
Abstract
Chronic progressive nephropathy (CPN) is the most commonly encountered spontaneous background finding in laboratory rodents. Various theories on its pathogenesis have been proposed, but there is a paucity of data regarding specific mechanisms or physiologic pathways involved in early CPN development. The current CPN mechanism of action for tumorigenesis is largely based on its associated increase in tubular cell proliferation without regard to preceding subcellular degenerative changes. Combing through the published literature from multiple biology disciplines provided insight into the preceding cellular events. Mechanistic pathways involved in the progressive age-related decline in rodent kidney function and several key inflexion points have been identified. These critical pathway factors were then connected using data from renal models from multiple rodent strains, other species, and mechanistic work in humans to form a cohesive picture of pathways and protein interactions. Abundant data linked similar renal pathologies to local events involving hypoxia (hypoxia-inducible factor 1α), altered intrarenal renin–angiotensin system (RAS), oxidative stress (nitric oxide), and pro-inflammatory pathways (transforming growth factor β), with positive feedback loops and downstream effectors amplifying the injury and promoting scarring. Intrarenal RAS alterations seem to be central to all these events and may be critical to CPN development and progression.
Keywords
Introduction
Chronic progressive nephropathy (CPN) is the most commonly encountered spontaneous background finding in laboratory rodents. Chronic progressive nephropathy has been reviewed extensively over the past 40 years, yet its pathogenesis remains controversial. 1 -8 It occurs in both male and female rats, but at a higher incidence and severity in males, and a similar syndrome is noted in several mouse strains. 9 In a retrospective review of GlaxoSmithKline toxicity studies, incidence rates varied widely among studies, by strain and with age in both rats and mice. Other investigators report incidences approaching 100% in some strains after 2 years, including wild rat strains. 5 Advanced stages of CPN include a spectrum of alterations in tubules, glomeruli, and underlying basement membranes. The hallmark features in rats include clusters of basophilic tubules, nuclear crowding, thickened basement membranes, and tubule casts. 10 -12 There are mononuclear cell infiltrates in the surrounding interstitium, varying in number and type, which are especially common in mice. As CPN progresses, severity increases and complex tubule changes develop, involving dilation and cyst formation, casts, and eventually hyperplasia. 13,14 Glomeruli are usually enlarged, associated with consistently increased mesangial matrix, collagen, and/or deposition of amorphous hyaline material. 7,15,16 The glomerular CPN lesions progress with advancing age leading to atrophy, with loss of glomerular podocytes, increased numbers of fibroblasts, contraction of capillary tufts, enlargement of the urinary space, and thickened basement membranes associated with glomerulosclerosis. There can also be accompanying changes in the epithelium of the renal pelvis, characterized by proliferation or hyperplasia of the epithelium lining the papilla. 3
For several years, various publications in the toxicologic pathology field, including those of the authors, have advocated that the underlying pathogenesis for CPN is unknown, or at least unproven, and probably multifactorial. The molecular pathways involved in early disease progression have generally been ignored in animal science, veterinary, and toxicologic pathology literature. Various theories on pathogenesis have been proposed and discussed in review articles and book chapters, but with surprisingly little hard data on the specific genes or proteins involved in the mechanisms or physiologic pathways related to CPN development. Recently, detailed reviews of CPN were published, which documented the results of a comprehensive list of studies concerning the later aspects of CPN pathophysiology. 2,3 With any review of the topic, the complexity of the CPN disease process is apparent and there is an obvious convoluted interrelationship between environmental factors and genetic background. While reviewing literature related to aging changes in rodent kidneys, the authors unexpectedly were confronted with evidence from a whole series of experiments that have identified specific proteins and cellular pathways common to spontaneous renal degenerative changes in the kidneys of rats and mice. Upon thorough review of pathology and available photomicrographs, it became clear that the spontaneous renal aging processes in many of these studies were compatible or identical to what the toxicologic pathology community has referred to as CPN. The objective of this article is to bring together this body of research and demonstrate that 5 key and interrelated proteins—renin, angiotensin II (AngII), hypoxia-inducible factor 1α (HIF-1α), transforming growth factor-β1 (TGFβ1), and nitric oxide (NO)—are essential components in the pathogenesis of the degenerative phase of CPN, based on their expression/alteration in rats with CPN lesions as compared to unaffected controls. It is not the objective of this article to provide a comprehensive review of all of the factors involved in CPN development and pathogenesis from initiation to end stage, nor is it possible to do so. After decades of research, the initiating causal events remain elusive, and for many of the earliest pathophysiologic events in CPN development, specific proteins have not been identified. However, this article is intended to provide evidence that once the initiating event of CPN has been unleashed (genetic/epigenetic) and enhanced via identified progression factors (nutritional, toxic, environmental, etc), the renin–angiotensin system (RAS) within the renal interstitium is activated (Figure 1). This results in changes to the NO/NO synthase (NOS) system regulating vascular tone and to renal parenchymal hypoxia effects and inflammatory pathways, regulated by HIF-1α and TGFβ, respectively (Figure 1). Eventually, well-established regenerative pathways in renal tubules are induced late in CPN disease, leading to the recognized association between CPN and tumorigenesis. Since these later regenerative phases have already been characterized, they are reviewed in this article, but we do not attempt to elucidate any new pathophysiologic factors in the late phase of the disease and there are no new additions or changes to the existing mechanism of action (MOA) regarding CPN-related tumor development.
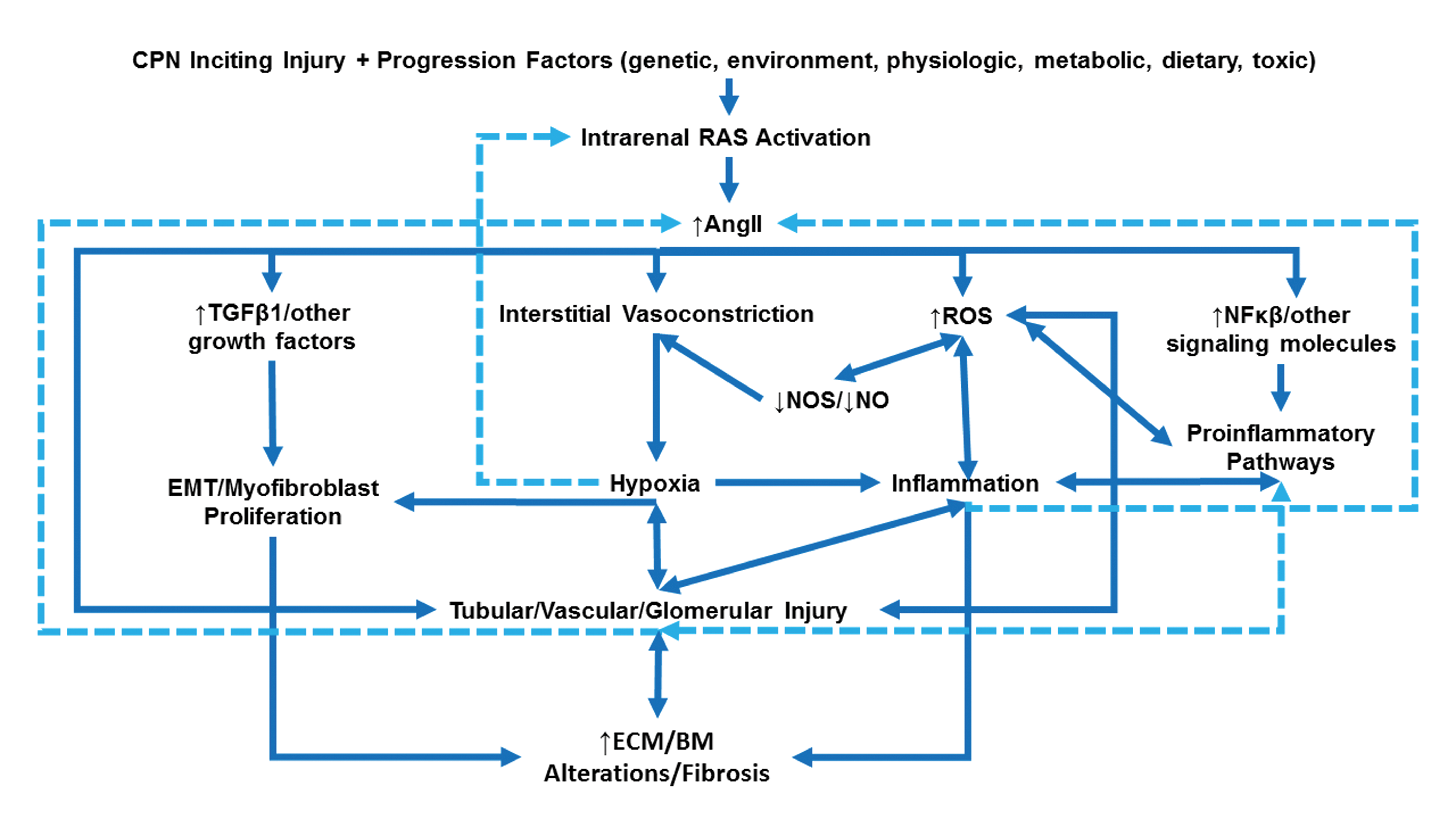
Intrarenal renin–angiotensin system (IRAS) and the associated pathways in the development and progression of chronic progressive nephropathy (CPN) in rats. Genetic/epigenetic and progression factors predispose rats to CPN development. The IRAS activation and increased intrarenal angiotensin II (AngII) expression result in (1) interstitial vasoconstriction with subsequent local hypoxia, (2) increased nuclear factor-κB signaling leading to activation of pro-inflammatory pathways, (3) increased transforming growth factor β1 signaling leading to activation of profibrotic pathways that promote epithelial–mesenchymal transition and myofibroblast proliferation, and (4) increased oxidative stress via reactive oxygen species (ROS) and decreased nitric oxide synthase (NOS) and nitric oxide (NO) generation. These pathways have multiple points of intersection (double-headed arrows) and feedback loops (dashed lines) that amplify IRAS and the downstream effectors, resulting in injury to tubular, glomerular, and vascular tissue components with subsequent increase in extracellular matrix (ECM) deposition, basement membrane (BM) alterations, and interstitial and glomerular fibrosis.
Chronic Progressive Nephropathy Pathogenesis From a Historical Perspective
Because some of the oldest reviews of CPN in rats indicated that the earliest lesions were thickening of glomerular basement membranes, particularly thickened Bowman’s capsule, it was originally assumed the initial lesion was centered on the glomerulus. 5,15,17 However, it has been shown since by both light microscopy and ultrastructural evaluation that precursor glomerular defects are not detectable prior to tubule lesions and several recent investigators question whether glomerular events truly initiate the complex events associated with the disorder. 3,12 Importantly in this regard, it has been demonstrated that the albuminuria that accompanies CPN may result less from changes in glomerular permeability but instead from failure to reabsorb albumin from proximal tubules, suggesting a tubular defect in megalin/cubilin transport. 18,19 Rats excrete considerable amounts of protein in urine, even without any significant glomerular injury. The protein in the filtrate is predominated by both intact and fragmented albumin and is normally reabsorbed in tubules. 20 Considerable changes have been noted in the excretion of albumin, independent of glomerular permeability changes, during the very early stages in rat models of nephropathy. 20 Presumably, similar changes in tubule function occur with CPN.
Another prevalent theory concerning CPN pathogenesis centers on the idea that hyperfiltration and functional overload of the glomerulus is the underlying basis of the disease. 21 Excessive intake of dietary proteins has long known to be associated with increased incidence of CPN in chronic rodent studies. Since high-protein, carbohydrate, or ad libitum diets were associated with enhanced CPN, and these perturbations were also known to increase renal blood flow (RBF) and glomerular filtration rate (GFR), it seemed logical that adaptive hemodynamic changes resulting in chronic elevations in perfusion and hyperfiltration would damage glomeruli. 21 However, in an elegant series of measurements, Baylis 22 showed that there is no elevation in glomerular capillary pressure with advancing CPN and no relationship to glomerular morphologic damage. This led to theories for a tubule-based insult as the primary or initial pathophysiologic location of injury. Defects of basement membranes, including alterations in composition, have been suggested as a cause of CPN based on thickened membranes noted as one of the earliest morphologic changes in tubules and glomeruli. 23,24 Basal laminae surrounding proximal tubules appear progressively wrinkled and folded with time. 5 Aberrant accumulation of extracellular proteins and increased epithelial–mesenchymal transition (EMT) have been demonstrated in the aging rat kidney. 25 –27 Age-related dysregulation of renal extracellular matrix (ECM) in rats has been shown to be associated with the loss of epigenetic gene silencing as well as with decreased matrix metallopeptidase 2 activity. 28 –30 However, it is difficult to determine whether alterations to the membrane are actually causal or simply an effect of early damage at the tubulointerstitial interface. Immunochemical investigations have revealed neither immune complexes nor serum antibody are responsible for basement membrane thickening, and serum from rats with advanced CPN are negative for immunoglobulin G (IgG) or IgM antibodies to kidney antigens. 31 This is strong evidence against an immunologic basis for the renal defects. Accumulation of advanced glycation end products (AGEs) has also been hypothesized as a cause of CPN, because AGEs accumulate within senescent rat kidneys and are known to induce ECM expansion in both the glomerular mesangium and the interstitium. 32,33 However, other effects noted with CPN seem to be independent of AGEs, including those in tubule epithelial cells.
Due to the ubiquitous nature of the disease in rats, most authors have presumed there is an underlying genetic basis for CPN, but no single gene appears to be responsible and multiple genes are thought to be involved. Rather than genetic or chromosomal inheritance-based causality, a recent publication 34 has suggested that the disease may represent proliferation of a clone of precursor tubular epithelial cells endowed with the ability to secrete pro-inflammatory and prosclerotic cytokines. The phenotype in this proposed mechanism is thus a result of somatic mutation or epigenetic changes as a first event in CPN pathogenesis. 34 Unfortunately, the author provided no concrete evidence for the existence of such a clonal population or any data to refute more straightforward chromosomal-based genetic etiology.
Although tubule cell atrophy and loss are indicative of a degenerative aging process, it is certain that CPN is also a regenerative disease based on studies labeling DNA synthesis as an index of cell proliferation, including tritiated thymidine, bromodeoxyuridine, or proliferating cell nuclear antigen. 35 –38 Tubule cell basophilia, noted from the earliest stages, is accompanied by increased mitoses and pyknosis and represents an increase in cytoplasmic endoplasmic reticulum content and hence increased messenger RNA (mRNA) associated with substantial cell turnover. 1 Small tubule regeneration, characterized by often pigmented and tightly coiled basophilic tubules within atrophic areas, are even noted in end-stage kidneys and likely represent persistent regenerative phenomena. 37 It is through these regenerative processes and pathways that CPN is associated with increased tumorigenesis in rodents. 2
In 2013, a consortium of experts from the Society of Toxicology published a guideline for evaluation of exacerbation of CPN as a valid MOA for rat renal tumors. 2 The framework for analysis of any MOA and human relevance/risk has been developed by the International Life Sciences Institute and International Program on Chemical Safety. 39,40 In the 2013 paper and other manuscripts, it has been stressed that there is no human analog for CPN and that CPN in rodents is a distinct entity with major differences from human renal diseases. 2,9 Some of the early criticisms of the MOA for CPN-associated renal tumors were based on misunderstanding of the scope of CPN relationship within a study (individual vs group) 41 and have since been dismissed. 2 Most scientists and experts in the field, including the authors, are in agreement with the 2013 guideline, but there are some gaps in early pathogenesis that could yet be filled. As long as the key overriding event is identified, it is not necessary to know the detailed etiology or specific underlying pathways for establishing an MOA for tumor formation. However, the rationale for an MOA is greatly enhanced with the identification of the molecular pathways and protein targets involved in the specific stages of tumor progression. In the case of CPN, the MOA is largely based on its associated increase in tubular cell proliferation and rightly focused on the molecular events associated with enhanced cell turnover, without regard to the subcellular degenerative changes that precede regenerative processes. Based on our extensive literature review, there appears to be sufficient information published in other fields of biology to fill in some gaps regarding the preceding cellular events in the degenerative phase of CPN development which will strengthen the case for the current CPN MOA and its interpreted lack of human relevance by providing an increased understanding of CPN pathogenesis in rodents.
Caveats
Chronic progressive nephropathy is universally encountered in animal research and is referred to in various literature sources under a host of pseudonyms and synonyms, including aging nephropathy, chronic nephritis, spontaneous or chronic nephrosis, protein overload nephropathy, spontaneous glomerular sclerosis, chronic progressive nephrosis, progressive glomerulonephrosis, glomerulosclerosis, rat nephritis, and dietary nephritis, among other more recent labels. 5 This project was initiated to assemble and review research from a variety of biologic fields (renal physiology, human nephrology, safety pharmacology, gerontology, hypertension research, renal injury models, and internal safety data). The authors combed through histologic descriptions and clinical data to assess whether renal changes in these divergent studies were consistent with and actually describing what the toxicologic pathology field refers to as CPN in INHAND (International Harmonization of Nomenclature and Diagnostic criteria for lesions in rats and mice) and SEND (Standard for the Exchange of Nonclinical Data) terminology. 10 Using CPN as a linchpin diagnosis, the biochemical, molecular, genomic, proteomic, and physiologic end point data were compared to build a body of evidence for identifying key mechanistic pathways in the progression of disease and deterioration of kidney function that accumulate with age in the rat. Importantly, several key critical pathway factors were identified where specific proteins known to be altered in “aged rodents” demonstrated kidney lesions compatible with CPN. These critical junctures were then connected using data from other renal models. The assimilation of this information into the toxicologic pathology lexicon is critical to providing the most accurate translational risk assessment of drug-related enhancement of rodent CPN to human patients.
Some of the information gathered in this review are taken from animal models. There is an inherent peril in utilizing such data to draw parallels to a spontaneous disease, such as CPN. Many years ago, Goldstein et al indicated that the proposed pathogenesis of CPN was largely derived from studies on experimental kidney disease models and therefore was suspect. 42 As Hard and Khan 1 have rightly pointed out, “despite clinical and morphologic similarities in these various entities, the assumption that the pathogenesis of disease progression is the same regardless of the mode of reduction in renal mass is unproven.” It is clear that simply demonstrating a pathway is present in a rat model of kidney disease with a different genetic defect does not prove the same factors would be expected in CPN pathogenesis. However, the authors have instead relied upon evidence from experiments in aged rodents with spontaneous kidney disease to identify which key proteins are actively involved. For each of the 5 key proteins highlighted (renin, AngII, HIF-1α, TGFβ1, and NO), we identified published experiments showing spontaneous rats or mice with CPN-like lesions had mRNA or actual protein levels modified as compared to controls. The review of the renal pathology was key in the process of validating that the spontaneous kidney lesions were not some other rodent renal disease. Since many pathophysiologic processes are shared across species, animal models have been utilized for decades to elucidate and identify specific mechanisms of disease and toxicity, and the physiologic pathways that unite these factors. Rodent models of kidney disease in this review were only utilized to show how each of the 5 proteins interact at the molecular level and to provide further weight of evidence for their mechanistic function in renal degeneration.
Some of the data described in this review also include mechanistic work in human kidneys or those from other animal species. In this regard, the focus of highlighting these various kidney diseases is to demonstrate parallel molecular pathways of injury at the cellular or molecular level, but in no way suggest that similar molecular pathways imply that CPN recapitulates acute or chronic kidney disease (CKD) in humans. Rather, it should be stressed that the initiating stimuli for CPN in rodents appears to be at the genetic and epigenetic level, and these inherited predilections and their characteristic phenotypic progression are not evident in human patients. Despite similar molecular pathways of injury within the tubulointerstitial compartment in some stages of renal disease, rodent and human kidney diseases fundamentally differ. 9 Histologically, the basement membrane thickening noted with degenerative and regenerative tubules in CPN is only noted in human kidney diseases associated with tubule atrophy, and this should signal that the pathogenesis of the 2 diseases are substantially different. 2 Another fundamental and important difference includes increased versus decreased size of the kidney in advanced CPN and end-stage CKD, respectively. In addition, the clinical course of the 2 diseases differ remarkably, as do accompanying clinicopathologic parameters, immunologic factors, and relationship to other modifiers such as diet, gender, or hormonal factors. 9 Similar functional responses to specific degenerative regulatory stimuli at the molecular level do not correspond to enhanced clinical risk. In particular, enhanced tumor progression in rodents with compounds that enhance CPN does not translate into increased human kidney tumor formation because these drugs or chemical agents do not appear to exacerbate clinical kidney diseases and, most importantly, proliferative pathways of CPN are not shared across species. Acknowledging these caveats and relying on a weight-of-evidence approach from spontaneous aging rodent kidneys, multiple models and multiple rodent strains, and human kidney disease molecular analysis, a cohesive picture of pathways and protein interactions was assembled.
Involvement of the Interstitium and Damage to Multiple Renal Compartments
Few authors have suggested the interstitium is the primary or initial focus of injury with CPN since interstitial involvement appears mild by microscopy, with only focal accumulations of mononuclear inflammatory cells and fibrosis at later stages of the disease. However, progressive expansion of the interstitial ECM is a constant feature of the disease in chronic phases, and fibronectin/thrombospondin accumulation within the interstitial space has been reported in rodents commencing at 12 months of age. 4 Accumulating data from a wide variety of sources indicates that the interstitium is involved in the development of many chronic renal diseases without apparent morphological change. It is perhaps novel that the interstitium may play such a central role in kidney disease, and some might question how interstitial changes could result in the complex array of alterations in the glomeruli and tubules that are reflected in CPN phenotypes. However, connections to and across the basement membrane result in functional interactions between tubules and interstitium, forming an interdependency between them. As such, the tubulointerstitial compartment has been considered one unit in nephrology for years. 43 This close relationship between renal tubule epithelium and interstitial cells was demonstrated in a recent study where laser-ablated tubular epithelial cell replacement was dependent on intimate communication with platelet-derived growth factor (PDGF) β receptor–positive interstitial cells. 44 Tubuloglomerular feedback has been known for several decades to be a source of interaction between disparate portions of the nephron, based on the function of the macula densa and juxtaglomerular apparatus. The renin–angiotensin–aldosterone system (RAAS) plays a key role in this cross talk between thick ascending limb (TAL), afferent and efferent arterioles, and juxtaglomerular cells. 45 However, the extent to which the interstitium can affect glomerular injury is only now being elucidated. Even mild preexisting tubulointerstitial injury sensitizes glomeruli to subsequent podocyte-specific injury. 45,46 Conversely, glomerular injury and abnormal filtration results in intrinsic toxicity to the proximal tubule and is a key factor in renal disease progression, including in CPN. 47 Protein overabsorption activates inflammatory and profibrotic pathways that result in scarring within the interstitium, so the interaction goes both ways. 48
Polyarteritis Nodosa and CPN
One of the first clues to potential factors involved in CPN pathogenesis came from observations in rodent carcinogenicity studies. It has been noted by several investigators that there is often a remarkably high correlation between the presence of CPN of moderate-to-high grade and the concomitant presence of polyarteritis nodosa (PAN) in the same individual rats or mice. 12,49 For instance, in a recent 2-year carcinogenicity study, 42 of 43 CD1 mice with PAN also had severe kidney lesions of grade 3 or higher (on a 0-5 scale; K.S.F., unpublished data, 2016). Review of 3 recent internal rat carcinogenicity studies using Han Wistar or Sprague Dawley strains demonstrated that over 93% of rats with vascular lesions consistent with PAN noted at the terminal necropsy also had CPN of moderate or higher grade. These kinds of associations lead one author to conclude that PAN may be caused by CPN. 49 The pathogenesis based on this premise might involve detrimental effects on the renin–angiotensin–aldosterone axis with severe kidney disease and hence resulting in systemic hypertension. On first glance, hypertension as a cause of CPN makes sense. Chronic hypertension is one of the 2 most common causes of chronic renal failure in humans and has even been shown to cause nephropathy in rodents. 50 Intraglomerular hypertension is a primary causal factor in the progressive glomerulosclerosis that characterizes diabetic nephropathy in humans. 51 The association of renal hypertension and PAN is not a new concept but one that was initially proposed over 75 years ago. 52 –55 Investigations into the cause of PAN using a transgenic rat model have demonstrated increased local renin release is a primary causal factor of PAN in rats. 56 However, it has been shown that most rats with mild-to-moderate CPN are not hypertensive, and in fact, if systemic hypertension develops in rats with CPN, it occurs only very late in the course of disease. 33 Glomerular blood pressure does not increase with advancing age in rats even up to 19 months and does not correlate with glomerular damage. 22,57 Glomerular volume in rats does increase with age, but it does not correlate with any glomerular damage. 22 Glomerular capillary pressures remain normal and there are no lesions in either afferent or efferent arterioles suggestive of glomerular hypertension, even in severe cases of CPN. 1,2,4 Further, there are certainly many animals with evidence of PAN that lack kidney lesions of any kind. 58 Interestingly, female Hatano low-avoidance rats (stress nonreactive) develop significantly higher incidences of CPN and PAN than female Hatano high-avoidance rats (stress reactive) while maintaining low systemic blood pressure. 59 These factors argue against the paradigm that CPN directly causes PAN though pressure effects and strongly argue against systemic or glomerular hypertension as causal factors in CPN.
Taking the reciprocal argument into consideration, could PAN be causally related to CPN pathogenesis? This is extremely unlikely for several reasons. First, the incidence of CPN is ubiquitous in rodents across strains and much greater than the incidence of vascular lesions or PAN, especially in rats. Rats with the earliest changes of CPN, including focal tubule basophilia and minimally thickened basement membranes, virtually never have any histologic evidence of vascular medial changes or PAN. In fact, PAN is rarely evident at 16 weeks of age in rats when early CPN lesions are relatively common, but minimal. Most importantly, although vascular lesions are commonly associated with CPN in elderly rats, it has been noted that even in 2-year studies there are very rarely any vascular lesions in the kidney of rats with CPN. 1,6 No investigators in this area have been able to demonstrate a direct link between morphologic vascular changes in renal vessels and CPN pathology. The only reasonable conclusion is that PAN does not cause CPN. Instead of a causal link, CPN and PAN may have an initiating factor common in their pathogenesis. Such a close association between 2 distinct disease entities deserves further scrutiny in order to elucidate any shared mechanistic features. Increased renin release and effects on AngII are known to be involved in PAN pathogenesis. 56 Therefore, we examined the possibility that factors related to local renal blood pressure control might be involved in CPN pathogenesis.
From pharmacologic study data, it is clear that systemic hypertension itself is not a factor in CPN pathogenesis, and glomerular hypertension does not appear to be involved. 22 Thus, a hypothesis was formulated that local factors related to interstitial blood pressure control at the capillary level, unrelated to systemic pressure, might be mechanistically linked to the process in some way.
Renin–Angiotensin System Is a Potential Factor in CPN Pathogenesis
Angiotensin II, the major effector of the RAS, is involved in important physiologic events in the vasculature and kidney. 60 The RAS is altered during aging, with reduced plasma renin activity (PRA) and decreased aldosterone levels, partially compensated for by increases in the density of AngII and vasopressin receptors in aging rats and in older human patients. 61 –65 Despite the decline in RAS activity with age, local renal secretion of AngII is markedly increased and responsiveness to AngII is enhanced. 66 –68 Recent work in the field of CKD in humans has focused on one arm of this axis, the intrarenal renin–angiotensin system (IRAS). Several comprehensive reviews of the IRAS have been published. 69 –71 Unlike the systemic RAS, IRAS acts as a local autocrine/paracrine system in the kidney, which involves both angiotensin-dependent and angiotensin-independent actions. 71 Intrarenal RAS alterations have been reported in animal models of hypertension, 72 –74 and inappropriate IRAS activation has been implicated in a variety of animal models of renal disease as both an initial response and a driver for disease progression without increased PRA or plasma AngII. 60,75,76 Similar to systemic RAS, AngII is the major product of the IRAS and activation of the AngII type 1 receptor (AT1R) pathway results in the production of pro-inflammatory mediators, intracellular formation of reactive oxygen species (ROS), cell proliferation, and ECM synthesis, all of which facilitate renal injury. 77 Intrarenal AngII levels are regulated locally and distinct from circulating AngII concentration. 78 –80 All of the components necessary to generate intrarenal AngII are present within the interstitial and intratubular compartments of the kidney involving multiple independent mechanisms, including classic RAS pathways, (pro)-renin receptors (PRRs), and chymase (Figure 2). 60,69,70,81 –110 In addition, intrarenal AngII positively augments other components of IRAS, such as proximal tubular angiotensinogen (AGT), collecting duct renin, and tubular AT1Rs. 69 Alternative processing of AngI or further processing of AngII by other enzymes (chymase, cathepsins, neprilysin, angiotensin-converting enzyme 2 [ACE2]) can lead to the formation of biologically active angiotensin-related peptides (AngIII, AngIV, Ang[1-7], Ang[1-9]) that activate alternative receptors in a series of signal amplification steps (AngII type 2 receptor [AT2R], Mas receptor [MasR], mas-related G-protein–coupled receptor [MrgD], AngII type 4 receptor [AT4R]). 96 Angiotensin II type 2 receptor activation by AngII or alternative angiotensin-related ligands generally counteracts classical AT1R/AngII actions, resulting in vasodilation, natriuresis, and anti-inflammatory tissue-protective effects. 111 The levels of AT2R are normally low in the kidney but may be increased during kidney injury. 111 Angiotensin-converting enzyme 2 is highly expressed in the kidney, where it mediates the degradation of AngII to other angiotensin-related peptides. 112 Studies in rodent kidney models suggest that ACE2 is an important counterregulatory enzyme in IRAS that may protect against renal injury by reducing renal levels of AngII. 112 –114 However, changes in IRAS occur with aging in rats, resulting in activation of the ACE/AngII/AT1R axis and inhibition of the ACE2/Ang(1-7)/AT2R/MasR axis. 66,90
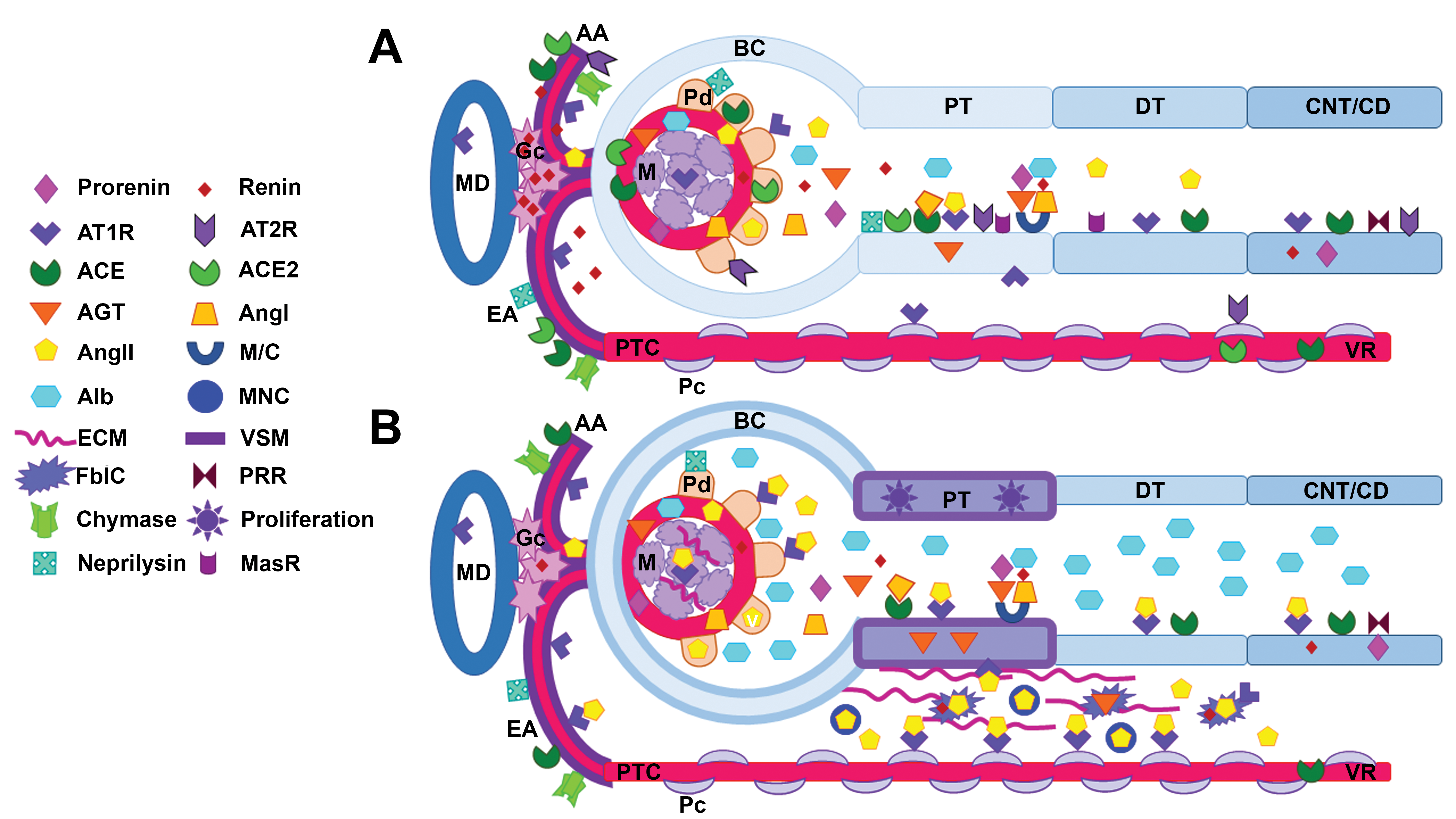
Intrarenal renin–angiotensin system (IRAS) components in (A) healthy young rat and (B) an aged rat with early chronic progressive nephropathy (CPN). Circulating angiotensin II (AngII) precursor molecules (renin, prorenin, and angiotensinogen [AGT]), (pro)-renin receptors (PRR), chymase, and/or neprilysin are necessary to generate intrarenal AngII and angiotensin-related peptides within the kidney. Megalin/cubulin (M/C) transporters in the proximal tubules (PT) can uptake filtered RAS components as well as albumin (Alb). Angiotensin-converting enzyme (ACE) and ACE2 are present in PT brush border and less so in distal tubules (DT). Renin is produced in the granular cells (Gc) in the juxtaglomerular region and afferent arteriole (AA). AngII vasoconstrictor (AT1R) and vasodilatory (AT2R and MasR) receptors are expressed in most tubular segments. Both ATR1 and ATR2 are also present in vascular smooth muscle (VSM), podocytes (Pd), and/or mesangial cells (M). B, In the aging rat with early CPN, IRAS components are altered in amount and/or activity (increased: AGT, AngII, ATR1; or decreased: renin, ATR2, MasR, ACE2) which contribute to interstitial peritubular capillary (PTC) vasoconstriction, leading to local hypoxia and increased oxidative stress. Growth factors and signaling molecules result in characteristic morphologic changes: basophilic PT with thickened basement membranes extending to adjacent Bowman’s capsule (BC), dilated Bowman’s space, podocyte slit diaphragm alterations with increased Alb/protein filtration, increased mesangial matrix, expansion of the peritubular interstitium with increased mononuclear cells (MNC), and extracellular matrix (ECM) synthesis by fibroblast-like cells (FblC). CD indicates collecting duct; CNT, connecting tubule; EA: efferent arteriole; MD, macula densa; VR, vasa recta.
Angiotensin II and renin have been shown to be involved in the progression of kidney disease in a variety of clinical syndromes and in rodent models. 66 Chronic IRAS activation also promotes end-organ damage associated with aging. 68,115 Renoprotection has been achieved by suppressing the IRAS axis in humans and rodents. 81,116,117 Angiotensin-converting enzyme converts AngI to AngII most commonly in the bloodstream, but it may also be produced within the brush border of the renal tubules normally. 60,69,118,119 Increased AngII responsiveness (in association with lowered renal renin and ACE) has been shown in the kidneys of aged rats with lesions comparable to CPN. 33,65,86 Intrarenal RAS activation (increased AT1/AT2 receptor ratio, ACE, and PRR) is evident in Sprague Dawley rats as early as 14 months of age, despite the presence of minimal renal injury, indicating that age-related activation of the IRAS may participate in the development and/or progression of age-related renal fibrosis. 61 Angiotensin II has also been shown to be specifically expressed within the renal interstitium in rodent models of kidney disease, such as the salt-sensitive hypertensive rat and renal ablation rats (but not in normal rats), and is considered integral to their pathogenesis. 60,120,121 It appears that AngII regulation may be critical during both the development and the advanced stages of CPN. This is reflected in studies showing ACE inhibitor administration prolongs the life span of rats and decreases the incidence of aging-related nephropathies. 62,122,123 Infusion of AngII into rats results in direct glomerular injury and proteinuria via binding to its receptors on podocytes. 124 Angiotensin II directly stimulates TGFβ1 and plasminogen activator inhibitor and as such contributes to profibrogenic pathways, leading to expansion of the ECM and interstitial fibrosis. 125 Angiotensin II and renin effects are synergistic. Reduced PRA has been shown to precede glomerular lesions in Fischer344 (F344) rats and severe CPN-related glomerulosclerosis has been noted in rats with the lowest renin values. 33 In a separate study comparing 3-, 12- and 15-month old Sprague Dawley rats, similar decreases in renin were noted with aging. 86 In yet another rat aging model in females, reduced PRA was documented to be due to reductions in renin synthesis within the juxtaglomerular apparatus, and this predisposed the rats to renal damage. 126 In all 3 of the previously mentioned studies, these alterations were limited to local renal tissue as neither increased systemic blood pressure nor changes in renal perfusion pressure were involved. Renin transcription decreased with age in rats in one study, but decreased synthesis and impaired renin release have been noted in several other studies with increasing age, suggesting impaired renin secretion is a primary mechanism. 86,93,127,128 Importantly, in a rodent model of progressive nephropathy, augmented renal AGT protein content and increased interstitial AngII and AT1R expression were observed without alterations in circulating PRA or plasma AngII levels. 60 The increased number of renal interstitial cells expressing AngII and AT1R were especially noted in regions associated with renal damage, which also paralleled lesion severity. 60 This indicates that renal interstitial vasoconstriction and subsequent renal injury can occur with local IRAS alterations, but without any evidence of systemic RAS activation in circulating blood. In another rat model of chronic NOS inhibition, local IRAS activation (increased ACE activity and AngII protein level) was also detected in the renal cortex. 100 These findings from rodent models suggest that local IRAS without systemic RAS activation may be a plausible mechanism involved in CPN development. In support of the relevant findings of increased intrarenal AngII and AT1Rs in rat models of progressive nephropathy, AT1α receptor knockout resulted in marked prolongation of life span in mice with reduced age-related renal changes and less oxidative damage than in wild-type mice with typical signs of age-related nephropathy. 129,130
Intrarenal RAS effects on the kidney extend beyond the interstitium to the glomerulus. It is well established that ACE and/or AT1R inhibitors may slow the course of renal disease, prevent glomerular injury, and decrease proteinuria in human patients as well as in rodent models of nephropathy and aging. 81,116,117,123,126,131 –133 Enalapril significantly decreased aging-related mesangial expansion, glomerulosclerosis, and interstitial fibrosis in mice, indicating a preventative effect on glomerular pathology and CPN severity. 133 However, it turns out that the renoprotective effects of ACE inhibitors related to the glomerulus may be associated with maintenance of the glomerular filtration barrier and slit diaphragm function rather than on effects on hydraulic pressures. 134,135 Podocytes have specific AngII receptors that appear to be involved in redistribution of important proteins in foot processes responsible for maintaining proper filtration diameters. 48,107,136 Hence, proteinuria associated with IRAS activation can be directly related to increased glomerular membrane permeability due to AngII-related altered pedicele molecular composition, with subsequent AngII-stimulated increases in glomerular mesangial matrix. 94,137 –139 Further injury to the kidney as a whole occurs as increased protein is delivered to tubules in the filtrate. 47,140 Taken together, this accumulation of data from a variety of laboratories is strong and provides direct evidence in rodents that IRAS is involved in CPN pathogenesis both in glomeruli and in the tubulointerstitium. Decreased renin and locally increased AngII utilize a series of interconnected signaling pathways to further damage the kidney and result in self-perpetuating stimuli. Although aldosterone is a key component of RAAS and there are documented changes in systemic aldosterone in humans with age, the lack of systemic effects on blood pressure associated with rodent aging or CPN incidence would suggest that aldosterone does not have a major impact on local IRAS in the kidney. 63,66
The kidney has dense sympathetic innervation that closely follows the renal arterial vasculature and forms a network of nerve fibers that not only predominantly innervate the cortex but also extend into the medulla. These fibers have varicosities at neuroeffector junctions as they traverse the vasculature, renin-containing granular cells, and tubules. 141 In the kidney, increased sympathetic tone decreases RBF and GFR, increases renin release, activates RAS, stimulates tubular sodium and water reabsorption, and potentiates glomerulosclerosis and proteinuria. 142 Activation of the renal nervous system can also result in higher levels of intrarenal AGT and AngII, independent of changes in renal function and systemic RAS. 141 Renal denervation as a therapeutic approach for hypertension and chronic renal disease has demonstrated decreased blood pressure and improved renal function in rodent diabetic, ablation, and fibrosis models and in some human clinical trials. 141,143 –147 Higher sympathetic nerve activity has been measured in the heart and liver of elderly humans, but not in the kidney. 148,149 However, the aged rat kidney has a decline in adrenergic responsiveness, a decrease in α-1 and β-2 receptors, and a decrease in sympathetic innervation. 150 –152 Prior to this decline in sympathetic innervation between 12 and 24 months of age, a significant increase in sympathetic innervation was noted between 3 and 12 months of age, suggesting a compensatory mechanism to counteract the impairment in renal function (CPN) that occurs in Sprague Dawley rats between youth and adulthood. 152 However, the role of the sympathetic nervous system in the initiation and/or progression of CPN has yet to be elucidated.
Downstream Mediators of IRAS
Nitric oxide is an important cell signaling molecule that helps modulate vascular tone, insulin secretion, airway tone, and peristalsis and is involved in angiogenesis. Both AngII and NO have antagonistic effects, most notably on vascular tone, but also on vascular remodeling and apoptosis. 153 However, they also regulate each other’s production and function. Angiotensin II stimulates NO release and influences the expression of all 3 NOS isoforms (Figure 1). Nitric oxide downregulates the AT1R and influences renin secretion. In the kidney, the distribution of the NOS isoforms parallels the expression of IRAS components in glomeruli and tubules. Hence, NO-AngII interactions influence both glomerular and tubular functions. 83,153 An imbalance of AngII and NO has been implicated in the pathophysiology of many vascular diseases, with a balance favoring AngII often resulting in oxidative stress injury. 153 In the early stages of renal disease, AngII specifically causes vasoconstriction of the efferent arteriole, leading to reduced blood flow to peritubular capillary network resulting in hypoxia. 154,155 Human CKD and animal models of acute kidney injury (AKI) both demonstrate significant imbalances in activities of AngII and NO, leading to high oxygen consumption and local tissue hypoxia associated with the progression of kidney disease. 156 Nitric oxide levels decrease in the kidney as rats age and this correlates with timing of morphologic aging changes characteristic of CPN. 157,158 In a long-term dietary study in male F344 rats, protein restriction maintained NO production in aged rats similar to young controls and delayed the progression of CPN. The decrease in NO was attributed to an accumulation of nitrogen metabolites in the blood with CPN progression, rather than a shortage of arginine as a substrate for NOS. 157 Dietary nitrate supplementation in aged rats partially compensated for age-related disturbances in renal NO generation and normalized the renal gene expression profile of AngII receptors (AT1/AT2 receptor ratio) without altering plasma renin or AngII. 159 Strain-specific protection against CPN age-dependent renal injury has been described in the F344xBrown Norway male rat and is dependent on maintenance of NOS. F344xBrown Norway rats fail to show signs of glomerulosclerosis or tubulointerstitial fibrosis compared to F344 male rats as they age, and this is associated with higher levels of renal NOS, thus reinforcing the hypothesis that progressive tubuloglomerular injury may be related to renal NOS deficiency. 160 Administration of inhibitors of NOS can result in increased severity of chronic renal disease and diabetic nephropathy in rats and enhanced onset of aging changes in the kidney that are consistent with CPN. 161 Other investigators have shown that decreased NO bioavailability may be a common to both vascular and glomerular dysfunction. 162 Aminoguanidine is an AGE inhibitor, and beneficial effects with long-term treatment include decreased age-related CPN-like lesions in the rat kidney. Advanced glycation end products are known to interfere with NO action and age-associated dilatory impairment could be attributed to NO quenching by vascular AGEs. 163 Strain, age, and disease model–associated alterations in NO/NOS appear to parallel CPN incidence and onset in rodents.
It is well known that CPN lesions are often more prevalent in males than in females and studies in aging rats have shown that androgens are responsible for this difference. 22,164 Interestingly, renal NOS production and/or bioavailability differences were noted between sexes in aged F344 rats, indicating that NO levels in the renal cortex may have an effect on CPN severity. 165 Similarly, renal neuronal NOS (nNOS) abundance and activity declines in the Sprague Dawley male rats with advancing age in parallel with increasing kidney damage, whereas nNOS is maintained in female rats during aging along with attenuated renal damage, suggesting that the renal NO deficiency in the aging male rat may contribute to the age-dependent kidney damage. 166,167 Other examples of sexual dimorphism in the renal NO system have also been reported in humans and rats. 168 Greater endothelial NOS and inducible NOS activity was detected in the renal medulla of female Sprague Dawley rats compared to their male counterparts, which was reduced by ovariectomy in females and unchanged by castration in males. 169 Similarly, in a separate rat model of aging, ovariectomy increased renal injury and this was related to decreased AT2R expression. 170 The ratio of AT2/AT1 receptor in the kidneys of SHR rats is higher in female rats and this corresponds to less severe hypertension and decreased renal lesions. 171 In a rat model of renal ischemia/reperfusion, male rats showed much greater sensitivity to AngII infusions, related to gender differences in RAS vasodilator receptors. 172 Similar gender differences were observed in Wistar rats treated with MasR blockers. 173 These strain-, gender-, and age-associated alterations in NO/NOS and AngII receptors, therefore, appear to be responsible for similar differences in CPN incidence and presentation that have long been recognized by toxicologic pathologists.
The combination of IRAS-mediated effects of AngII and NO/NOS on renal interstitial vasoconstriction can lead to transient epithelial hypoxia. The outer medulla is particularly vulnerable to reduced oxygen supply due to an imbalance between energy demand (active electron transport) and limited medullary blood flow. 174,175 Rats exposed to normobaric hypoxia (10% O2) for 2 weeks produced focal damage in the kidney in the corticomedullary region. 110 The findings were reminiscent of early CPN lesions and were characterized by basophilia, dilated tubules, mild interstitial inflammatory cell infiltrates, and peritubular fibrosis. In addition to ACE which produces AngII, neprilysin also degrades vasoconstrictive peptides including AngII to generate vasodilatory products Ang(1-7). Decreased neprilysin expression and increased local ACE accompanied tubular injury at the outer medullary region in the hypoxic model. This suggests that hypoxia induces a hypertensive phenotype due to an imbalance of vasoactive peptides and that hypoxic injury shares similar mechanistic features with CPN. 110 Other ischemic models also have histologic lesions mimicking CPN, suggesting similar pathogenesis. 97,174,176 –178 It has been proposed that a common pathway of local hypoxia creates a vicious circle that initiates and maintains focal renal injury in these models. Similarly, early hypoxic events may initiate and perpetuate CPN progression in rodents.
Since the introduction of the “chronic hypoxia hypothesis,” 177 an increasing number of studies in animal models and humans have shown an association between hypoxia and chronic renal disease, with early declines in tissue oxygenation suggesting a causal relationship. 174,179 –183 Similarly, hypoxia has been detected in aging rat kidneys, suggesting a causal role in age-related fibrotic renal lesions. Aged transgenic “hypoxia-sensing” rats demonstrated tubulointerstitial hypoxia in the cortex and, to a lesser degree, in the medulla when compared to young transgenic rats. The degree of hypoxia was positively correlated with age-related tubulointerstitial injury and also associated with activation of HIF and upregulation of HIF-regulated genes. 184 Cellular responses to hypoxia are controlled by HIF genes. Downstream target genes are upregulated that protect the cell from hypoxia by promoting neovascularization (vascular endothelial growth factor), erythropoiesis (erythropoietin), and glycolysis (glucose transporter 1) and by decreasing oxidative stress (heme oxygenase 1). 185 Under conditions of renal hypoxia, HIF-1α accumulates in tubules and in papillary interstitial cells, whereas HIF-2α is induced in peritubular endothelial cells and fibroblasts. 180,186 Several factors relevant to renal injury and fibrosis, including NO, tumor necrosis factor α (TNFα), AngII, interleukin-1, insulin, and insulin-like growth factors, have been shown to induce stable HIF-1α. 181,187 –189 Paradoxically, intrarenal HIF signaling initially meant to combat hypoxia may actually contribute to disease progression by promoting interstitial fibrosis. Deletion of HIF-1α in tubular epithelial cells has been shown to reduce fibrosis in the unilateral ureteral obstruction model of CKD. It is thus increasingly clear that renal epithelial HIF-1 signaling is intimately associated with the development of chronic renal disease and renal fibrosis, by increasing expression of ECM-modifying factors and by facilitating EMT. 190
Kidney ischemia/hypoxia can be further influenced by changes in the rate of oxygen consumption associated with CKD progression and renal tubule damage. 191,192 Intrarenal hypoxia has been shown to be present before structural changes of kidney disease. 181 Increased oxygen demand has been noted in animal models of renal mass reduction, hypertension, and diabetes, all of which can be ameliorated by IRAS blockade or HIF activation. 193,194 Rats were administered dinitrophenol (a mitochondrial uncoupler) for 30 days, which resulted in intrarenal tissue hypoxia, increased urinary protein excretion, vimentin expression in tubular epithelium, and infiltration of inflammatory cells. 195 Acute NOS inhibition or chronic AngII infusion increased renal oxygen consumption in normal animals and resulted in similar renal pathology. 193,196 In a 5/6th remnant kidney model, reduced renal NO was associated with high AngII activity. 158,197,198 Angiotensin II inhibition normalized the increase in oxygen consumption, suggesting that AngII regulates oxygen utilization independent of hemodynamic effects. 193 In a separate experiment in adult Wistar rats, AngII-induced oxidative stress resulted in defective renal oxygenation. 196 The close relationship between intrarenal NO/NOS and AngII signaling pathways has been previously highlighted, but these close interactions therefore also extend to hypoxia/HIF signaling pathways, and activation or inhibition of one pathway immediately affects others. Hence, all are likely to be involved in CPN pathogenesis despite the lack of direct evidence from pharmaceutical toxicity studies.
Reactive oxygen species can be generated throughout damaged kidneys. 199 Pericytes surrounding the vasa recta in the renal medulla are AngII responsive and, under IRAS activation, result in vasoconstriction leading to ischemic generation of ROS by damaged tubules. 199 There appears to be a close intercellular communication between the TAL and the vasa recta pericytes which involve ROS and NO scavenging. 199 The TAL constitutively produces ROS, which is normally kept in check by local interstitial NO production. 199 However, ROS and AngII interactions are not limited to the interstitium. With glomerular injury, podocytes can undergo increased ROS production, resulting in increased local IRAS amplifying tubuloglomerular injury. 199,200 Nuclear and mitochondrial AngII receptors have been identified in renal cells that modulate intracellular oxidative stress. 101,201 Angiotensin II induces excessive release of vascular superoxide radicals generated by stimulation of nicotinamide adenine dinucleotide/NADphosphate oxidase that inactivates NO. 202,203 Inhibition of AngII formation attenuates mitochondrial oxidative stress and, through NO, improves vasodilation. 204 This has downstream effects on inflammation, cytokine and growth factor production, fibrosis, and hence on overall renal function. 66
Angiotensin II affects many genes and proteins, but investigators have attempted to find the initial effectors in kidney. After AngII administration to mice for 7 days, a single gene, fatty acid binding protein 1 (Fabp1), was identified that was associated with the very earliest stage of AngII-induced renal injury. 205 Fatty acid binding protein 1 plays a critical role in renoprotection through regulating oxidative stress and free fatty acid metabolism, and it has been shown to be a good urinary biomarker for tubular injury in both humans and rodent models. 206 A transgenic mouse model that overexpressed human Fabp1 in proximal tubules demonstrated reduced oxidative stress and tubulointerstitial damage following either AngII administration or unilateral ureteral obstruction, further supporting the close association between AngII-induced oxidative stress and renal injury. 207,208 A few animal models have shown improvement when antioxidants were initiated prior to the development of renal injury. 209 Zucker Diabetic Fatty rats demonstrated augmented oxidative stress and enhanced intrarenal AGT, which preceded renal injury and was ameliorated by antioxidant treatment. 210,211 In a recent study in aged mice, resveratrol antioxidant treatment for 6 months not only attenuated oxidative stress but it also decreased inflammation and fibrosis in the aging kidney and modulated age-associated changes in IRAS components. 109,212,213
Inflammation and Fibrosis in CPN
Alterations in IRAS from a variety of causes will lead to renal interstitial vasoconstriction, which may explain the variety of other factors that can hasten the onset or severity of CPN in rodents. Endothelial cells in the interstitial microcapillaries, especially in the outer medulla and corticomedullary area, undergo activation after ischemic insult. 176 This results in loss of vascular patency and upregulation of leukocyte adhesion molecules. Under these circumstances, AngII is able to activate many intracellular pathways that induce cellular proliferation and amplify the inflammatory response in mesangial, glomerular, and tubular tissues (Figure 1). 214 Reactive oxygen species also augment ischemic epithelial damage and amplify these inflammatory signals. The tubule epithelial cells in the pars recta are particularly sensitive to ischemia 215 and, under conditions of interstitial vasoconstriction and AngII stimulation, release a variety of mediators, chemokines, and pro-inflammatory cytokines such as TGFβ1, monocyte chemoattractant protein 1 (MCP-1), TNFα, and interleukin 6, which amplify the inflammatory response. 66,216 In addition, increased proximal tubule MCP-1 mRNA and protein expressions are upregulated by exposure to levels of albumin equivalent to those seen in proteinuria of CPN and CKD, which would further enhance interstitial inflammation. 217 There is a pronounced paracrine effect of TGFβ1 on the surrounding interstitium, resulting in conversion of resident fibroblasts to myofibroblasts and synthesis of profibrotic growth factors such as PDGF and connective tissue growth factor (CTGF) leading to the production of collagens. 218 –220 If tubule degeneration is severe enough to result in separation of the basement membrane and disruption of laminar adhesion molecules attached to tubule epithelium, TGFβ1, PDGF, and CTGF may also promote EMT, resulting in the generation of additional local myofibroblast populations and enhanced interstitial fibrosis. 221 –224
Chronic progressive nephropathy is associated with high levels of albumin in the urine filtrate and the protein itself may augment degenerative processes. Angiotensin II has been shown to activate nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) in the kidney. 225,226 Both AngII and endothelin-1 (ET-1) have been shown to increase NF-κB activity in vivo and in vitro in rat proximal tubules exposed to albumin overload and this activation was blocked with AngII and ET-1 receptor antagonists. 227 In fact, NF-κB activation occurs very quickly (24 hours) following bovine serum albumin administration to rats and prior to the appearance of renal infiltrating cells. 227 Nuclear factor κ-light-chain-enhancer of activated B cell activation leads directly to RAS activation in proximal tubule cells and is mediated by the activation of NADPH oxidase and ROS formation. 228 In a rat model of albumin overload, renal AngII was increased in a NF-κB-dependent manner via AGT induction and decreased ACE2. 229 An IRAS-positive feedback mechanism has been postulated and NF-κB may be involved in this response. 230,231 Further, inhibitors of NF-κB have been shown to reduce tubulointerstitial injury in proteinuric rat models. 232 This is due in part to well-known NF-κB effects of induction of pro-inflammatory cytokines 233 but also reflects the important role NF-κB plays in IRAS activation. This suggests that NF-κB may be responsible for perpetuating the IRAS signal in rodents with CPN and may be involved in the initial stimuli, leading to IRAS activation that is important early in this disease.
Inflammation is variably present in kidneys from rodents with CPN, and the nature of the infiltrate differs widely, although lymphocytes and macrophages predominate. Expression levels of molecules associated with the activation of inflammasomes and their downstream signaling pathways are markedly increased during the renal aging process in rats. 234 Stimulation of the NF-κB, TGFβ1, and mitogen-activated protein kinase pathways may be ultimately responsible for pro-inflammatory signals at the molecular level. However, there are a host of stimuli which can help recruit leukocytes to the renal interstitium, including hypoxia, disrupted basement membranes, tubule degenerative products (proteins, lipids, etc), and stimulation of parallel pathways. 233,235 As noted previously, protein overloading at the tubule interface will invariably result in interstitial inflammation through NF-κB signaling, regardless of the type of glomerular injury. 140,229 This effect of proteinuria would be limited to the later chronic stages of CPN where casts are prominent. Inflammation plays a major role in AKI and progressive renal disease across species and therefore may have a limited role in early CPN pathogenesis by amplifying other degenerative signals. 216,235 Inflammation is likely to increase the severity of epithelial and basement membrane damage, especially if neutrophil proteases are involved, and leukocytes secrete cytokines and ROS that will also augment the ROS load. 236 Importantly, inflammation will amplify the profibrotic response in the interstitium, resulting in increased collagen deposition. 237
The Inciting Cause of CPN
This review is not intended to define specific steps initiating CPN or what would be considered the “ultimate cause” at the biochemical or chromosomal level. Given the difference in phenotype and onset between strains, it is likely that there are multifactorial genetic or epigenetic predispositions to CPN. Since there are no known associated disease states involved in initiating lesions, this is further evidence that CPN has a genetic basis or predisposition. In most rat strains, a large majority of males will have significant features of CPN by 20 months of age. In that sense, CPN could be considered a consequence of aging, but given the differences in age of onset, phenotype, sex, and strain susceptibility, and the variety of environmental, dietary, or toxic factors that modify its expression and onset, it should be stressed that CPN is a disease process and not simply an aging change associated with kidney senescence. In the aging rat kidney without evidence of CPN, histologic alterations are limited to simple thickening of glomerular and tubule basement membranes without alterations to glomerular mesangial cells or tubule epithelial cells. 238
Although progression factors such as diet, metabolism, and drug administration can alter the age of onset and severity of CPN, they are unlikely to be related to the actual “initiating cause.” Mechanisms involved in clinical renal aging have been identified and include genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, altered intercellular communication, and stem cell exhaustion. 239,240 It is unclear if any or all of these play a role in CPN initiation in rodents, but it is likely that genetic or epigenetic effects on any of these functions would predispose rodents to earlier onset of CPN. There is now an established link between epigenetic alterations noted with aging and renal dysfunction. 241 Certain genes have been identified that are highly responsive to epigenetic modification. 242,243 The regulation of aging processes is common across species and demonstrates epigenetic changes influenced by nutrition, inflammation, gut microbiome, and environmental factors. 241 In a recent study of aged rats given an inhibitor of mammalian target of rapamycin complex 1 (mTORC1), decreased CPN severity and renal inflammation corresponded with decreased c-Myc expression as compared to kidneys of untreated aged rats. 242 Interestingly, Myc haploinsufficient mice have increased life span, decreased age-related pathology, and decreased activation of mammalian target of rapamycin pathways compared to wild-type mice. 243 These studies suggest that these 2 genes, mTORC1 and c-Myc, may be involved in regulating age-related renal changes through effects on epigenetic mechanisms. 242
Other specific genes responsible for CPN in rats have not yet been identified, nor have there been extensive genomic comparisons of rats with CPN to age-matched lesion-free controls to prove the pathways described here are valid. However, these data may not be as helpful as one might think. Transcriptomic data from human kidneys showed remarkable similarity between renal aging and those associated with CKD. 244,245 Transcriptomic data from a wide range of renal studies in rats showed similar results that tend to be nonspecific as to disease process, and hence, proteomic or genomic data from renal studies are not particularly enlightening as to mechanism. 246,247 In short, degenerative kidneys tend to show similar molecular changes across species and the pathways involved generally overlap regardless of cause.
In this article, we described evidence for multiple proteins and molecular and cellular pathways that are involved in the pathogenesis of CPN in the degenerative phase in rodents. The initiation of CPN at the genetic level remains a mystery. At present, we could find no evidence in the literature for a gene or set of genes that are definitively responsible. To avoid the pitfalls of previous genomic studies of CPN pathogenesis, renal sampling should be undertaken in the degenerative phase of the disease when mechanistic pathways do not overlap with other kidney diseases. Although future experimental work is necessary to elucidate the inciting cause at the chromosomal level, temporal investigations in the most susceptible rat strains beginning very early in CPN development (4-6 weeks) would be required. Subsequently assessing genomic and proteomic differences compared to unaffected or resistant strains may provide a list of suspect genes. These investigations might include (1) whole-genome sequencing, (2) mRNA expression analysis of subgross regional and/or laser microdissected specific renal structures and subsequent in situ hybridization, (3) quantitative protein analysis of the same kidney regions and subsequent immunohistochemistry, and (4) intrarenal blood flow and perfusion measurements via advanced imaging modalities. In fulfillment of Koch’s postulate, gene knockout/knockin experiments could definitively identify potential genes of interest. This proposed work will not be a trivial undertaking but could further affirm the rodent specificity of this disease entity.
Conclusion
Despite the dogma in toxicologic pathology literature that CPN pathogenesis is unknown and controversial, there appears to be abundant data tying similar histologic alterations to specific molecular pathways in aged rats. We have identified 5 key proteins in rodents with CPN linked to intrarenal events, involving vasoconstriction, hypoxia, and ROS generation. Decreased renin, increased AngII, decreased NO/NOS, increased NF-κB/TGFβ1, and increased hypoxia/HIF pathways within the renal interstitium appear to be critical inflexion points in the early degenerative phase of the disease (Figure 1). Based on an extensive review of the available literature, it seems highly likely that local IRAS alterations (Figure 2) may be critical to CPN progression. Similar pathways are shared between CPN and ischemic animal models of kidney disease where IRAS has demonstrable involvement. Interactions of many intersecting pathways form the basis of a positive feedback loop involving local vasoconstriction and ischemia, (pro)-renin/AngII release, and renal parenchymal oxidative stress injury that are responsible for the progressive nature of the disease. Downstream effectors such as growth factors and pro-inflammatory signals amplify the injury and promote scarring in both glomerular and tubulointerstitial compartments.
Although several of the proteins and pathways noted in CPN are similar and shared with those in human renal diseases and respective animal models, there are important distinctions. For instance, primary drivers for renal injury associated with diabetic nephropathy in humans or rodents involve cytotoxic effects of glucose overload on the tubule epithelium and downstream sheer stress responses in the glomerular mesangium, 248,249 which are lacking in CPN. Hypoxic responses in AKI in humans result in dramatic, severe oxygen depletion that occurs in minutes or hours, whereas the vasoconstrictive changes and HIF-1α-related responses in CPN are much more subtle and localized to the renal interstitium, occurring over several weeks to months. 250 Peracute vascular compromise also occurs in several rodent models that differ dramatically with those in CPN. 251,252 Drug-induced kidney injury (DIKI) in human patients or in renal models, such as in rodents given cisplatin, doxorubicin, or gentamicin, is associated with overt necrosis, apoptosis, and/or degeneration of the renal tubule epithelium as a primary event. 247 Effects on mitochondrial oxidation, adenosine triphosphate synthesis, membrane stability, or metabolism are important initiating alterations in DIKI, 253 which, if they occur at all in CPN, are limited to very late in the course of the disease in individual tubules. Interstitial nephritis is a primary inflammatory disease, and both the severity/distribution and consequences of the inflammatory response fundamentally differ by an order of magnitude. 254,255 Although the primary location of molecular events is shared (interstitium), the histologic picture and molecular pathogenesis fundamentally differ between CPN and interstitial nephritis. Most importantly, there are clear differences in the pathogenesis of human CKD, from CPN, because the changes in CKD are related to systemic involvement of the RAAS. 256 Although local IRAS is key to CPN pathogenesis, changes in AngII and renin levels are limited to the local renal interstitial environment and, as we have noted previously, are not associated with any systemic alterations in blood pressure or changes in plasma levels of angiotensin, renin, or aldosterone. This highlights the pronounced difference in causal origin of the 2 diseases (hypertension vs genetic/epigenetic factors).
The MOA associated with the particular molecular alterations related to IRAS is limited to the degenerative changes of CPN and separates from the later proliferative pathways that are responsible for tubule hyperplasia and tumor formation in rodents. Hence, the previously published MOA for CPN tumor pathogenesis remains valid, and the information assimilated here simply augments the information on CPN pathophysiology. Although the ultimate cause of CPN appears to be at the genetic/epigenetic level, specific genes responsible for disease initiation remain undetermined.
Animals
Virtually all studies referenced in this review were conducted externally. All internal animal procedures were conducted in an American Association for the Accreditation of Laboratory Animal Care–accredited facility at GlaxoSmithKline (GSK) in accordance with GSK policies on the care, welfare, and treatment of laboratory animals, and they were reviewed and approved by GSK’s Institutional Animal Care and Use Committee as appropriate.
Footnotes
Acknowledgments
The authors thank Daniela Ennulat, Jessica Caverly-Rae, and John Kreeger for their helpful review and suggestions concerning the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential, real or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.