
Abstract
The Port Delivery System with ranibizumab (PDS) is an investigational drug delivery system designed to provide continuous intravitreal release of ranibizumab for extended durations. The PDS consists of a permanent, surgically placed, refillable intraocular implant; a customized formulation of ranibizumab; and ancillary devices to support surgery and refill procedures. A toxicology program was conducted to evaluate the ocular toxicology and biocompatibility of the PDS to support its clinical development program and product registrational activities. PDS safety studies included a 6-month chronic toxicology evaluation in minipigs as well as evaluation of nonfunctional surrogate implants (comprised of the same implant materials but without ranibizumab) in rabbits. Biocompatibility of the implant and ancillary devices was evaluated in both in vitro and in vivo studies. Implants and extracts from implants and ancillary devices were nongenotoxic, noncytotoxic, nonsensitizing, and nonirritating. Ocular findings were comparable between implanted and sham-operated eyes, and no systemic toxicity was observed. The results of this nonclinical toxicology program demonstrated that the PDS was biocompatible and that intravitreal delivery of ranibizumab via the PDS did not introduce any new toxicology-related safety concerns relative to intravitreal injections, supporting ongoing PDS clinical development and product registrational evaluation.
Keywords
Introduction
Neovascular age-related macular degeneration (nAMD) is a chronic, progressive retinal disease, and a leading cause of vision loss in developed countries. 1,2 Treatment of nAMD requires long-term, repeated, and frequent delivery of effective therapy to the back of the eye. Intravitreal anti–vascular endothelial growth factor (VEGF) therapy is the standard of care for treating nAMD. 3 –6 Direct, local intravitreal delivery of anti-VEGF agents, such as ranibizumab (Lucentis; Genentech, Inc), into the vitreous humor maximizes retinal exposure and minimizes systemic exposure. However, intravitreal injections are required as often as once per month to achieve optimal vision outcomes, and this creates a considerable burden for patients, their caregivers, and health care providers. 7 –10 In real-world scenarios, there is significant undertreatment, and visual outcomes for patients with nAMD fall short of those attained in clinical trials. 11 There is a significant unmet need for continuous drug delivery to the back of the eye to alleviate the frequent intravitreal injections and monitoring associated with treatment, as well as to improve patient outcomes.
The Port Delivery System with ranibizumab (PDS) is an investigational intraocular drug delivery system that aims to address treatment burden and the effects of undertreatment in nAMD. The PDS consists of a surgically placed, permanent, refillable intraocular implant uniquely designed for continuous delivery of a customized formulation of ranibizumab into the vitreous (Figure 1). 12 The PDS also includes 4 ancillary devices that facilitate surgical implantation into the pars plana, initial fill with ranibizumab, in-office refill-exchange procedures, and explantation if removal is deemed clinically necessary (Figure 1). 12 In the phase 2 Ladder trial (NCT02510794) in patients with nAMD, the PDS was generally well tolerated. In the primary analysis population (N = 220), the median time to first implant refill was 8.7, 13.0, and 15.0 months in the PDS 10, 40, and 100 mg/mL arms, respectively. Furthermore, in the PDS 100 mg/mL arm, nearly 80% of patients went ≥6 months without requiring an implant refill, and vision and anatomic outcomes were comparable to those of monthly intravitreal ranibizumab 0.5 mg treatment. 12 The Archway phase 3 trial (NCT03677934) is ongoing and recently met its primary endpoint showing noninferior and equivalent visual outcomes compared monthly ranibizumab injections. The data also demonstrated that over 98% of PDS patients were able to go 6 months without needing additional treatment. 13 The Portal extension study (NCT03683251) is recruiting for patients with nAMD.
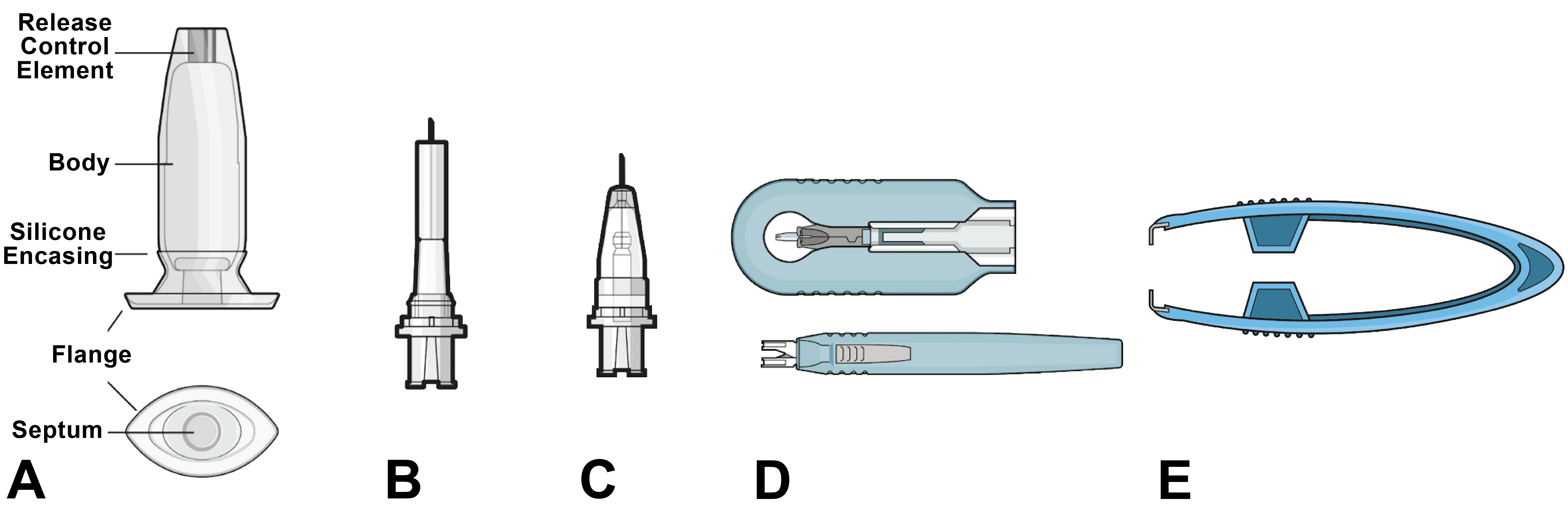
Illustrations depicting the devices in the Port Delivery System with ranibizumab: (A) implant, (B) initial fill needle, (C) refill needle, (D) insertion tool assembly with implant and handle, and (E) explant tool. The images and their relative proportions are not to scale. Genentech, Inc, used by permission.
To support clinical development of the PDS and registration in patients with nAMD, a comprehensive nonclinical toxicology program was designed to characterize the safety profile of the PDS and 4 ancillary devices (initial fill needle, insertion tool, refill needle, and explant tool). Ranibizumab, administered by intravitreal injection, is approved for the treatment of nAMD. 14 Here, we describe the comprehensive nonclinical toxicology and biocompatibility testing battery conducted to support the clinical development and registration of the PDS in patients with nAMD.
Materials and Methods
Ocular implant toxicology and biocompatibility studies were conducted in compliance with US Food and Drug Administration (FDA) regulations for Good Laboratory Practice (GLP) for Nonclinical Laboratory Studies. For in vivo studies, animal care and procedures complied with the Animal Welfare Act, the Guide for the Care of Laboratory Animals, and the Office of Laboratory Welfare regulations, as well as relevant local, state, and federal laws. All in vivo animal study protocols were approved by the local animal care committee (Covance or NAMSA contract research organizations) in accordance with the Association for Research in Vision and Ophthalmology’s Statement for the Use of Animals in Ophthalmology and Vision Research.
Toxicology and Biocompatibility Testing Strategy
Because there is no specific regulatory guidance for ocular toxicology and biocompatibility battery of testing for a permanent and refillable drug/device combination (ie, the implant and ancillary devices), a comprehensive testing strategy was based on a combination of International Conference for Harmonisation guidelines, International Organization for Standardization (ISO; https://www.iso.org/standards.html) medical device testing standards ISO 10993 (biological evaluation of medical devices) and ISO 11979 (ophthalmic implants–intraocular lenses), and American National Standards Institution (ANSI) Z80.7 standards (Supplemental Table S1). The ISO guidance for intraocular lenses was modified for evaluation for a drug-eluting device and comprised the ISO 10993 biocompatibility evaluations (Supplemental Table S1). The Center for Devices and Radiological Health device classifications that were considered in development of the testing strategy for the PDS devices were class 2 for the needles and explant tool and class 3 for the implant and insertion tool assembly.
6-Month GLP Ocular Implant Study in Rabbits
The clinical route of the PDS is intraocular implantation. Thus, all ocular toxicology studies were conducted using ocular implantation. New Zealand White rabbits were selected as an appropriate species to assess long-term implant tolerability based on ISO 10993 standards in a 6-month study (to enable phase 2). To match the smaller size of the rabbit eye, scaled one-third–size surrogate implants were made from the same materials as the clinical implant. The scaling process rendered the implants used in the rabbit study nonfunctional.
Surgical implantation of the device into the pars plana was conducted in anesthetized animals using a 1.8-mm angled Xstar Slit Knife (Beaver-Visitec, Inc). Before surgery, animals were anesthetized with ketamine (20-50 mg/kg), dexmedetomidine (0.0315 mg/kg), and glycopyrrolate (0.1 mg/kg). For all eyes (untreated, implanted, and sham surgery), a topical anesthetic (0.5% proparacaine) was instilled before surgery. The eyes and periorbital region were cleaned with a dilute ∼1% povidone–iodine solution. A suture was placed to close the conjunctival flap, taking care not to place the suture near the insertion site. A sham implantation procedure included the creation of a conjunctival flap to expose the sclera, and a scleral incision as above. The incision and conjunctival flap were subsequently closed with sutures. Upon recovery from anesthesia, tramadol was administered via oral gavage at a dose level of 4 mg/kg, and buprenorphine sustained release was administered at a dose level of 0.2 mg/kg via subcutaneous injection. At least 6 hours after the initial administration, a second dose of tramadol at the same dose level was administered. Females also received bland ophthalmic ointment in the eyes ≥6 hours after the initial administration of tramadol and buprenorphine.
Male and female rabbits (N = 11) received surrogate implants in the right eye, and a sham surgical procedure was performed on the left eye on day 1. A subset of male and female animals was necropsied at the 3-month treatment period (interim necropsy, 3 animals/sex/group), and the remaining animals were observed until terminal necropsy at 6 months (8 animals/sex/group). Assessment of toxicity was based on mortality, clinical observations, body weights, ophthalmic examination (OE; slit-lamp biomicroscopy and indirect ophthalmology), intraocular pressure (IOP) measurements, full-field electroretinography, and clinical and anatomic pathology.
In Vitro and In Vivo Biocompatibility Evaluation of Implant and Ancillary Devices
In vitro and in vivo biocompatibility studies were chosen based on direct or indirect interaction of the implant or 4 ancillary devices (insertion tool, initial fill needle, refill needle, and explant tool) with ocular tissues. Biocompatibility testing was performed consistent with ISO 10993-3, ISO 10993-5, ISO 10993-6, ISO 10993-10, ISO 11979-5, and ANSI Z80.7 standards using the finished implant (full sized and surrogate, sterilized, and packaged), and the 4 ancillary devices (Table 1). Long-term testing for direct and indirect interactions was performed on the implant itself and on extracts from the implant, and for short-term direct and indirect interactions on extracts from the ancillary device tools. Briefly, methodologies for the implant and 4 ancillary devices are described below.
Biocompatibility Studies and Results.
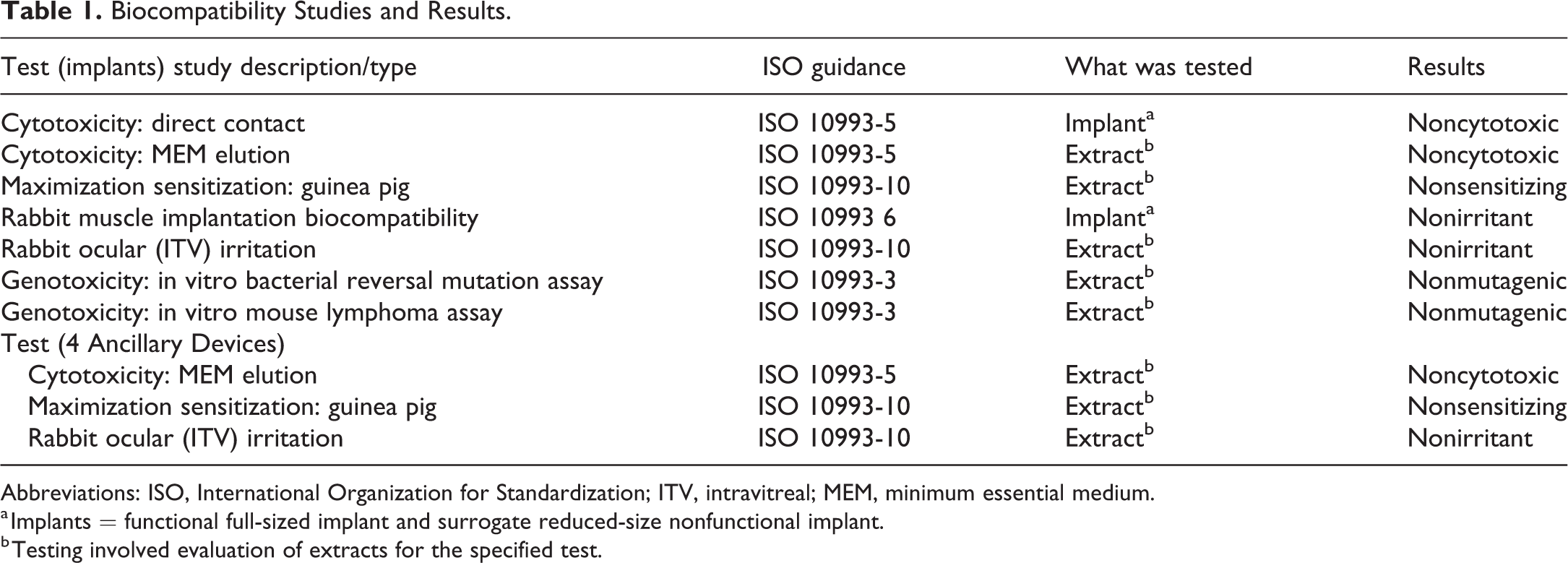
Abbreviations: ISO, International Organization for Standardization; ITV, intravitreal; MEM, minimum essential medium.
a Implants = functional full-sized implant and surrogate reduced-size nonfunctional implant.
b Testing involved evaluation of extracts for the specified test.
Implant
Direct cytotoxicity of the implant was evaluated in L-929 mouse fibroblast cells, with direct placement of 3 implants over a subconfluent cell monolayer for 24 hours. Indirect cytotoxicity was evaluated by extraction of 46 implants into minimum essential medium supplemented with serum to optimize extraction of both polar and nonpolar components. The implant extract was placed over a subconfluent L-929 mouse fibroblast cell monolayer for 48 hours.
The dermal sensitization potential of implant extracts was evaluated in male guinea pigs in vivo. Forty-six implants were used to generate extracts in sesame oil and saline (0.9% sodium chloride), which were each tested in 10 animals.
Local tissue responses to the implant were evaluated in muscle tissue of male New Zealand White rabbits over a 12-week period. Four implants were surgically implanted under anesthesia into the paravertebral muscle of each of 3 rabbits on day 1. Animals were observed and then necropsied at 12 weeks. Assessment of tolerability was based on evaluation of mortality, clinical observations, body weights, and anatomic pathology of implant sites. Briefly, for histopathology studies, the eye was trimmed in a sagittal plane of section (superio–inferior), with a cut on either side of the optic disc to divide the eye into a central segment (containing the optic disc), temporal, and nasal calottes. The temporal calotte was further subdivided along a horizontal plane (temporonasal) to produce a superior segment (containing the implant) and an inferior segment. The central segment, nasal calotte, and inferior segment of the temporal calotte were paraffin embedded as standard. The temporal superior segment (containing the implant) was processed and embedded in EXAKT system technovit plastic enabling sectioning of the implant in situ.
Ocular irritation following a single intravitreal injection of implant extract was evaluated in the eyes of male New Zealand White rabbits (n = 6). Forty-six implants were used to prepare a balanced salt solution extract that was administered to the right eye as a single 0.2-mL intravitreal injection on day 1. The left eye of each animal served as a control and was administered a single 0.2-mL intravitreal injection of balanced salt solution on day 1. Animals were observed until necropsy on day 3. Assessment of toxicity was based on mortality, clinical observations, body weight, OE (slit-lamp biomicroscopy and indirect ophthalmoscopy), and vitreous white blood cell count.
The mutagenicity of the implants was assayed in multiple strains of Salmonella typhimurium (TA98, TA100, TA1535, and TA1537) and Escherichia coli (WP2uvrA) in the presence and absence of an exogenous mammalian metabolic activation system (S9), in accordance with ISO 10993-3. Forty-six implants were used to prepare 95% ethanol and saline extracts, respectively. Mutagenic potential was evaluated using the mouse lymphoma forward gene mutation assay in accordance with ISO 10993-3. Forty-six implants were used to prepare 95% ethanol extracts and 365 implants were used to prepare serum-free cell culture medium (RPMI0) extracts. The mouse lymphoma L5178Y/TK+/− cell line, heterozygous at the thymidine kinase (TK) locus, was treated with these extracts for 4 hours in the presence and absence of metabolic activation (S9) and 24 hours in the absence of metabolic activation.
4 Ancillary Devices
Extracts of ancillary devices also underwent 3 biocompatibility tests, indirect cytotoxicity, in vivo sensitization, and in vivo ocular irritation testing as described above for relevant implant biocompatibility tests.
6-month GLP chronic toxicology/TK evaluation of the PDS with ranibizumab in minipigs to support phase 3 and registration
The Yucatan minipig was selected in a chronic toxicity/TK study (to enable phase 3 and registration) as the appropriate species to assess the PDS because the eye of a minipig (compared with nonhuman primates) is large enough to assess a functional, full-scale clinical implant. 15 The implants were packaged and sterilized by ethylene oxide, the same method used for the clinical implant. Female Yucatan minipigs (Sinclair Research Center) were used in this study (Table 2). Animals were fasted for ≥2 hours before sedation and flunixin meglumine was administered at a dose of 2 mg/kg by intramuscular injection. They were subsequently anesthetized with a combination of midazolam, dexmedetomidine, glycopyrrolate, and butorphanol. A topical anesthetic (0.5% proparacaine) was instilled in each eye before surgery. The eyes and periorbital region were cleaned with a 1% povidone–iodine solution. A conjunctival incision was made in the superotemporal quadrant of the eye. Using a 3.2-mm slit knife (Beaver-Visitec Inc), a full-thickness scleral incision (parallel to the limbus in the supratemporal quadrant) was created at the pars plana, ∼4 mm posterior to the limbus. The implant was inserted into the vitreous cavity through the scleral incision so the proximal end of the device fit tightly within the scleral incision and sat flush with the sclera. One horizontal mattress suture consisting of 9-0 nylon was passed through the sclera on either side of the implant to reduce the risk of implant extrusion because it was not optimized for porcine eyes. Then, the conjunctiva was sutured at the limbus using a simple continuous pattern with 7-0 Vicryl. The limbal traction suture was removed. A reversal agent (atipamezole) was administered following the procedure. Upon recovery from anesthesia, buprenorphine-sustained release was administered at a dose of 0.07 mg/kg by subcutaneous injection to provide analgesia. An antibiotic (100 µL of 400 mg/mL cefazolin) was administered via subconjunctival injection in each eye following completion of the implantation procedure. Topical triple antibiotic and dexamethasone ophthalmic ointment (Neo-Poly-Dex, neomycin–polymyxin B-dexamethasone; S.A. Alcon-Couvreur N.V.) were administered to both eyes upon recovery from anesthesia and once daily through study day 3.
Study Design for 6-Month Chronic Toxicology/TK Evaluation of the PDS in Female Yucatan Minipigs.

Abbreviations: NA, not applicable; PDS, Port Delivery System with ranibizumab; TK, thymidine kinase.
Each animal (n = 5) received a vehicle-filled implant in their right eye, which was refilled with vehicle monthly for 7 total doses (initial implant fill at the time of surgery plus 6 refills); a drug-filled implant (2 mg dose) that was not refilled during the study (n = 3); or a drug-filled implant (2 mg dose), which was refilled (2 mg/dose) monthly for 7 total doses (n = 7; Table 2). All animals underwent sham surgery on their left eye. More animals were included in implant refill groups because of the anticipated challenges of nonspecific immune-mediated ocular inflammation due to the administration of humanized protein.
Ocular toxicity outcomes evaluated following surgery were clinical observations; OE and IOP at days 1 to 15 and months 1 to 6; spectral-domain optical coherence tomography, full-field electroretinography, and fluorescein angiography at months 3 and 6; serum toxicokinetics; antidrug antibodies (ADAs); and histopathology (as described in the methods above) following sacrifice at study end.
For toxicokinetic studies, serial blood samples were collected from the minipigs over the course of the 6-month study and processed for serum. Ranibizumab concentrations were assessed by an enzyme-linked immunosorbent assay (ELISA) designed to quantify serum exposure in minipigs. In this assay, prediluted serum samples were incubated with biotinylated recombinant human VEGF and a monoclonal anti–ranibizumab-VEGF complex antibody. These were subsequently transferred to a streptavidin-coated ELISA plate and incubated for an additional 2 hours. The monoclonal anti–ranibizumab-VEGF complex antibody reagent binds specifically and with high affinity to ranibizumab-VEGF complexes, which are then captured onto the surface of the well via the interaction between biotin and streptavidin. After washing, enzyme-conjugated goat polyclonal antibody against mouse immunoglobulin G was added for detection, followed by washing and addition of enzyme substrate for color development. The assay sensitivity was 30.0 pg/mL. Use of excess concentration of biotinylated recombinant human VEGF in the first step of the assay ensured that effectively no interference on quantitation of total ranibizumab concentration would be exerted by endogenous VEGF. Serum exposure, as described by the maximum concentration and area under the concentration-time curve, as well as the accumulation ratio following multiple dose administrations was determined by noncompartmental analysis utilizing Phoenix WinNonlin (Certara L.P.). Serum concentrations were pooled by group for the analysis.
Antidrug antibodies were detected in serum from study animals using a bridging ADA ELISA. In this assay, biotin-labeled drug was used to capture antiranibizumab ADAs onto streptavidin-coated plates, with digoxigenin-labeled ranibizumab used for detection via addition of enzyme-labeled antidigoxigenin antibody, followed by enzyme substrate and color development.
Results
In Vivo Evaluation of Ocular Implant in Rabbits
In the 6-month rabbit GLP study, the one-third–scaled nonfunctional surrogate implant was well tolerated after implantation into the rabbit eye. Ocular implantation had no effect on clinical signs, body weight, IOP, electroretinography, or clinical pathology. Nonadverse procedure-related ocular findings (mild conjunctival hyperemia or chemosis localized over the surrogate implant) were comparable between implanted right eyes and sham surgery left eyes and persisted generally up to 1 month after surgery, resolving with dissolution of absorbable sutures in the conjunctiva. These nonadverse findings were procedure related and attributed to aseptic preparation of the ocular surface, the creation of conjunctival or scleral incisions with their attendant wound healing response. The implant remained in place in 23 of 24 eyes. The extrusion of the implant in one eye into the subconjunctival space was considered to be because the scaled nonfunctional implant was not optimized for the thickness of the rabbit sclera.
Microscopic findings at the implantation sites were similar to sham surgery sites. For histologic preparation of the eyes containing the implants, the appropriate portion of the eye was embedded in plastic and sectioned with the implant in place, allowing visualization of the ocular tissues in association with the implant. Microscopically, implantation sites were characterized by a disruption through the temporal sclera, choroid, and retina (findings were considered related to the surgical procedure and to the implant itself). At the 3-month interim necropsy in 2 implanted eyes, a thin layer of fibrous connective tissue aligning the surface of the intraocular portion of the implant was noted, and in 1 eye, it was accompanied by very small numbers of mixed inflammatory cells and multinucleated giant cells. These microscopic findings were observed at the 3-month interim necropsy only, were considered related to a normal biological response to the implant, and were not considered adverse. No microscopic findings were detected in the 6-month terminal necropsy animals. Microscopic findings at the implantation sites were similar to the sham surgery sites. There were no other ocular microscopic findings. Microscopic findings in nonocular tissues were consistent with common incidental findings in this species; no implant-associated findings were observed.
Overall, implantation of a scaled nonfunctional implant into the vitreous of male and female New Zealand White rabbits was well tolerated with findings limited to those related to the surgical implantation procedure. Microscopic findings at the implantation sites were similar to the sham surgery sites.
Biocompatibility of the PDS Implant
Results of the implant biocompatibility evaluations are summarized in Table 1. The implant and implant extracts were determined to be nongenotoxic, noncytotoxic, nonsensitizing, and nonirritating and were therefore considered biocompatible.
No cell lysis or decolorized zones were observed in the subconfluent cell monolayer in direct cytotoxicity testing. In indirect cytotoxicity testing, 48 hours of incubation of cells with the implant extract also did not cause cell lysis or decolorized zones in the subconfluent cell monolayer. The implant was therefore considered not to be directly or indirectly cytotoxic.
No delayed dermal contact sensitization was observed in guinea pigs exposed to implant extracts in either sesame oil or saline. Implantation into the paravertebral muscle in rabbits resulted in no effects on mortality, clinical signs, or body weight after 12 weeks compared with control animals. Histologically, the implant was well tolerated and resulted in muscle reactions comparable to the implantation of a US Pharmacopeia high-density polyethylene control, and therefore, was not considered irritating. Intravitreal injection of implant extract had no effect on mortality, clinical signs, vitreous white blood cell count, or body weight. The single injections of extract were well tolerated and noninflammatory, and therefore, considered nonirritating.
In the bacterial genotoxicity tests, the ethanol and saline implant extracts did not cause increases in the mean number of revertant bacteria per plate with any of the tester strains with or without metabolic activation. The implant extracts were therefore not considered mutagenic. The ethanol and RPMI0 implant extracts did not cause a ≥2-fold increase in the mean mutant frequency in the mouse lymphoma cell line tested, either in the presence or absence of metabolic activation and therefore, they were not considered mutagenic.
Biocompatibility of Ancillary Devices
Results of the 4 ancillary device biocompatibility evaluations are also summarized in Table 1. The extracts of ancillary device tools were determined to be nongenotoxic, noncytotoxic, nonsensitizing, and nonirritating and were therefore considered biocompatible.
In the minimum essential medium elution cytotoxicity tests, extracts from ancillary devices tested did not cause cell lysis or decolorized zones, and were therefore, considered noncytotoxic. In the dermal sensitization evaluations, none of the devices tested caused delayed dermal contact sensitization and therefore, were not considered sensitizing. In the rabbit intraocular irritation model, single intravitreal injections of extracts from the ancillary devices were well tolerated and noninflammatory, and therefore, were considered nonirritating.
Chronic GLP Toxicology of the PDS Implant in Minipigs to Support Phase III and Registration
In this minipig study (using a clinically functional PDS implant), the implant was initially filled with 2 mg/eye of ranibizumab in 1 group of animals or another group of animals refilled monthly for 7 total doses (initial implant fill at time of surgery plus 6 refills; Table 2) and was well tolerated in 9 of 10 animals.
In all dose groups, including vehicle control refilled monthly (6 refills), procedure- and implant-associated ophthalmic findings of mild conjunctival inflammation, localized over the implant, were observed. In addition, commencing within the first week of implantation, blood on the distal surface of the implant and in the vitreous, which sometimes severely degraded the ability to visualize the fundus, was observed. These clinically meaningful findings decreased by day 29 and resolved without consequence by day 71 in most eyes. Ophthalmic findings also included implant protrusion/extrusion above the sclera, traumatic cataracts secondary to contact between the intraocular portion of the implant and the posterior lens capsule, and fibrosis over the implant surface. Because these findings also occurred in vehicle control animals, they were considered to be related to the surgical implantation procedure because the implant was not optimized for use in the pig eye and could not be securely seated in the sclera (Figure 2).
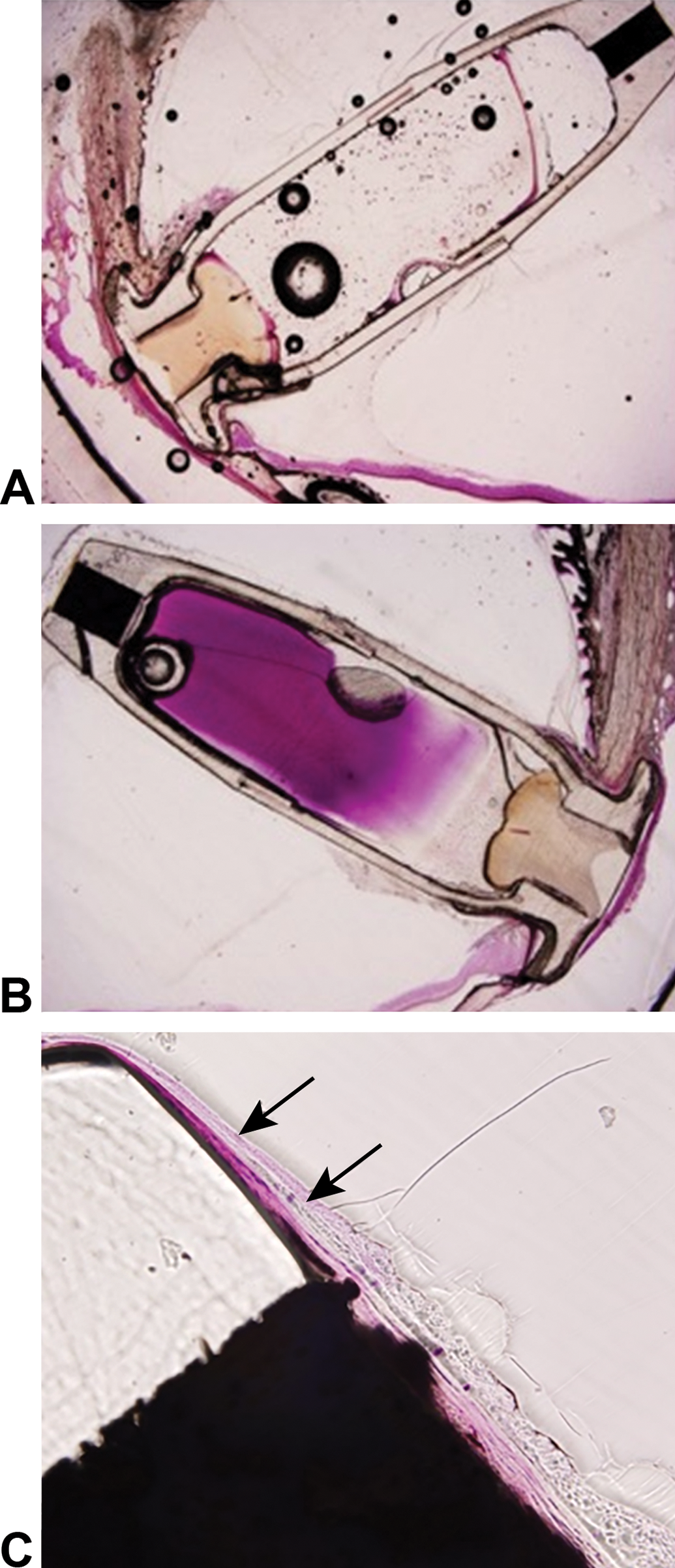
Ocular histopathology in the region of the Port Delivery System with ranibizumab at the end of the 6-month study in Yucatan minipig eyes. Representative photomicrographs of (A) control implant (vehicle dosing only); (B) an implant with only a single dose of ranibizumab and no refilling during the study; and (C) an implant refilled with repeat doses of ranibizumab. Arrows indicate fibrosis around the implant. The bubbles and retinal detachment observed in the images were artifacts of the processing, embedding, and sectioning processes.
Anterior and posterior segment inflammatory responses were more pronounced in eyes administered multiple implant refills than in eyes administered only a single implant fill of ranibizumab, and correlated with optical coherence tomography, fundus photography, and fluorescein angiogram findings of perivascular sheathing, epiretinal membrane formation, venous dilation, vascular leakage, capillary microaneurysms, optic nerve swelling, and increased hyperreflective spots in the vitreous. Vitreous optic nerve traction or retinal detachment was observed only in eyes administered multiple doses of ranibizumab via the PDS. One animal refilled with monthly ranibizumab developed severe mixed-cell panophthalmitis, necessitating unscheduled euthanasia after the fifth refill. Microscopic intraocular findings in this animal included slight to marked infiltrates of plasma cells, neutrophils, lymphocytes, and macrophages. The findings in this animal, along with its highest ADA titer among all treated animals in this study, were considered to be secondary to an immune-mediated response to a heterologous protein (ranibizumab).
Systemic toxicity evaluations, including clinical observations, body weight, toxicokinetics, clinical pathology, and gross and microscopic pathology, were included in the chronic study. No systemic toxicity was observed in minipigs following a single fill or monthly PDS refills for a total of 7 doses with 2 mg/eye for up to 6 months.
Exposure to ranibizumab was similar in the single-dose and monthly refill groups during the first 28 days (maximum concentrations of 2.1 and 1.7 ng/mL, respectively). Ranibizumab accumulated 15- to 20-fold following monthly PDS refill. As commonly seen in animals exposed to humanized therapeutic protein, ADAs were detected in the serum of all (10/10) ranibizumab-administered animals and in none (0/5) of the animals in the control group. The presence of ADAs potentially resulted in increased serum concentrations of ranibizumab due to the ADA–fragment antigen binding (Fab) complex having a longer systemic half-life than the Fab alone. 16
Overall, the procedure for implant insertion with or without ranibizumab was associated with local external inflammation, vitreous hemorrhage that sometimes prohibited fundus visualization, and movement (protrusion/extrusion) of the implant (not optimized for the porcine eye) leading to traumatic cataracts or retinal pigment epithelium mottling. Administration of single or multiple doses of ranibizumab via the PDS was associated with increased severity of ocular inflammation, which necessitated unscheduled euthanasia for 1 animal. All of the ranibizumab (via the PDS)-related ophthalmologic and histological findings were considered either directly or secondarily related to an immune-mediated response to a foreign protein, as evidenced by presence of ADAs in all animals administered ranibizumab.
Discussion
Here, we describe a comprehensive overall toxicology program conducted to characterize the safety profile of the PDS, and a biocompatibility battery of the implant and 4 ancillary devices to support the clinical development program and registration in patients with nAMD. The results demonstrated that the implant and 4 ancillary devices were well tolerated and biocompatible in vitro and in vivo. A chronic 6-month toxicology/TK study in minipigs with the PDS did not show any toxicology-related safety concerns relative to the implant or a combination of the implant and ranibizumab.
The comprehensive biocompatibility and toxicology tests were undertaken in vitro and in appropriate animal species to assess the biocompatibility of the implant and ancillary devices before conducting a clinical trial program of the PDS in patients with nAMD. Although the efficacy and safety profiles of intravitreal ranibizumab injection for the treatment of nAMD have been established, 14,17 the toxicology and biocompatibility of the PDS implant and its ancillary devices had not been evaluated. Although some long-acting drug delivery systems have been approved to treat ophthalmologic conditions, including retinal diseases, none of them are permanent or refillable. 18 –20 The design of the PDS toxicology program therefore presented a number of challenges, foremost among which was the lack of regulatory guidance for a permanent, refillable implant for the back of the human eye. Developmental challenges, including the selection of appropriate toxicology species, the unique nature of immunogenicity within the eye, and designing a drug/device combination for optimal intraocular delivery, were also faced.
Several critical factors should be considered when designing a comprehensive toxicology and biocompatibility testing strategy for a permanent, refillable ocular drug/device combination. In the absence of specific national or international guidelines and standards for evaluation of intravitreal implant system devices, International Conference for Harmonisation guidelines and ISO and ANSI standards were utilized as a starting point. The Center for Devices and Radiological Health device classifications considered in development of the testing strategy for the PDS devices were class 2 for the needles and explant tool and class 3 for the implant and insertion tool assembly. For ocular toxicology studies, and drug/device combinations specifically, species selection and relevance for human risk assessment are the most important elements. For the PDS, the minipig eye is large enough for a functional implant and was the only relevant species for evaluation in a chronic drug/device combination toxicology/TK study. No systemic toxicity was observed in this study. Fibrosis adjacent to the implant was observed in both treated animals and controls (Figure 2), and thus appeared to be due to instability of the implant, which is not optimized for the structure of the pig eye. 15,21 Furthermore, the implant also required suturing to hold it in place, exacerbating the local response. Overall, the PDS implant and associated procedures, such as refilling, were well tolerated in this species. Inflammation was observed in some animals treated with ranibizumab. All ophthalmologic and histological findings related to the administration of ranibizumab (via the implant) were considered related to an immune-mediated response to a humanized protein (ranibizumab) in those animals. This was supported by the presence of systemic ADAs in all animals receiving ranibizumab in the present study and in nonhuman primates receiving intravitreal ranibizumab in previous studies. 22 This observation does not raise safety concerns, because immunogenicity in nonclinical species is not predictive of immunogenicity in humans. 23 As with all biologic therapies, there is a potential for recipients to have an immune response to the agent and to produce ADAs. 14 The clinical significance of ranibizumab ADAs in patients with nAMD is currently unclear 14,24 because there have been no reported differences in efficacy or tolerability of ranibizumab in patients with ADAs. 24 Notably, intravitreal ranibizumab (Lucentis) has demonstrated a favorable benefit–risk profile in global postmarketing experience. 25 –27 The PDS phase 3 data also demonstrated a favorable benefit–risk profile. 13
A comprehensive biocompatibility evaluation strategy was devised based on a range of ISO 10993 (biological evaluation of medical devices) standards that were relevant to the implant and materials (https://www.iso.org/standards.html), ISO 11979 (ophthalmic implants–intraocular lenses), and ANSI Z80.7 (ophthalmics–intraocular lenses). These standards were selected as they are the closest available standard, anatomically and procedurally, for the setting in which the implant would be utilized. Long-term evaluation of the implant was undertaken in a rabbit model; however, because the functional implant was too large for implantation into the rabbit eye, a one-third–sized nonfunctional surrogate was used, which was otherwise identical in every respect to the functional implant. Hence, any results obtained with the surrogate in this model would be directly applicable to the implant. As mentioned in the Results section, in a 6-month implantation study in the rabbit model, the surrogate implant was well tolerated.
A battery of in vitro and in vivo biocompatibility tests of the implant determined that the implant was noncytotoxic, nongenotoxic, nonsensitizing, and nonirritating. No inflammatory or other local responses were noted when the implant was implanted into muscle tissue of rabbits. Extracts of ancillary devices were determined to be noncytotoxic, nonsensitizing, and nonirritating and were therefore considered biocompatible.
The goal of a comprehensive toxicology program and biocompatibility testing for a drug/device combination product is to assess patient risk and support clinical development and product registration not only for device-related components (ie, implant and 4 ancillary devices) but also for ranibizumab. These 2 topics (drug delivered over an extended period of time and using a permanent, refillable device) need to be considered together. Lack of ocular-specific guidelines presents a unique challenge for ophthalmic drug development in general, and more specifically, for a permanent, refillable drug/device combination such as the PDS. Lack of ocular-specific guidelines prompted the team to seek an interaction with the health authority focusing on gaining acceptance regarding the nonclinical toxicology program for this drug/device combination product. At the completion of the 6-month toxicology study in rabbits with the nonfunctional implant and the biocompatibility program, and before initiation of phase 2, at a preinvestigational new drug meeting with the FDA (Center for Drug Evaluation and Research and Center for Devices and Radiological Health), a question was asked about the adequacy of the completed toxicology and biocompatibility program to support clinical development and registration. The FDA recommended a chronic 6-month toxicology/TK study of the PDS in an appropriate animal model with multiple refills to enable evaluation of the functional implant before phase 3, while phase 2 was conducted. A 6-month chronic toxicology study of the PDS in minipigs was initiated with the clinical implant because it fits in the eye of this animal model. The clinical implant is not optimized for the minipig eye, as described in the Materials and Methods section; therefore, at the time of surgery, the implant was secured via a horizontal mattress suture in order to reduce the risk of implant extrusion. As described in the Results section, in the 6-month chronic toxicology study of the PDS in minipigs, procedure-related postsurgical vitreous hemorrhage was observed in the majority of animals. Since vitreous hemorrhage resolved without intervention within a month of implantation in the majority of animals, and did not limit assessment of safety endpoints during the toxicology study, we developed a minipig surgical model of vitreous hemorrhage where various surgical techniques were evaluated. 15
Another challenge for a drug/device combination product for retinal diseases is related to biocompatibility testing standards, such as ISO 10993. Because ISO 10993 does not apply to ocular implants, it was utilized as a starting point, along with ISO 11979 and ANSI Z80.7 standards. As a general guidance for permanent tissue medical devices such as implants, ISO 10993-1 specifies evaluation of biological effects related to tissue response following implantation (ISO 10993-6) and a test for irritation potential (ISO 10993-10). Typically, health authorities recommend a muscle implant and a test for irritation in rabbits as acceptable biocompatibility studies for evaluation of local effects. Even though such biocompatibility tests were performed as described in the Results section, ocular implantation is the most relevant to human risk assessment of the implant rather than rabbit muscle tissue response or a test for irritation in rabbits. Although not explored in this program, we believe that ocular implantation evaluation would have been acceptable with the health authorities and the 2 in vivo tests, muscle implant and irritation in rabbits, omitted.
In conclusion, nonclinical development of a drug/device combination product requires a thorough understanding of the requirements and early interaction with health authorities on the acceptance of either proposed or completed comprehensive toxicology and biocompatibility testing. The program of nonclinical toxicology and biocompatibility testing for the PDS described here found no evidence of toxicity or biocompatibility concerns for the PDS implant or its ancillary devices. In addition, delivery of ranibizumab into the vitreous via the PDS implant did not introduce any new toxicology-related safety concerns compared with intravitreal injections of ranibizumab. These findings enabled phase 2 clinical development; further testing of the PDS in the Archway phase 3 trial in patients with nAMD (NCT03677934) is ongoing to support product registration.
Supplemental Material
Supplemental Material, sj-docx-1-tpx-10.1177_0192623320968079 - Nonclinical Toxicology and Biocompatibility Program Supporting Clinical Development and Registration of the Port Delivery System With Ranibizumab for Neovascular Age-Related Macular Degeneration
Supplemental Material, sj-docx-1-tpx-10.1177_0192623320968079 for Nonclinical Toxicology and Biocompatibility Program Supporting Clinical Development and Registration of the Port Delivery System With Ranibizumab for Neovascular Age-Related Macular Degeneration by Vladimir Bantseev, Joshua Horvath, Giulio Barteselli, Shrirang Ranade, Mauricio Maia, Daniela Bumbaca Yadav, Chris Schuetz, Amy Shelton and Helen S. Booler in Toxicologic Pathology
Footnotes
Acknowledgments
The authors acknowledge the valuable support of Signe Erickson, Jay Stewart, Evan Thackaberry, Cindy Farman, T. Michael Nork, Ellison Bentley, Paul E. Miller, and Ewa Budzynski.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: V.B., J.H., G.B., S.R., M.M., D.B.Y., C.S., A.S., and H.S.B were employees of Genentech, Inc, South San Francisco, CA, at the time that this work was conducted.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding was provided by Genentech, Inc, South San Francisco, CA, for the study and third-party writing assistance, which was provided by Ashley Sizer, PhD, of Envision Pharma Group.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.