
Abstract
Changing the physical state from crystalline to amorphous is an elegant method to increase the bioavailability of poorly soluble new chemical entity (NCE) drug candidates. Subsequently, we report findings from repeat-dose toxicity studies of an NCE formulated as a spray-dried amorphous solid dispersion (SD-ASD) based on hydroxypropyl methylcellulose acetate succinate (HPMC-AS) in rats. At necropsy, agglomerates of SD-ASD were found in the stomach and small intestine, which in reference to literature were termed pharmacobezoars. We interpreted the pH-dependent insolubility of HPMC-AS in the acidic gastric environment to be a precondition for pharmacobezoar formation. Gastric pharmacobezoars were not associated with clinical signs or alterations of clinical pathology parameters. Pharmacobezoar-correlated histopathological findings were limited to the stomach and consisted of atrophy, erosion, ulcer, and inflammation, predominantly of the nonglandular mucosa. Pharmacobezoars in the small intestines induced obstructive ileus with overt clinical signs which required unscheduled euthanasia, prominent alterations of clinical pathology parameters indicative of hypotonic dehydration, degenerative and inflammatory processes in the gastrointestinal tract, and secondary renal findings. The incidence of pharmacobezoars increased with dose and duration of dosing. Besides the relevance of pharmacobezoars to animal welfare, they limit the non-observed adverse effect level in nonclinical testing programs and conclusively their informative value.
Keywords
Background
Nonclinical toxicological studies are an essential part of drug development requested by international guidelines (e.g., ICH M3). Following molecular design of new chemical entities (NCEs) to reach and increase pharmacological activity against disease-relevant biological targets in vitro, NCEs have to be tested in vivo in one rodent and one nonrodent species to get insights into the complex interplay of metabolism and drug exposure. These studies are the basis to characterize the toxicity profile as well as for the definition of the non-observed adverse effect level.
Therefore, it is indispensable to achieve adequate plasma levels of the NCE in the respective animal species via the same route of administration as intended in humans. For oral administration, an NCE dose resulting in blood plasma levels 50 times higher than the intended clinic systemic exposure, or an NCE dose of 1000 mg/kg/day resulting in an exposure 10 times higher than the clinical exposure, is considered acceptable as the maximum dose for toxicological studies, next to the maximum tolerated dose or the maximum feasible dose. 1 While such drug plasma levels may be readily achieved with highly soluble and permeable NCEs, a great share of NCEs exhibit poor aqueous solubility and a low dissolution rate leading to low bioavailability in vivo.2,3 Thus, nonclinical formulation developers are often the first to face the challenge of reaching solubility levels of NCEs sufficient to enable adequate plasma concentration levels in vivo when adequate bioavailability cannot be achieved with suspensions of the crystalline NCE. 4
Even though the variety of solubility-enhancing approaches, so called “bioenabling formulations,” is broad, effects of required additives on physiologic parameters or toxicity limit the available options for use in toxicological studies with laboratory animals. 5 By changing the physicochemical state of the crystalline test item to amorphous, the lattice energy as the main energy barrier for the dissolution of the crystalline form can be circumvented. This can rise the apparent solubility of the NCE by a multiple compared with the saturation solubility of the crystalline form, provided that thermodynamically favorable recrystallization from the supersaturated solution can be prevented.
A common method for preparation of amorphous solid dispersions of NCEs for nonclinical safety studies is spray drying. For this purpose, the test item is dissolved together with a suitable carrier polymer in an organic solvent. This solution is then dispersed into a fine spray, where the fine drops dry rapidly in the surrounding hot gas. The resulting spray dried amorphous solid dispersions (SD-ASDs) are fine powders that can be administered to laboratory animals as suspensions.
In combination with its high glass transition temperature and great capability of interactions between NCEs and polymers, hydroxypropyl methylcellulose acetate succinate (HPMC-AS) is often the polymer of choice for the formulation of NCEs via spray drying for use in nonclinical safety testing in vivo.6 -9 A further advantage compared with other carrier polymers such as polyvinylpyrrolidone is the pH-dependent solubility. At pH > 5.5, deprotonation of succinate substituents enables dissolution of the polymer and the amorphously incorporated NCE to supersaturated levels.10 -12 Considering the physiological intraluminal pH gradient, HPMC-AS–containing SD-ASDs are insoluble in the acidic environment of the stomach, whereas favorable supersaturated solutions of NCEs are formed in the small intestine.13,14 Therefore, thermodynamically favorable recrystallisation can be prevented until the supersaturated solution of test item reaches the site of absorption of most NCEs, the small intestine.
To prevent dissolution before administration to the test species, for example, via an oral feeding canula for rats, SD-ASDs are usually suspended in an acidic vehicle (e.g., 0.01-N hydrochloric acid [HCl]). In some cases, surfactants like sodiumdodecylsulfate (SDS) or viscosity enhancers such as hydroxyethylcellulose (HEC) may be added to the vehicle to improve wettability of the SD-ASDs and homogeneity of the suspension.5,15
HPMC-AS has been examined with regard to acute, subchronic, and chronic toxicological effects in a battery of studies by Hoshi et al.16 -20 As HPMC-AS was found to be only poorly absorbed and toxicologically relevant effects were absent at doses up to 2500 mg/kg/day in rats, HPMC-AS was recognized as safe for use in nonclinical studies. For pharmaceutical use, for example, as a carrier for ASDs, it even gained the GRAS status provided by the United States Food and Drug Administration. 21
In this work, we report findings induced by an HPMC-AS–based SD-ASD formulation of the NCE drug candidate BI 1026706. The BI 1026706 molecule has Biopharmaceutical Classification System (BCS) class II properties with a water solubility of 0.04 mg/mL, an intrinsic dissolution rate of <50 µg/cm²/min in 0.1-N HCl or McIlvaine buffer pH 2.2 to 7.4, and an apical-to-basolateral permeation rate in Caco-2 cells of 5.7 × 10−6 cm/s. BI 1026706 had no toxicologically relevant effect on gastric emptying or intestinal transit. An initial exploratory toxicity study with a conventional crystalline suspension of BI 1026706 revealed insufficient drug plasma levels to enable a meaningful nonclinical safety testing program. Subsequently, this study was extended by a second part with an HPMC-AS–based SD-ASD that resulted in substantially higher maximum plasma levels and exposure (AUC0-24 hours). However, findings in subsequent toxicological studies revealed formation of agglomerates that, in reference to reported descriptions of indigestible concretions in the gastrointestinal tract of other species, were termed as “pharmacobezoars.”22 -26 Pharmacobezoars consisting of SD-ASDs were found in the stomach and small intestine of rats orally dosed in repeat-dose toxicity studies with a spray dried formulation containing 70% BI 1026706 and 30% HPMC-AS. Pharmacobezoars induced by an SD-ASD formulation in rats have, to our knowledge, not been reported before.
Materials and Methods
Animal Studies
Repeat-dose toxicity studies and embryo-fetal development (EFD) studies conducted with oral gavage administration of the SD-ASD formulation of BI 1026706 in rats are listed in Tables 1 and 2. The studies were conducted in accordance with animal welfare regulations of Boehringer Ingelheim and local authorities (Regierungspräsidium Tübingen, Baden-Württemberg, Germany). RccHan:WIST rats were obtained from Envigo RMS Limited (United Kingdom) for the 26-week oral toxicity study and from Charles River Laboratories, Research Models and Services Germany GmbH (Germany), for all other studies. Animals were aged 7 to 9 weeks at study start. Besides mated female rats housed individually in studies on embryo-fetal development, rats were housed in groups of 5 of the same sex per cage in a barrier system with target ranges of climate conditions according to standards established in the respective laboratory of study conduct. Bedding material (soft wood granules) was changed once per week, and wooden gnawing sticks as well as plastic shelters were provided for environmental enrichment. Pelleted food (Kliba No. 3808; Provimi Kliba SA, Kaiseraugst, Switzerland, or Teklad 2014 Diet; Teklad Diets, Madison, WI, USA, for the 26-week study) was available ad libitum.
Incidence of gastric PBs in rats from repeat-dose toxicity studies and studies on embryo-fetal development with BI 1026706 administered by oral gavage as SD-ASD suspension; sacrifice at end of dosing.
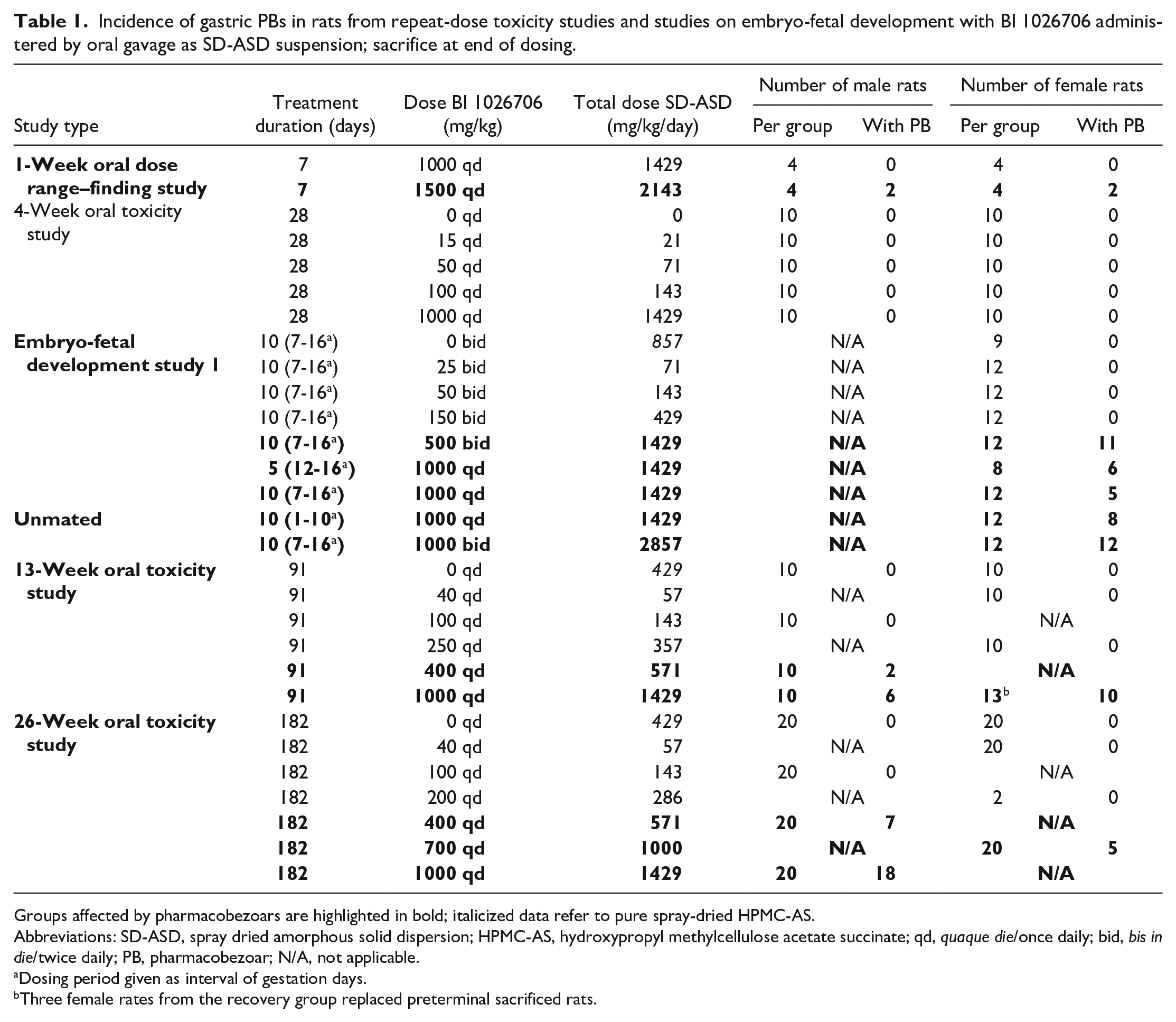
Groups affected by pharmacobezoars are highlighted in bold; italicized data refer to pure spray-dried HPMC-AS.
Abbreviations: SD-ASD, spray dried amorphous solid dispersion; HPMC-AS, hydroxypropyl methylcellulose acetate succinate; qd, quaque die/once daily; bid, bis in die/twice daily; PB, pharmacobezoar; N/A, not applicable.
Dosing period given as interval of gestation days.
Three female rates from the recovery group replaced preterminal sacrificed rats.
Incidence of gastric PBs at necropsy in rats from repeat-dose toxicity studies and studies on embryo-fetal development with BI 1026706 administered by oral gavage as an SD-ASD; sacrifice at end of dosing-free recovery period.
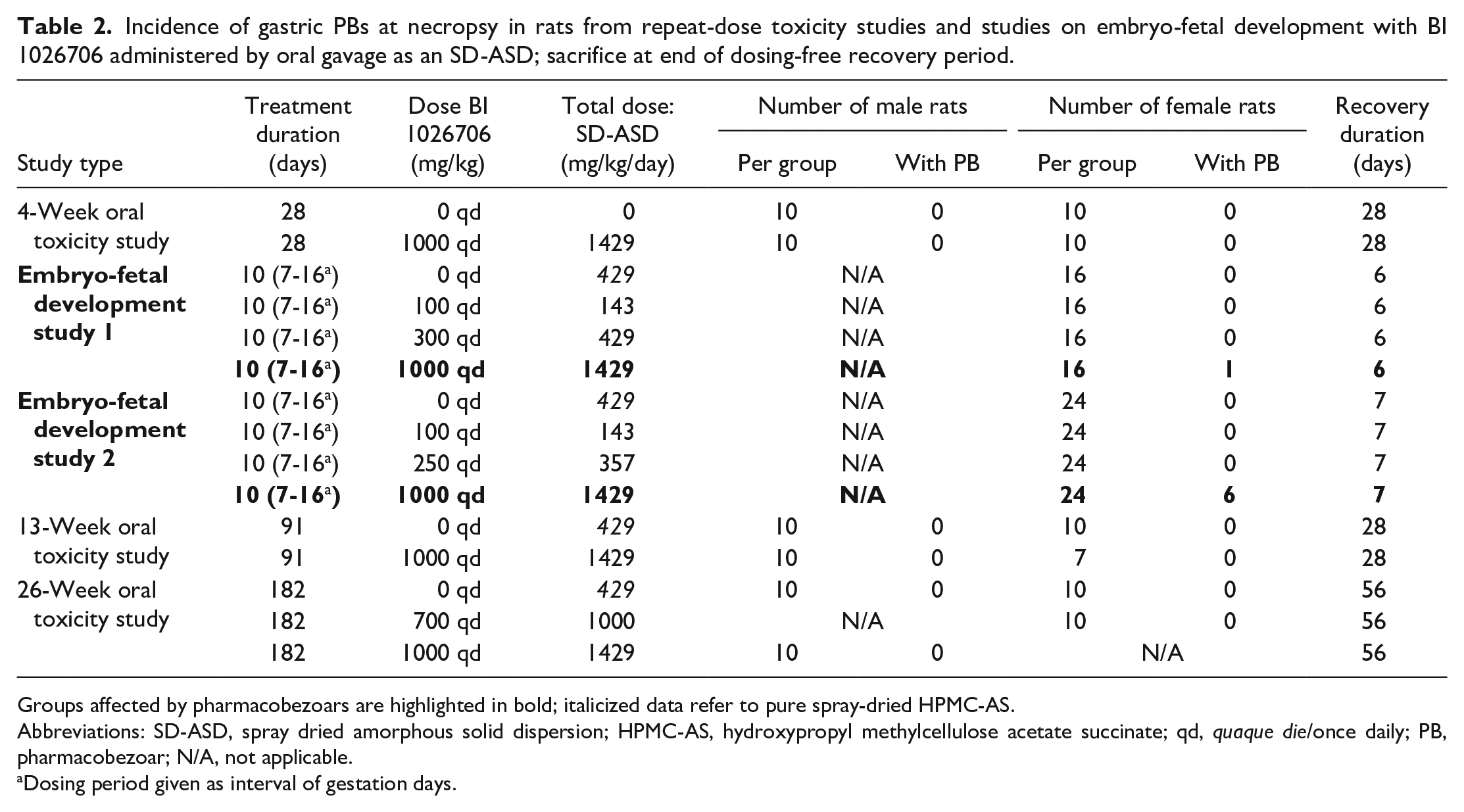
Groups affected by pharmacobezoars are highlighted in bold; italicized data refer to pure spray-dried HPMC-AS.
Abbreviations: SD-ASD, spray dried amorphous solid dispersion; HPMC-AS, hydroxypropyl methylcellulose acetate succinate; qd, quaque die/once daily; PB, pharmacobezoar; N/A, not applicable.
Dosing period given as interval of gestation days.
Preparation of Suspensions for In Vivo Studies and Dosing
Different batches of SD-ASDs incorporating nominally 70% of amorphous BI 1026706 into an HPMC-AS polymer matrix were prepared by spray drying. The batch-wise deviation from the target composition was less than 2%. The resultant conversion factor of the BI 1026706 dose to the dose of the SD-ASD formulation was approximately 1.43 (Tables 2 and 3) and adjusted for content deviations. Before administration, the appropriate amount of SD-ASD was weighed in a glass beaker. A paste was formed by the addition of a small amount of vehicle, and then the remaining amount of vehicle was added. The formulation was mixed with a magnetic stirrer until visual homogeneity. During the administration period, continuous stirring was maintained. Control groups of EFD studies as well as 13-week and 26-week oral toxicity studies received suspensions containing an amount of spray dried HPMC-AS equivalent to the share of the polymer of a high dose. Concentration and homogeneity of samples were tested by taking samples before, during, and after administration. The vehicle used to prepare suspensions consisted of 0.1% SDS and 0.5% HEC (Natrosol 250HX; Ashland, Wilmington, USA) in 0.01-N aqueous HCl. In the 1- and 4-week oral toxicity study, no HEC was added to the vehicle that, in both cases, has been prepared every 4 days by weighing the appropriate amount of additives into a glass beaker and bringing to volume with 0.01-N HCl. Rats were dosed via an oral feeding canula at a dose volume of 10 mL/kg body weight.
Incidence of findings and PBs in the stomach of rats from 26-week repeat-dose toxicity study with BI 1026706 administered by oral gavage as SD-ASD suspension; sacrifice at end of dosing.
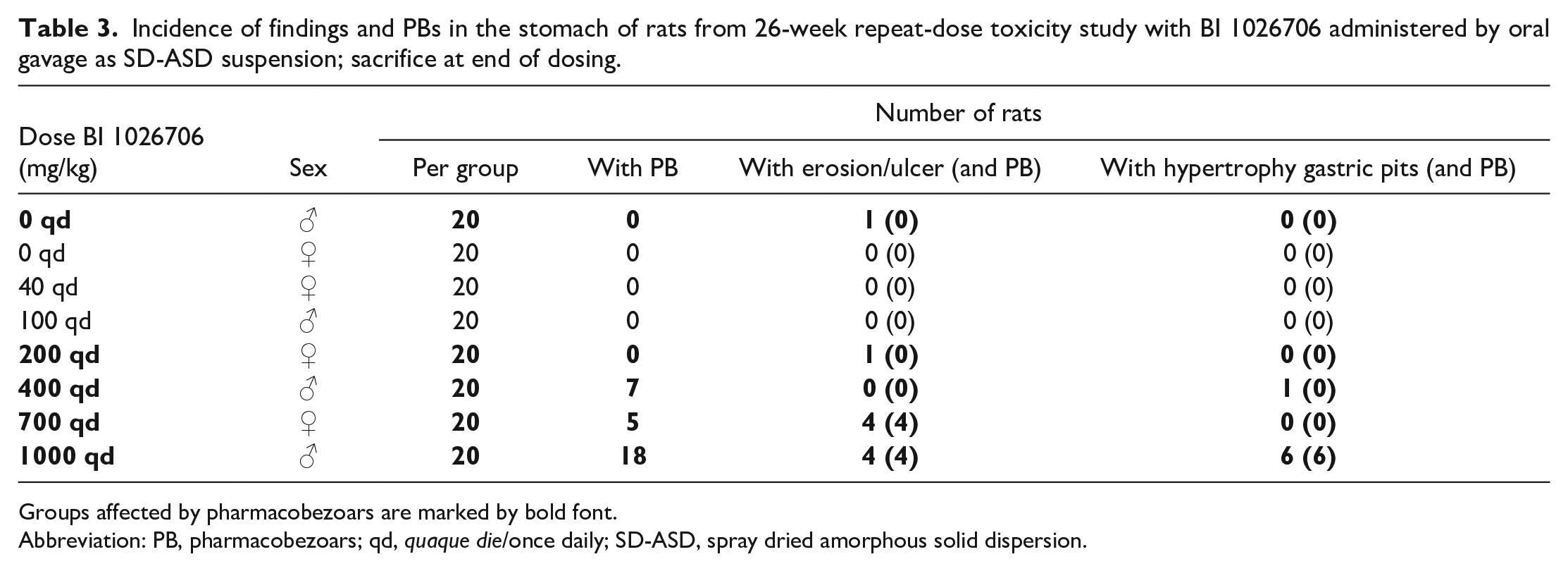
Groups affected by pharmacobezoars are marked by bold font.
Abbreviation: PB, pharmacobezoars; qd, quaque die/once daily; SD-ASD, spray dried amorphous solid dispersion.
Results
One-Week Oral Dose Range–Finding Study
At necropsy, pharmacobezoars were found in the stomach of 2 of 4 female and 2 of 4 male rats of the high-dose group (1500 mg/kg qd), whereas no pharmacobezoars were found in the 1000-mg/kg qd group (Table 2). Pharmacobezoars were easily identifiable by color, shape, and consistency: They were firm and irregularly spheroid with a longest diameter of 3 to about 15 mm. Larger pharmacobezoars sporadically entrapped few hair fragments or small food particles and usually contained an irregular cavity. As the BI 1026706 SD-ASD itself, pharmacobezoars were of light yellow color (Figure 1), and analytical investigations revealed a similar composition of pharmacobezoars and SD-ASD. Besides intragastric pharmacobezoars, intestinal pharmacobezoars were found in 3 female rats. During the in-life period, the affected animals appeared clinically normal and showed normal body weight development and food consumption. Urinanalysis and hematology was not performed. Clinical pathology on plasma were within normal limits. Based on the potential of bezoars to cause intestinal obstruction,27 -29 these findings limited the dose for the subsequent 4-week oral toxicity study to 1000 mg/kg qd. Remarkably, pharmacobezoars were not observed in the 1000-mg/kg qd group of the following 4-week oral toxicity study in rats but occurred again in studies on embryo-fetal development as well as in the subsequent 13-week and 26-week oral toxicity studies.

Round-shaped to irregular-shaped pharmacobezoars from rats repeatedly dosed with BI 1026706 spray-dried amorphous solid dispersion for 24 days in an investigational follow-up study (details not shown).
Embryo-Fetal Development Studies
EFD study 1 has been conducted as a dose range–finding study to assess the tolerability of BI 1026706 in pregnant female rats. In the first part, groups of 16 pregnant female rats were dosed 0, 100, 300, and 1000 mg/kg qd for 10 days, from gestation day 7 to 16, and necropsied on gestation day 22, which equalled a 6-day recovery period from dosing. Occurrence of pharmacobezoars was limited to one female rat in the high-dose group (Table 2).
A second part of the study was conducted to assess whether twice daily dosing may increase the BI 1026706 exposure. Six groups of 12 female rats each were dosed 25, 50, 150, 500, or 1000 mg/kg bid, respectively. In parallel, 12 female rats received 1000 mg/kg qd, and 9 female rats served as control group. Eight female rats were dosed for a shorter duration of 5 days after gestation with 1000 mg/kg qd, and a group of 12 unmated female rats received the same dose of SD-ASD for 10 days to allow evaluation of a possible pregnancy effect on systemic exposure.
The lowest dose per administration leading to pharmacobezoar formation in this study was 500 mg/kg bid with 11 of 12 female rats affected. Nominally the same dose per day for the same duration, but administered once per day, led to pharmacobezoars in 5 of 12 mated and 8 of 12 unmated female rats. A comparable incidence of pharmacobezoars was found in the 1000-mg/kg qd group dosed for 5 days as well (6 of 8 female rats affected). The 1000-mg/kg bid group had the highest incidence with pharmacobezoars found in all 12 female rats.
Based on the knowledge on pharmacokinetics gained from the EFD study 1, the main EFD study 2 has been conducted by once-daily oral gavage administration of 0, 100, 250, or 1000 mg/kg of BI 1026706 to groups of 24 mated female rats. Pharmacobezoars with diameters between 4 and 8 mm were found during necropsy in 6 of 24 mated female rats of the high-dose group when necropsied on day 22, that is, one week after the cessation of dosing. The pharmacobezoar morphology was similar to that found in the 1-week oral toxicity study. In both EFD studies, no relevant differences in body weight, food consumption, or litter parameters between control and test item-treated groups were observed, besides two late litter resorptions at 1000 mg/kg qd. Investigations on hematology, blood chemistry, and histopathology were not performed.
Thirteen-Week Oral Toxicity Study
Groups of 10 male rats each were dosed with either vehicle or the SD-ASD at dose levels of 100, 400, or 1000 mg/kg qd. Groups of 10 female rats each received either vehicle or dosages of 40, 250, or 1000 mg/kg qd. Additional 10 male and 10 female rats received either vehicle or 1000 mg/kg qd for 13 weeks and were maintained for a 4-week treatment-free recovery period before necropsy.
None of the 10 female rats receiving the 40-mg/kg qd dose nor the 10 male rats receiving the 100-mg/kg qd dose or the 10 female rats receiving the 250-mg/kg qd dose were affected by pharmacobezoars. With 2 of the 10 affected male rats at a dose of 400 mg/kg qd, pharmacobezoars were observed at the lowest dose in the sequence of studies so far. The incidence of pharmacobezoars in the 1000-mg/kg qd group without recovery was 6 out of 10 in male rats and 10 out of 13 in female rats. This included 3 female rats that were sacrificed unscheduled due to deteriorated clinical condition during the treatment period. These animals were replaced by 3 female rats from the 4-week recovery group, treated with the same dose for the same duration. Pharmacobezoars were not found in the remaining 7 recovery female rats.
A The three premature decedents were found with distended abdomen, hunched back, rough fur, and cold on touch. Their condition deteriorated rapidly, requiring unscheduled sacrifice on the day after onset of signs. Blood samples were taken on the day of necropsy for clinical pathology investigations. This revealed slightly high white blood cell counts, neutrophils, monocytes, and large unstained cells. Clinical chemistry on plasma revealed increases exceeding the upper limit of the 95% percentile of laboratory internal historical control data from age-, sex-, and strain-matched controls from the past two years (ULN). High glutamate dehydrogenase activity (≤8.53X ULN/18.43 units per liter), high glucose concentration (≤4.30X ULN/10.71 mmol/L), total cholesterol (≤1.74X ULN/2.00 mmol/L), creatinine (≤2.16X ULN/31.23 mmol/L), and urea (≤8.20X ULN/7.32 mmol/L) were observed. Low plasma concentrations have been measured for albumin (≥0.81X lower limit of normal (LLN)/4.17 g/L), sodium (≥0.86X LLN/138 mmol/L), and chloride (0.80X lower limit of normal (LLN)/98.01 mmol/L). High concentrations have been measured for magnesium (≤2.27X ULN/0.98 mmol/L), phosphate (≤1.64X ULN/2.34 mmol/L), and for calcium in one animal (1.13X ULN/2.73 mmol/L).
During necropsy of these 3 female rats, elongated pharmacobezoars were found that led to complete obliteration of their small intestines (Figure 2). The stomach was markedly distended by a brown liquid content. The esophagus and pharynx were distended by intraluminal food, considered as a consequence of regurgitation due to the intestinal obstruction.
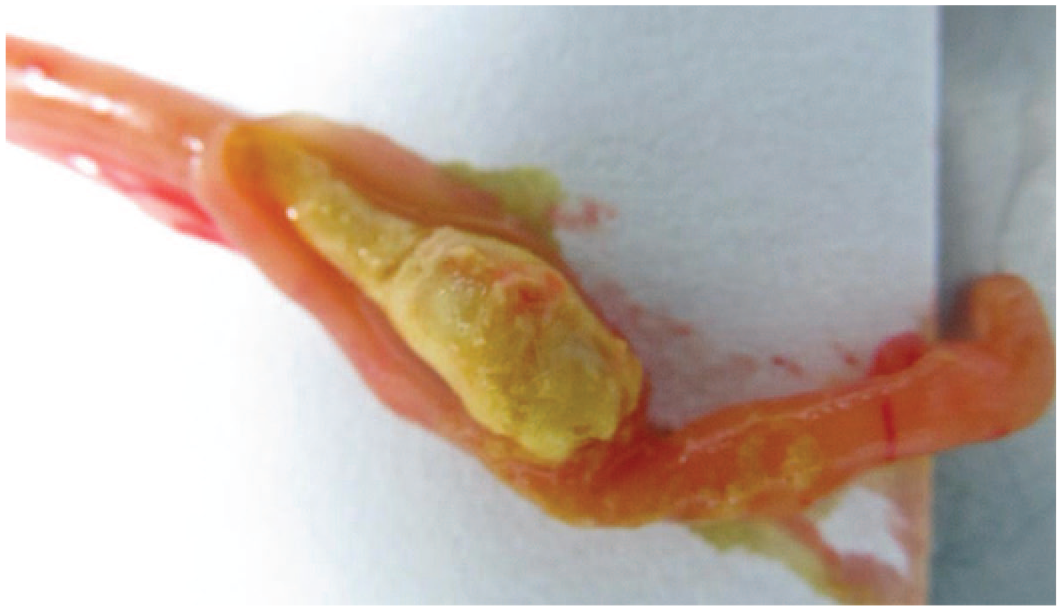
Pharmacobezoar in the jejunum of female rats repeatedly dosed with BI 1026706 spray-dried amorphous solid dispersion in a 13-week oral toxicity study until unscheduled sacrifice on day 60; at its largest diameter, the pharmacobezoar fully obliterated and distended the jejunum.
Histopathology of the preterminal decedents revealed findings in the gastrointestinal tract and the kidneys. The nonglandular stomach was affected by minimal to marked erosion and ulcer, minimal or mild hemorrhage, mild or moderate multifocal epithelial atrophy, moderate or marked submucosal edema, and minimal to moderate mural or submucosal mixed cell infiltration (Figures 3 and 4). The small intestine in the area and proximal to the obstructive pharmacobezoar was moderately to markedly dilated. The esophagus of one animal was in the area of luminal distension affected by severe acute transmural necrosis and inflammation.
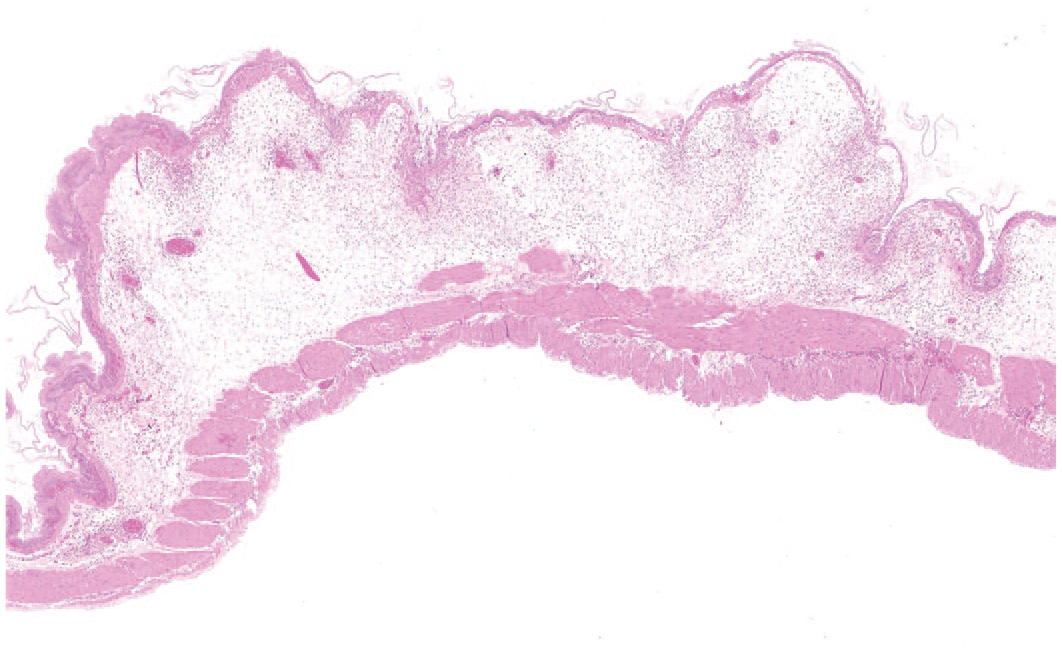
Nonglandular stomach of a female rat from the 1000-mg/kg qd dose group of the 13-week study sacrificed prematurely on day 63; prominent epithelial flattening, multiple ulcers, and submucosal edema with inflammatory cell infiltration; hematoxylin & eosin, original objective 2X.
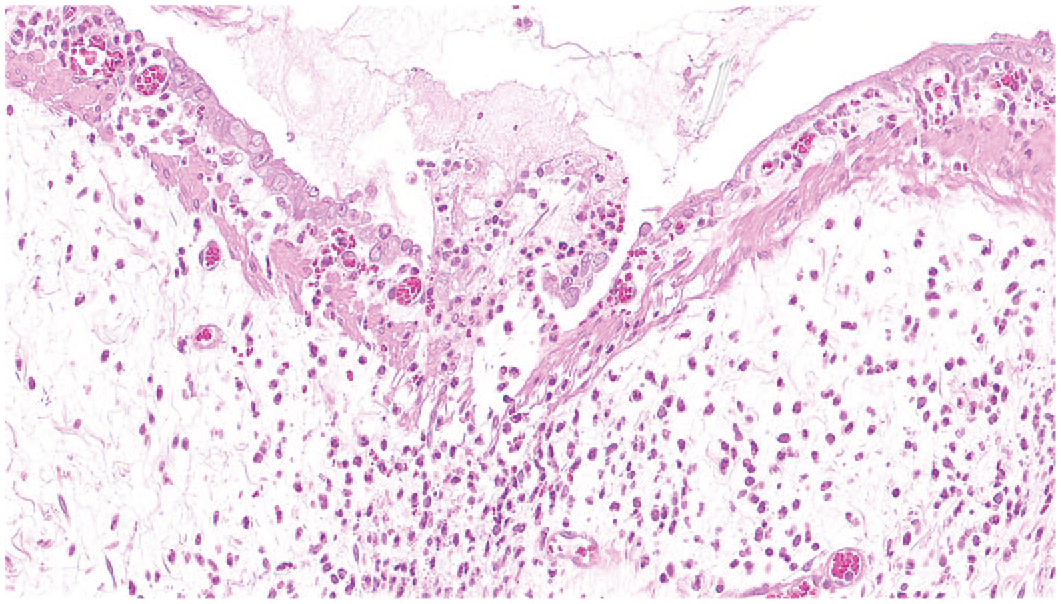
Detail of Figure 2 demonstrating a small ulcer with prominent mucosal and submucosal mixed cellular inflammation; note the markedly atrophic squamous epithelium to both sides of the ulcer. hematoxylin & eosin, original objective 31X.
Microscopically, the kidneys were affected in all three preterminal decedent female rats: Predominantly in medullary rays, intratubular gray material and tubular basophilia were present. These findings were interpreted as the consequence of dehydration, leading to precipitation of the test item or metabolite(s) and subsequent local tubular irritation. Other findings in the preterminal decedents were interpreted as secondary to the deteriorated general condition and included adrenal cortical hypertrophy, depletion of lymphocytes in thymus and spleen, as well as reduced zymogen granules in pancreatic acini.
Animals with pharmacobezoars sacrificed at the end of the dosing period showed no clinical abnormalities. Clinical pathology and anatomic pathology findings differed between animals, with pharmacobezoars exclusively in the stomach and one female that had an additional pharmacobezoar in the jejunum. The latter animal showed clinical pathology and histopathology findings similar to those described for the preterminal decedent female rats including ulcers of the gastric nonglandular mucosa and histopathological findings of the kidneys that were similar to those described for the unscheduled sacrifices. In all other animals, clinical pathology parameters were within normal limits, and histopathology revealed no findings that could be attributed to the presence of pharmacobezoars. The pharmacobezoar morphology was again similar to that observed in the 1-week study.
Twenty-Six-Week Oral Toxicity Study
Groups of 20 male rats each were dosed once daily at 0, 100, 400, or 1000 mg/kg qd, and groups of 20 female rats each were dosed at 0, 40, 200, or 700 mg/kg qd. Additional 10 male and 10 female rats received doses equal to those of control groups or the high-dose group, followed by an 8-week treatment-free recovery period after the cessation of dosing. The high dose was limited in female rats to 700 mg/kg qd because of the unscheduled sacrifices of 3 female rats in the 13-week study. Even though this dose led to formation of pharmacobezoars in 5 of 20 female rats without recovery, no deterioration of well-being was observed. Besides the 700-mg/kg qd dose group, 7 of 20 male rats of the 400-mg/kg qd dose group and 18 of 20 male rats of the 1000-mg/kg qd dose group were affected. Pharmacobezoars were again absent in the 10 male rats that were necropsied after receiving 1000 mg/kg qd for 26 weeks followed by an 8-week recovery period as well as in the 10 female rats that received 700 mg/kg qd for the same study and recovery duration. No pharmacobezoars were found at necropsy in rats that 0, 40, 100, or 200 mg/kg qd, nor did they cause unscheduled sacrifices in the high-dose groups. In dose groups affected by pharmacobezoar formation, ulcer of the stomach mucosa and hypertrophy of gastric pits were, with the exception of one male rat of the 400-mg/kg qd group, restricted to animals with pharmacobezoars, especially at increased incidence and severity in male rats given a dose of 1000 mg/kg qd (Table 3).
Overall, body weight development was stable in all groups of the BI 1026706 nonclinical testing program, and individual data did not show a decrease of body weight in animals with pharmacobezoars compared with unaffected animals. In all EFD studies and repeat-dose toxicity studies, all treatment-related findings, besides local irritant effects in the stomach, were observed with similar incidence and grades in animals with and without pharmacobezoars. Predominantly increased extramedullary haematopoiesis and follicular cell hypertrophy in the thyroid glands were classified as minor or nonadverse and found to be reversible during recovery.
Discussion
We describe, to the best of our knowledge, for the first time the observation of pharmacobezoar formation in nonclinical toxicity studies. The data presented reveal that the formulation type, daily dose, and the duration of dosing are factors influencing the formation of gastric pharmacobezoars of an SD-ASD formulation containing 70% BI 1026706 and 30% HPMC-AS. Comparing incidences of groups treated with the same dose per day, bid dosing seems to favor pharmacobezoar formation. Daily treatment of rats with the crystalline form of BI 1026706 at dose levels up to 2000 mg/kg for 14 days induced no pharmacobezoars, while pharmacobezoars were found after daily dosing of the SD-ASD formulation at a BI 1026706 dose level of 1500 mg/kg/day for 7 days. When analyzed chemically, pharmacobezoars consisted of BI 1026706 and HPMC-AS in a comparable ratio as in the SD-ASD formulation. Remarkably, pharmacobezoars did only occur sporadically in the course of other nonclinical safety studies with SD-ASDs containing formulations of follow-up compounds at similar dose levels and not in all studies using SD-ASDs containing HPMC-AS as a carrier polymer (data not shown). While the reasons for the particularly enhanced pharmacobezoar formation potential of this HPMC-AS–based SD-ASD–containing BI 1026706 are not understood so far, the pH-dependent insolubility of the carrier polymer below pH 5.511 is believed to be a prerequisite for pharmacobezoar formation in the acidic environment of the rodent stomach. However, hydroxypropylmethylcellulose acetate succinate (HPMC-AS) is due to its physicochemical properties combined with its toxicological safety, commonly the preferred and established choice for formulation of SD-ASDs for preclinical testing. Principally, a change to a polymer with pH-independent good solubility would be a more promising approach to prevent pharmacobezoar formation than changing the polymer to another pH-dependent polymer. Practically, both these options are unfavorable. Other pH-dependent soluble polymers are often less effective regarding the level of supersaturation or inhibition of recrystallization, 30 and SD-ASDs containing respective carrier polymers would not dissolve in the stomach as well. Application of pH-independent well-soluble polymers would make the preparation of suspensions for oral administration generally impossible as suspended particles would immediately dissolve in the vehicle. Resulting supersaturated solutions for oral administration would be associated with negative effects such as the risk of recrystallization of an indefinite amount of NCE prior to reaching the site of absorption. The propensity of the SD-ASD formulation of BI 1026706 formation to form pharmacobezoars increased with dose and duration of treatment. The only outlier was the 4-week oral toxicity study, where even the highest daily dose of 1429 mg/kg did not result in pharmacobezoar formation.
The nonclinical testing program of BI 1026706 comprised mice, rabbits, and cynomolgus monkeys in addition to rats. All studies were conducted with the same SD-ASD formulation containing HPMC-AS. Apart from rats, only mice, but none of the other species, was affected by formation of pharmacobezoars. This species specificity of pharmacobezoar formation might be due to smaller ratios of SD-ASD dose to stomach size in other species along with anatomical specifics of the rodents’ stomach that may facilitate pharmacobezoar formation. Separated by the limiting ridge from the muscle-walled glandular part, the thin-walled nonglandular part mainly serves as room for food storage and does not show relevant motility. Instead, a constant tonus pushes the chymus forward over the limiting ridge into the glandular stomach. 31 Even though the pharmacobezoar formation process is not understood so far, such a storage room unique to rodents might be the compartment where pharmacobezoars can grow in size by every single dose without being broken down by gastric motility.
The lowest dose leading to pharmacobezoar formation was 400 mg/kg qd administered for 13 or 26 weeks. In studies where food and water consumption were measured, no alteration was observed as long as occurrence of pharmacobezoars was restricted to the stomach. Pathological findings were restricted to local irritant effects on gastric mucosa, mainly of the nonglandular mucosa (Table 4). The development of these local irritant effects was time-dependent and observed first in animals of the 13-week study. Effects may have been induced physically due to the solid pharmacobezoar in permanent direct contact to the gastric mucosa, due to a permanent local irritant effect of the test item the pharmacobezoar consists of, or by both processes.
Incidence of clinical signs, clinical pathology, and anatomic pathology findings in rats with gastric or intestinal PB when compared to animals without pharmacobezoar in both study groups where intestinal PBs occurred.
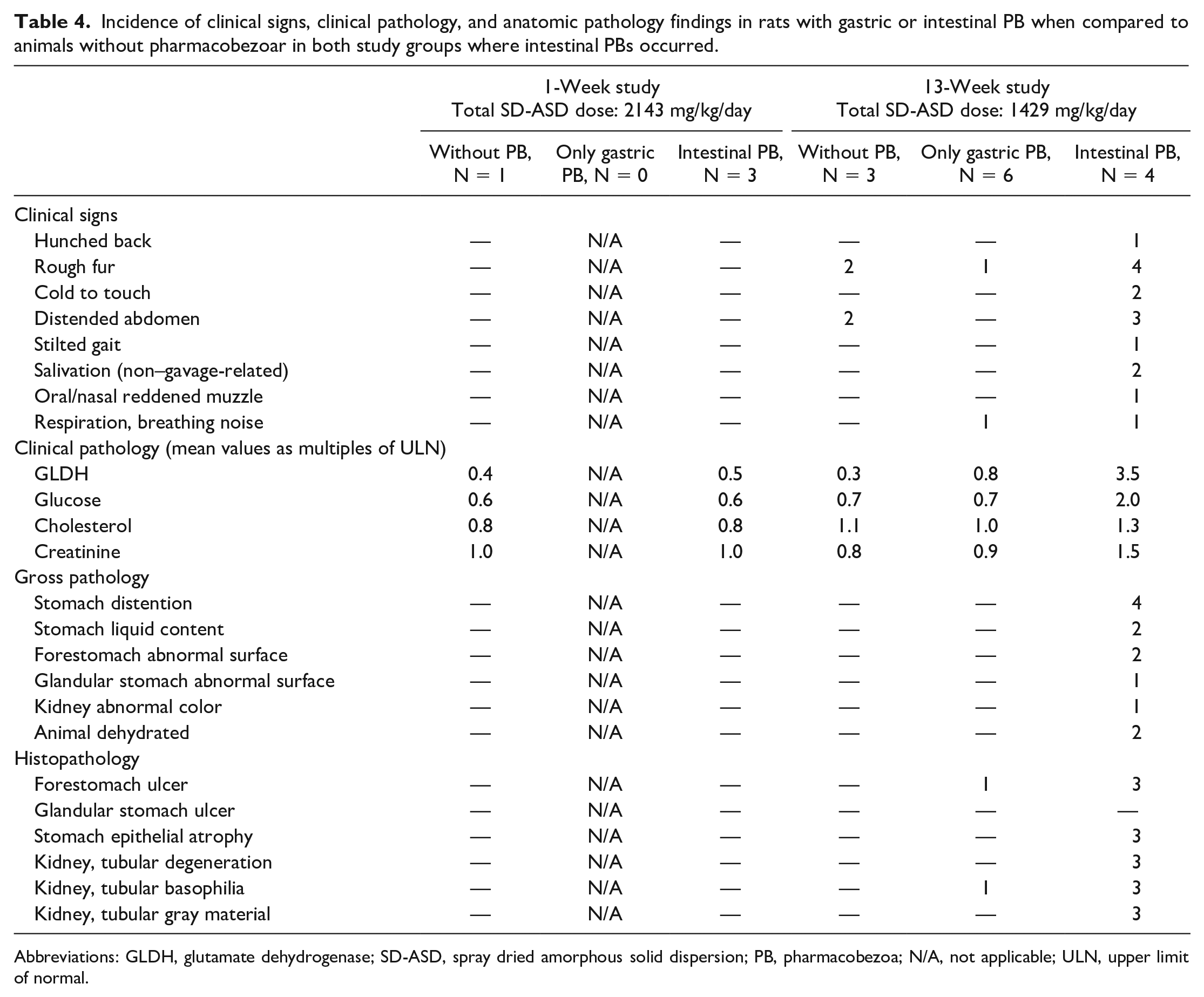
Abbreviations: GLDH, glutamate dehydrogenase; SD-ASD, spray dried amorphous solid dispersion; PB, pharmacobezoa; N/A, not applicable; ULN, upper limit of normal.
Within a dose group, there was generally a higher incidence of pharmacobezoars in female rats than in male rats. The reasons for this sex difference cannot be derived from the presented data but might be versatile and influenced by differences in the weight-dependent total SD-ASD dose, food consumption, or organ size. 32 In line with the higher incidence of gastric pharmacobezoars, observations of intestinal pharmacobezoars were restricted to female rats as well (Table 4). Besides in one animal in the 1-week study, intestinal pharmacobezoars were accompanied by gastric pharmacobezoars. Pharmacobezoars found in the jejunum are, due to the intraluminal pH enabling dissolution of the carrier-polymer in this location, expected to have passed the pylorus despite their size bigger than physiological maximal emptiable particle size 33 instead of having been formed in the small intestines. While small SD-ASD particles will dissolve in this location to supersaturated solutions as intended, these pharmacobezoars were obviously too big to dissolve before inducing intestinal obstruction and resulted in rapid deterioration of general condition as evidenced by rough fur, decreased body temperature, distended abdomen, and non–gavage-related salivation as signs of abdominal inconvenience in the 13-week study. Clinical pathology parameters of female rats with intestinal pharmacobezoars in the 13-week oral toxicity study, receiving 1000 mg/kg qd until preterminal sacrifice on day 51, 60, and 63, confirmed observations of physical impairment. Data indicated that pharmacobezoar-induced intestinal obstruction led to sequestration of fluid and electrolytes with subsequent hypotonic dehydration, as evidenced by low plasma sodium and chloride concentrations. The slightly low plasma albumin concentration is considered secondary to a likely marked reduction of food intake. The increase of plasma activity of glutamate dehydrogenase indicates hepatocellular alteration, which may be mediated by the entry of endotoxins and microbiota via the markedly altered mucosa of the nonglandular stomach. Furthermore, endotoxins activate leukocytes and increase gluconeogenesis 34 as reflected by the clinical pathology findings from preterminally sacrificed female rats. Increased creatinine and urea levels were considered indicative of a loss of renal function, 35 in line with the degenerative findings in renal tubules present in these animals. It is unclear whether the slightly high concentrations of magnesium, calcium, and phosphate were the consequence of increased intestinal absorption or impaired renal secretion.
The potential life-threatening consequences of pharmacobezoars in female rats emphasize the need of insights into the pharmacobezoar formation potential of SD-ASDs prior to in vivo administration in order to prevent corresponding impairments in the future.
The incidence of pharmacobezoars was reduced in recovery groups when compared with groups of animals treated at the same dose level and sacrificed on the day after the cessation of dosing. This suggests that pharmacobezoars, as long as they are retained in the stomach, may be slowly degraded after the cessation of dosing.
Conclusion
The use of SD-ASD formulations in toxicity studies is a way to substantially increase bioavailability of poorly soluble test items without the use of vehicles with limited tolerability like Tween 80 or HP-beta-cyclodextrin. However, pharmacobezoars were found during necropsy of rats administered HPMC-AS–based SD-ASDs in nonclinical toxicological studies. The incidences were increased by high doses and long duration of the studies. Pharmacobezoars found in the stomach during necropsy have not induced physical impairments or reduction of food and water consumption. Contrary, pharmacobezoars expelled into the small intestine were observed to result in potentially fatal obstructive ileus. Thus, the rodent-specific formation of pharmacobezoars from SD-ASDs is of relevance regarding animal welfare. Additionally, they may limit the maximum dose of an HPMC-AS–based SD-ASD formulation of an NCE, thereby giving adequate characterization of its toxicity profile. To further benefit from the properties of HPMC-AS as a carrier polymer for SD-ASDs in nonclinical studies, we take the occurrence of pharmacobezoars as a starting point for further investigations on pharmacobezoar formation in order to maximize the usability of SD-ASD formulations in nonclinical safety testing.
Footnotes
Acknowledgements
The authors acknowledge the excellent conduct and evaluation of the toxicity studies by the staff of the veterinary care, in vivo toxicology, reproductive toxicology, and pathology groups of Boehringer Ingelheim, Biberach, in particular to the study director and study pathologist of the 13-week study in rats, Dr Julia Schlichtiger and Dr Gabriele Pohlmeyer-Esch. The study director of the reproductive toxicology studies was Dr Bernd Baier. The authors are equally thankful for Labcorp staff for conducting and evaluating of the 26-week study under the auspices of Susan Cooper (†) as the study director and Vasanthi Mowat as the study pathologist.
Author Contribution
All authors contributed to the design and evaluation of the investigations described here. The first draft of the manuscript was written by Hannes Gierke. All co-authors reviewed the manuscript and agreed to its final version.
Declaration of Conflicting Interests
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Teresa Pfrommer, Kerstin Schäfer and Thomas Nolte are employed by Boehringer Ingelheim Pharma GmbH & Co. KG, the organization that funded the conduct of the toxicity studies with BI 1026706.
Funding
The toxicity studies conducted with BI 1026706 were funded by Boehringer Ingelheim Pharma GmbH & Co. KG.