
Abstract
A therapeutic option for monogenic disorders is gene therapy with ex vivo-transduced autologous hematopoietic stem cells (HSCs). Safety or efficacy studies of ex vivo-modified HSCs are conducted in humanized mouse models after ablation of the murine bone marrow and transfer of human CD34+ HSCs. Engrafted human CD34+ cells migrate to bone marrow and differentiate into various human hematopoietic lineages. A 12-week study was conducted in NSG™ mice to evaluate engraftment, differentiation, and safety of human CD34+ cells that were transduced (ex vivo) with a proprietary lentiviral vector encoding a human gene (BMRN-1) or a mock (green fluorescent protein) vector. Several mice intravenously injected with naive CD34+ cells or transduced CD34+ cells had variable lymphohistiocytic inflammatory cell infiltrates and microgranulomas in the liver and lungs consistent with graft-versus-host disease (GVHD). Spleen, bone marrow, stomach, reproductive tract, but not the skin had similar inflammatory changes. Ex vivo viral transduction of CD34+ cells did not impact engraftment or predispose to xenogeneic GVHD.
Genetically modified, autologous hematopoietic stem cells (HSCs) represent a new and promising therapeutic approach to treat genetic diseases. Recently, the first gene therapy product in this class was approved by the European Medicines Agency for the treatment of patients with severe combined immunodeficiency due to adenosine deaminase deficiency (Carriglio et al. 2017). Preclinical studies to support clinical trials for ex vivo-modified autologous HSCs rely on transplantation of transduced CD34+ cell in humanized immunodeficient mouse models like the NSG™ mouse that lack T/B lymphocytes and NK cells (Shultz et al. 2005). A preclinical safety study was conducted in female NSGTM mice to characterize viability, engraftment, and biodistribution of human G-CSF mobilized peripheral blood (MPB) CD34+ cells from healthy donors that were either transduced (ex vivo) with a proprietary lentiviral vector or a mock vector coding for green fluorescent protein (GFP). In this report, we describe microscopic findings consistent with graft-versus-host disease (GVHD) that were observed in NSG mice injected with transduced as well as nontransduced CD34+ cells.
The study comprised an ex vivo phase and an in vivo phase. In the ex vivo phase, MPB CD34+ HSCs were obtained by leukapheresis from healthy human donors under full informed consent. Human MPB CD34+ cells were transduced with the proprietary vector BMRN-1 (test article) or transduced with an unrelated vector carrying an expression cassette for a GFP. In the in vivo phase (Table 1), CD34+ cells (transduced and nontransduced) were transplanted to NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) (NSG™); supplied by Charles River Labs, Bar Harbor, Maine, USA). This phase was conducted at the Genethon animal facility at CERFE (Evry, France) in accordance with all national (France) and European ethical guidelines using 9-week-old mice. To allow engraftment of human CD34+ HSCs (transduced or nontransduced), mice were irradiated using an X-ray source (Faxitron CP160) at a rate of 0.84 Gy/min two to three hours prior to transplantation. Cells were administered intravenously by retro-orbital injection. A cohort of untreated NSG™ mice or mice transplanted with umbilical cord blood CD34+ cells (untransduced) served as controls. The presence of human cells (chimerism) in the recipient mouse organs was detected by qPCR for the ALB gene and also by flow cytometry for human CD45 antigen in blood and tissues at 8 and 12 weeks posttransplantation. Mice were euthanized 12 weeks after transplantation, and a complete set of tissues were collected and processed for histologic examination.
Experimental Groups and Treatment.
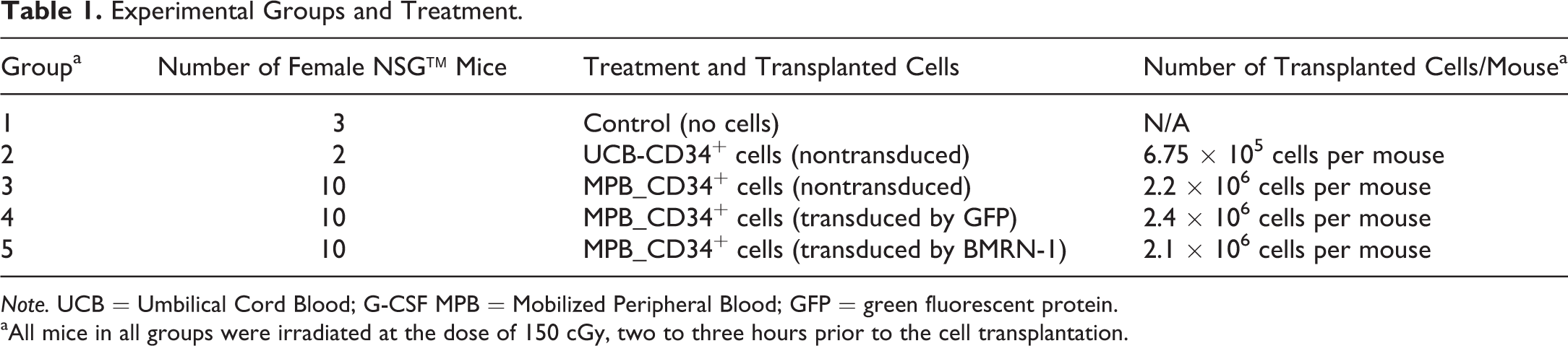
Note. UCB = Umbilical Cord Blood; G-CSF MPB = Mobilized Peripheral Blood; GFP = green fluorescent protein.
aAll mice in all groups were irradiated at the dose of 150 cGy, two to three hours prior to the cell transplantation.
Three mice that were euthanized early between day 40 and day 60 had microscopic features consistent with GVHD (described below). Engraftment efficiency expressed as percentage of human/total cells (chimerism) in blood and hematopoietic organs was at the expected levels for human/NSG mouse chimeras with slightly higher engraftment in the lymphoid organs (at 8 and 12 weeks). Engraftment was similar among the groups indicating transduction with a vector (BMRN-1 or GFP) had no impact on engraftment and differentiation capacity of human CD34+ cells in vivo. Histopathologic evaluation also confirmed the expected engraftment with lymphoid follicles in the spleen and a diffuse infiltrate of lymphocytes in thymus compared to naive NSG mice that had mostly stromal cells in the thymus and no lymphoid follicles in the spleen (Figure 1A–D). Human T and B lymphocytes were confirmed in the spleen and thymus of all mice (transplanted with HSCs) by IHC staining of tissue sections for the CD3 and CD20 markers (data not shown). There were no T or B cells in spleen and thymus from nontransplanted irradiated NSG™ mice. In addition to these expected microscopic features, several NSG mice transplanted with nontransduced cells as well as transduced (GFP or BMRN-1) CD34+ cells had minimal to occasionally moderate inflammatory changes in the lungs, liver, and spleen. Inflammatory changes varied from minimal to moderate perivascular, periportal, or peribronchiolar infiltrate of lymphocytes and macrophages in the liver and lungs to focal aggregates of epitheliod macrophages (microgranulomas) (Figure 2A–D). The latter finding (microgranuloma) was also present in the spleen with multinucleated giant cells. Less frequently hepatocellular apoptosis/single-cell necrosis, submucosal lymphocytic infiltrates in the gastrointestinal tract, uterus/vagina, and hypocellular bone marrow with large macrophages were present in a few mice. Overall, the incidence of inflammatory changes in individual tissues and across the three (groups 3, 4, and 5) cohorts was variable. Hepatic changes were present in 6/10, 4/10, and 5/10 mice in groups 3, 4, and 5, respectively, while the inflammatory changes in the lungs was observed in 2/10, 1/10, and 5/10 mice in groups 3, 4, and 5, respectively. Splenic microgranulomas were present in 5/10, 3/10, and 8/10 animals in groups 3, 4, and 5, respectively. A notable observation in this study was the lack of inflammatory changes in the skin in any treatment group, a microscopic feature reported in a few GVHD studies (Lockridge et al. 2013; Sonntag et al. 2015). There were no in-life observations of cutaneous changes as well.
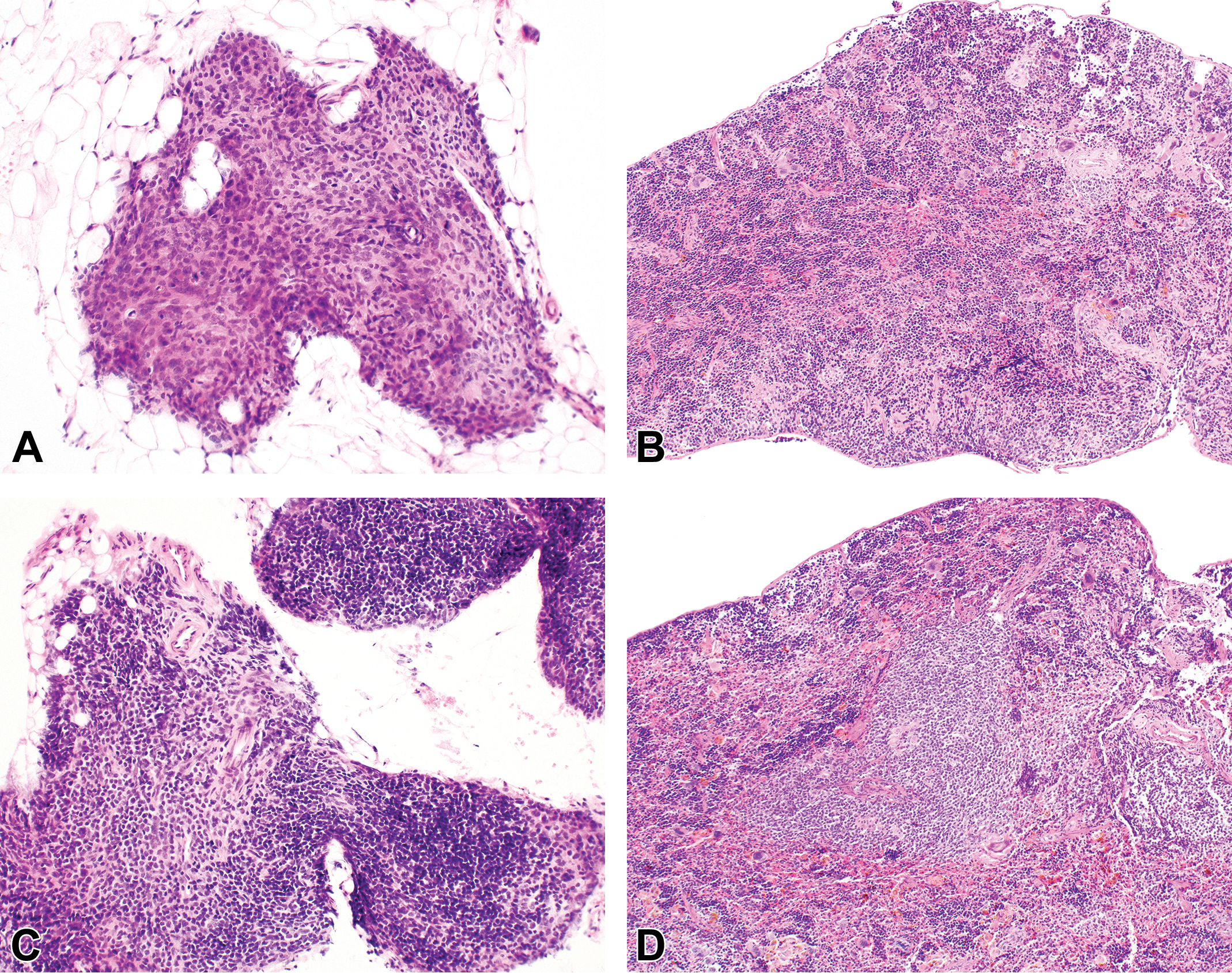
Thymus and spleen from naive (A and B) or human CD34+ cells transplanted (C and D) NSG™ mice. Thymic lobe from a naive (no CD34+ cell transplantation) NSG™ mouse is primarily composed of stromal cells (A). Engraftment of lymphocytes in a NSG™ mouse transplanted with human CD34+ hematopoietic stem cells (C). Lymphoid follicles in the spleen of a NSG™ mouse after transplantation of human CD34+ cells (D) compared to naive NSG mouse with no lymphoid follicles (B). Hematoxylin and eosin.
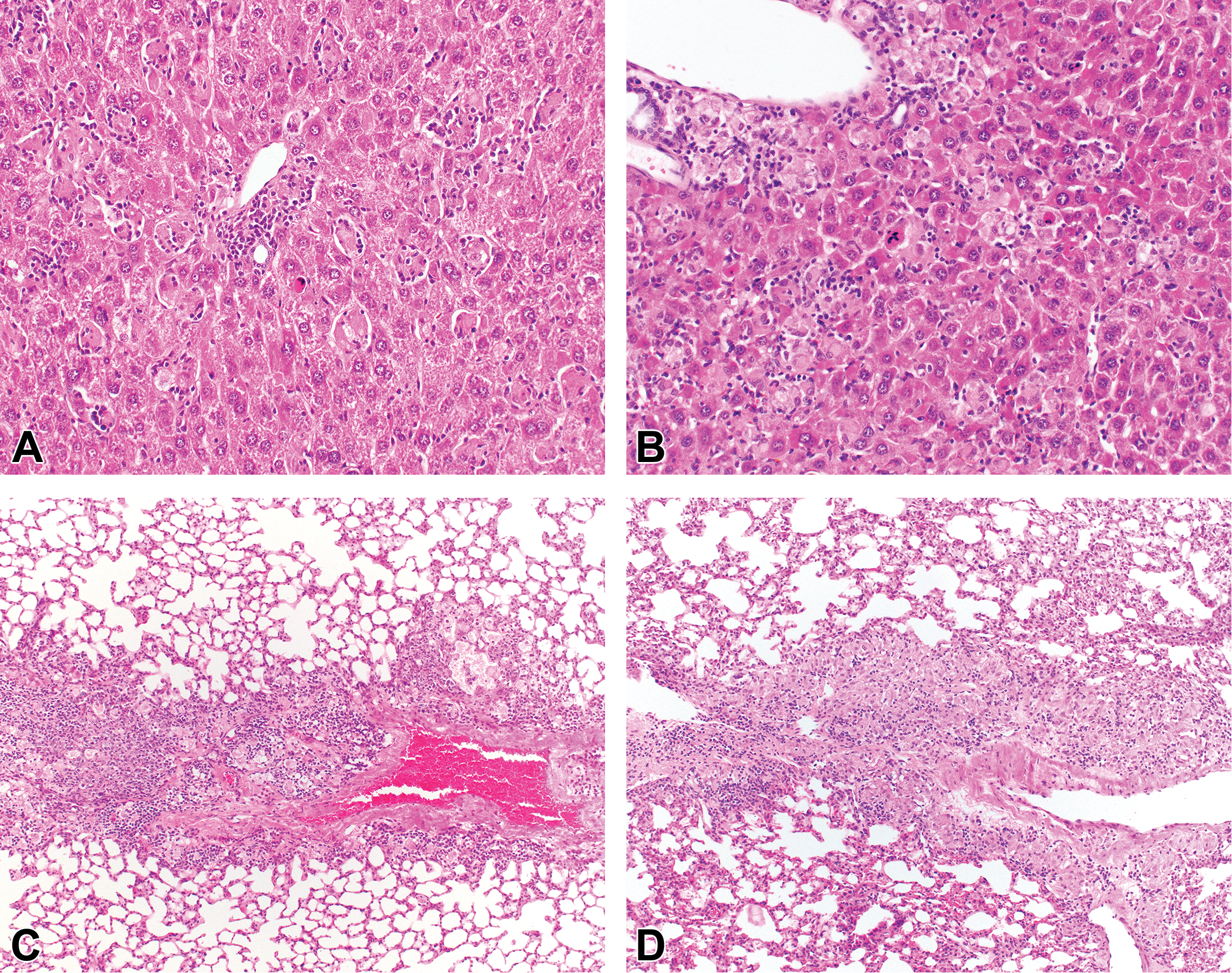
Graft-versus-host disease in the liver and lungs of NSG™ mice transplanted with nontransduced CD34+ cells (A and C) as well as CD34+ cells transduced CD34+ cells (B and D). Inflammatory changes are similar between mice injected with transduced or nontransduced CD34+ cells with perivascular, periportal, or peribronchiolar infiltrate of lymphocytes and macrophages. Note portal microgranulomas and hepatocellular apoptosis/single-cell necrosis. Hematoxylin and eosin.
NSG mice are often the recipient strain to generate mice with a “humanized” immune system. Engrafted (human CD34+) cells migrate to bone marrow and differentiate to all lineages of the mature immune system in these mice (Shultz et al. 2005). GVHD in the liver and lungs similar to that observed in our study have been reported in NSG™ mice transplanted with human PBMC or human bone marrow, fetal liver, and thymus (BLT-NSG model; King et al. 2009; van Rijn et al. 2003; Greenblatt et al. 2012; Lockridge et al. 2013) and in mice transplanted exclusively with CD34+ cells similar to our study (Sonntag et al. 2015; Fujii et al. 2015). Although the inflammatory changes in the liver and lungs were typical of GVHD reported in humanized mouse models, the inflammatory changes in other tissues (GIT/reproductive tract) observed in the current study perhaps reflect variants of the same disease entity. Cutaneous changes in mice reported previously were not evident in the current study as skin lesions may take longer to develop (mice in the current study were euthanized 12 weeks after transplantation). GVHD, although well-known and often elicited intentionally to study the pathogenesis of the disease, has not been reported in preclinical safety (or efficacy) gene or cell therapy studies utilizing NSG mice transplanted with CD34+cells (Scaramuzza et al. 2013; Visigalli et al. 2016; Carriglio et al. 2017).
The occurrence of GVDH depends on several factors including strain susceptibility, environmental pathogens, irradiation dose or chemotherapy (chemical ablation), subset and numbers of infused donor T cells, source/dose of transplanted CD34+ cells, and ex vivo manipulations, if any (Gorin et al. 2002; MacDonald, Hill, and Blazar 2017). There is no correlation between the number of CD34+ cells injected and the incidence of GVHD and also between the percentages of CD3+ T cells in the peripheral blood of NSG mice and the gender of the donor.
Translation and extrapolation of these murine preclinical findings to human safety should be considered by pathologists and toxicologists involved in the design of these gene therapy studies, and it is crucial to validate all new models and to be aware of caveats when interpreting and translating experimental data. Whereas the clinical program is intended for autologous transplantation, the preclinical study was a xenogeneic transplantation of human CD34+ cells to NSG™ mice.
Footnotes
Acknowledgments
We would like to thank the assistance provided by Ms. Valerie Ackley, Mr. Todd Oppeneer, and Ms. Paula Larkin at BioMarin Pharmaceutical Inc. In addition, we would like to thank the key scientists at Genethon (Drs. Fulvio Mavilio and Sabine Charrier) and Biodoxis (Dr. Virgile Richard).
Author Contribution
Authors contributed to conception or design (SC, PC, CF, CO); data acquisition, analysis, or interpretation (SC, PC, CF, CO); drafting the manuscript (SC); and critically revising the manuscript (SC, PC, CF, CO). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential, real, or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.