observed in the DIO mice compared with the lean mice. Generation of toxic metabolites by xenobiotic metabolizing enzymes is a key mechanism underlying liver injury induced by hepatotoxicants, such as carbon tetrachloride, 41 thioacetamide,42,43 valproic acid, 44 and acetaminophen.10,11,31 In a few hepatotoxicants, such as lipopolysaccharide, xenobiotic metabolism is less involved in the development of hepatotoxicity; lipopolysaccharide-induced hepatotoxicity is considered to be mediated by increased production of cytokines and reactive oxygen species by Kupffer cells/macrophages. 45 Further studies using such direct hepatotoxicants on the C57BL/6NTac DIO mouse would provide a better insight into the mechanism of altered susceptibility to hepatotoxicity in the fatty liver.
In conclusion, the C57BL/6NTac DIO mouse model appears to be more tolerant to APAP-induced hepatotoxicity than control lean mice after acute APAP exposure. The susceptibility to APAP-induced hepatotoxicity varies between animal models, but a differential susceptibility to APAP-induced hepatotoxicity in this NAFLD mouse model is probably due to altered xenobiotic metabolism in the fatty liver. These findings also have relevance for the potential differential pharmacologic effects in obese patients with NAFLD compared with lean patients. Animal models more relevant to human NAFLD, in terms of metabolic (obesity, dyslipidemia, and insulin resistance), pathologic (mitochondrial dysfunction and inflammation), and xenobiotic (alterations in xenobiotic enzymes and hepatic GSH stores) phenotypes, are necessary to investigate the mechanisms of altered toxicology and pharmacology in human patients with NAFLD.
Footnotes
Acknowledgements
We dedicate this manuscript to the memory of Ms. Natasha Clayton, our beloved colleague and friend who has suddenly passed away in December 2021.
Author Contributions
TI and AP designed the study; TI, GT, RC, NC, and RS generated the data; TI, GT and AP interpreted the data, and TI and AP wrote the manuscript with contributions from GT, RC, NC, and RS. All authors (except for NC) read and approved the final manuscript. Unfortunately, NC unexpectedly passed away during the preparation of this manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research has been supported by DNTP/NIEHS intramural program (ES103319-06 (2021) and ES102505-14 (2021)). We acknowledge the expert technical assistance provided by the Clinical pathology and Pathology support core laboratories in the Comparative and Molecular Pathogenesis Branch, DTT/NIEHS as well as the Comparative Medicine Branch, NIEHS. In addition, this research was also partly supported by JSPS (Japan Society for the Promotion of Science) Fund for the Promotion of Joint International Research (Grant No. 17KK0158).
Data Availability
The datasets analyzed for the study are available from the corresponding author on request.