
Abstract
Thorough morphologic evaluations of medical devices placed in or near the nervous system depend on many factors. Pathologists interpreting a neurologic device study must be familiar with the regulatory framework affecting device development, biocompatibility and safety determinants impacting nervous tissue responses, and appropriate study design, including the use of appropriate animal models, group design, device localization, euthanasia time points, tissue examination, sampling and processing, histochemistry and immunohistochemistry, and reporting. This overview contextualizes these features of neurologic medical devices for pathologists engaged in device evaluations.
Keywords
Medical devices applied to the nervous system are used to treat an ever-expanding array of disorders such as epilepsy (Jobst et al. 2010), movement disorders such as Parkinson’s disease (Fang and Tolleson 2017), migraine (Riederer, Penning, and Schoenen 2015), stroke (Dawson et al. 2016), pain (Wolter 2014), tinnitus (Tyler et al. 2017), and genetic diseases including enzyme replacement therapy for lysosomal storage diseases (Vuillemenot et al. 2016). Non-neurologic diseases have also been targeted for treatment using neurologic medical devices, including obesity (Gobel, Tronnier, and Munte 2017), rheumatoid arthritis (Koopman et al. 2016), sleep apnea (Strollo et al. 2014), chronic heart failure (De Ferrari et al. 2011), fibromyalgia (Lange et al. 2011), and many others. Burgeoning technologic expansion in neurologic medical device development (Parastarfeizabadi and Kouzani 2017; Jackson and Zimmermann 2012; Capogrosso et al. 2016; Nishimura, Perlmutter, and Fetz 2013; Adewole et al. 2016; Zhao 2017) will bring an ever-increasing variety of such devices in preclinical studies to be routinely evaluated by pathologists.
Neurologic medical devices employ various modes of therapy delivery. Devices can be “active” devices that produce a therapeutic property (i.e., electrical or thermal energy) or “passive” devices that solely deliver medications or drain cerebral spinal fluid (i.e., catheters coupled with pumps). The terms “active” and “passive” are used throughout this review to provide a framework for the discussion of neurologic devices. However, not all devices fit into these categories. For example, a combination device designed to deliver both electrical stimulation and drug therapy has both active and passive roles; this review does not explore the intricacies of combination devices in depth.
Before beginning assessment of a neurologic device, the pathologist should become familiar with the specific features of the device (i.e., dimensions and design), the method of device implantation, and the therapy intended to be delivered. These factors can influence tissue responses. Pathologists evaluating preclinical animal studies for neurologic medical devices should be familiar with current preclinical toxicologic neuropathology methods (see the January 2011 issue of Toxicologic Pathology that focuses on this topic). Test article effects and relevance are best determined by the study pathologist (Mann et al. 2012). This article provides an overview of general features of neurologic medical devices pertinent for successful biocompatibility and/or safety assessments.
Regulatory Framework
Because of the many types of devices and therapies, there is no specific sequence of preclinical studies suitable to obtain regulatory approval. Biocompatibility testing focuses on identifying tissue reactions incited by device components. Safety assessment focuses on evaluating the effects of the therapy administered via the device. In the United States, the Food and Drug Administration (FDA) classifies devices into Classes I–III, dependent on risk associated with the device. Most neurologic medical devices are considered FDA Class II (external devices which pose risks from use, i.e., transcutaneous electrical nerve stimulation units) or Class III (implantable devices, i.e., deep brain stimulator [DBS] systems) based on their moderate to high risk, respectively (FDA 2018). Biocompatibility and safety risks should be identified and may include hypersensitivity to materials, chemical toxicity, mechanical damage, and potential changes in material composition due to manufacturing processes, in vivo degradation, or electrochemical reactions, such as metal dissolution secondary to charge-driven processes (Cogan et al. 2004).
Because of the risks associated with devices, FDA guidance suggests evaluations of medical devices should be performed within a risk management framework informed by the International Organization for Standardization (ISO) Guidelines 10993-1 and ISO 14971 (ISO, I.O.F.S 2007, 2018). In addition, a toxicity risk assessment that includes chemical characterization of the finished device, ISO 10933-17, should guide biocompatibility and safety testing and should be accessible to the pathologist (ISO, I.O.F.S 2002).
Biocompatibility testing (not safety testing) is directed by ISO 10993, and the FDA has issued a guidance on the use of ISO 10993 (FDA 2016). ISO Guidelines 10933-6 are particularly pertinent as they focus on the evaluation of local effects at the device–tissue interface in or near the nervous system (ISO, I.O.F.S 2016). ISO-10993-4 is applicable to neuro-interventional devices that only contact the vessel wall, not nervous tissue directly (not discussed further here [ISO, I.O.F.S 2017]). In the case of neurologic medical devices, biocompatibility testing should include implants placed in clinically relevant locations (brain, spinal cord, and nerves), though supplemental muscle or subcutaneous implants may be helpful in some cases (FDA 2016). In addition to biocompatibility testing, any preclinical safety studies evaluating effects of delivered therapies are an important component of the regulatory process and should be provided to the evaluating pathologist, if available. In the United States, depending on the primary mode of action, a device may be reviewed by the FDA Center for Devices and Radiological Health, the FDA Center for Drug Evaluation and Research, and/or the FDA Center for Biologics Evaluation and Research.
Device Features Impacting Biocompatibility and Safety
Device biocompatibility assessment is typically performed without active therapy delivery, and the tissue responses in such studies result from the device materials and/or trauma due to surgical implantation. Cleaning and sterilization techniques can also impact biocompatibility. Device safety assessments evaluate tissue changes associated with the delivered therapy. Neurologic device biocompatibility and safety depends on many factors including device materials, design, and use (Sahyouni et al. 2017b; He and Bellamkonda 2008; Helmus, Gibbons, and Cebon 2008; Grill 2008; Adewole et al. 2016). Close collaboration between the groups responsible for these various factors is essential in the process of development, approval, and postmarket surveillance of devices.
Device Materials
In the context of the currently available neurologic devices and their uses, an ideal device material would be soft, nontoxic, noncarcinogenic, nonantigenic, nonmutagenic, nonpyrogenic, pro-healing, and (potentially) electrically conductive (Kim et al. 2008; Helmus, Gibbons, and Cebon 2008; FDA 2016). Currently, an ideal material does not exist.
Without an ideal material, many approaches are used to maximize biocompatibility and safety while deploying the desired therapy. The selection of appropriate neurologic medical device materials depends on application. For instance, since a common application of neurologic devices is electrical stimulation, materials can often be categorized as to whether they are nonconductive or conductive. Following are some lists of commonly used materials and coatings. Discussion of each is beyond the scope of this article, but prior to assessing a medical device, the pathologist should become familiar with reactions that have been reported to occur in response to the device components.
Intraparenchymal/intrathecal catheters that do not need to conduct electricity can be composed of nonconductive materials such as polyurethane (Hovland et al. 2007; Felice et al. 2011; MacAllister et al. 2016), polyethylene (Yaksh et al. 2003), or silicone (Nagel et al. 2017). In contrast, electrical neurologic medical devices (stimulators/recorders) must transmit currents, requiring use of conductive elements, metals, or conductive polymers.
Conductive elements used in neurologic medical devices include silicon (Polikov, Tresco, and Reichert 2005), carbon (Wang et al. 2006), and potentially graphene (Kuzum et al. 2014). Conductive metals may transmit electricity via capacitative conduction or faradaic conduction; capacitative conduction is less apt to cause tissue damage than faradaic conduction because charge species are not generated or destroyed during stimulation (Sahyouni et al. 2017b). Metals used in electrodes include platinum–iridium alloys (most common; Grill 2008), platinum (Orlowski et al. 2017), iridium (Sahyouni et al. 2017a; McCreery, Pikov, and Troyk 2010), stainless steel (Bijak et al. 2001; Koller et al. 1992; Nicolelis et al. 2003), tungsten (Nicolelis et al. 2003; Hachmann et al. 2013), gold (Polikov, Tresco, and Reichert 2005), and titanium (Mofid et al. 1997; HajjHassan, Chodavarapu, and Musallam 2008). Conductive polymers may also be used in neurologic medical devices including the commonly used poly(3,4-ethylenedioxythiophene) (PEDOT; Sahyouni et al. 2017b; Kim et al. 2008). While conductive polymers can form the body of the device, they can also be applied as a coating.
Surface coatings are often used to improve device biocompatibility and safety via a number of mechanisms. They can decrease faradaic reactions and oxide production, lower electrode impedance, increase cellular adhesion, promote cellular ingrowth, reduce immune responses, mitigate elastic modulus gradients, insulate to prevent focal temperature spikes, and/or deliver pharmacologic agents (Kim et al. 2008; Sahyouni et al. 2017b; He and Bellamkonda 2008; Cogan et al. 2009; Wolf 2008; Polikov, Tresco, and Reichert 2005; Zhong and Bellamkonda 2007). Coatings commonly used in neurologic medical devices include PEDOT (as mentioned above), sputter-coated iridium oxide (Wilks et al. 2017), polytetrafluoroethylene (teflon; Bijak et al. 2001; Nicolelis et al. 2003), polyimide (Hachmann et al. 2013; Rousche et al. 2001), hydrogels, and polyurethane (Rizzi et al. 2015). Various forms of silicone can be used as a covering (Koller et al. 1992) or adhesive (Bijak et al. 2001).
While coatings offer benefits, they also risk increasing the distance between electrodes and neurons, which may lead to reduced electrical device function due to increased current required for capture of the desired response (Kim et al. 2008). Potential delamination or corrosion of coatings can introduce foreign material into tissues. In some cases, such foreign material can be analyzed from the histology slide by scanning electron microscopy/energy dispersive spectroscopy to determine the source. The pathologist assessing biocompatibility must consider that the entire pathway of implantation may contain exfoliated device materials with potential inflammatory reactions. For devices with subdural implantation sites, cerebrospinal fluid (CSF) redistribution can lead to foreign material deposition in locations distant from the pathway of implantation. Addition of new features on the device surface, such as coatings, can impact biocompatibility and require additional biocompatibility testing.
Device Design
Several components of device structure including size, shape, and surface quality may impact tissue reactions (Grill 2008; He and Bellamkonda 2008; Polikov, Tresco, and Reichert 2005), and review of known changes associated with a design is important prior to device assessment. The required size of an electrical device may depend, at least in part, on the number of neurons that are being stimulated and/or recorded (He and Bellamkonda 2008). Smaller electrodes have greater electrical impedance but incite less tissue response (He and Bellamkonda 2008) and have a higher safety threshold for charge density (Butterwick et al. 2007). Increased electrical impedance is deleterious in current-controlled systems because it increases battery drain and in voltage-controlled systems because it may diminish neural capture of the stimulation (based on Ohm’s law).
Device shape impacts tissue reaction by affecting mechanical forces and, for electrical devices, charge density (Grill 2008). Shapes can be quite variable. For example, extraneural peripheral nerve stimulators can be annular/cuff/ring (Koller et al. 1992), helical (Agnew et al. 1989), slotted, flat, straight cylindrical, or coiled (Ortiz-Catalan et al. 2012; Sahyouni et al. 2017b). Intraneural peripheral nerve leads can be longitudinal or transverse (Sahyouni et al. 2017a, 2017b; Ortiz-Catalan et al. 2012). These shapes, as well as any coupled anchoring systems, can have an impact on the mechanical forces applied to tissue. Helical electrodes were designed to circumscribe nerves but cause less constriction than cuff electrodes (Sahyouni et al. 2017b). Elongate electrodes can incite varying tissue reactions along their length with more reactions at the electrode tip than along the shaft, potentially due to mechanical forces (Edell et al. 1992). Similarly, the greatest tissue reactions to intrathecal catheters typically occur at the catheter tip (Butt 2011). For electrical devices, shape can affect distribution of charge density on the surface of the device, which can lead to variations in the damaging electrochemical reactions that occur with stimulation (Grill 2008).
Surface texture can impact integration, as rough surfaces may enable better tissue adherence and reduced mechanical interface strain than smooth surfaces (He and Bellamkonda 2008). For conductive polymers, a rough surface can reduce electrical impedance and reduce the elastic modulus gradient between device and tissue (Kim et al. 2008).
Device Use
In addition to device materials, device use parameters impact biocompatibility and safety. For devices used in drug delivery (e.g., catheters), tissue responses may vary with route of delivery (epidural vs. intrathecal vs. intraparenchymal), delivery pump settings (bolus vs. infusion, infusion rate), dose volume, and drug concentration (Yaksh 2011).
For electrical devices, tissue damage can be impacted by multiple factors including the charge per phase, frequency, pulse width, amplitude, and the charge density (Figures 1A and B; Grill 2008; McCreery et al. 1990; Butterwick et al. 2007). Electrical stimulation can damage tissues via several mechanisms. Stimulation can generate electrochemical reaction products that can damage tissues and electrodes (Grill 2008). Stimulation can also damage neurons by inducing nonphysiologic levels of excitation and by increasing neuron excitation thresholds (Grill 2008; McCreery, Pikov, and Troyk 2010). Increased excitation thresholds can increase electrical stimulation requirements and incite damaging positive feedback loops (Grill 2008). Electrode heating can cause thermal damage (Figure 1C–F; Butterwick et al. 2007). Efficacy of electrical neurologic devices may be impacted by differences in electrical impedance and current generation between nervous tissue and device components. Nervous system tissues generate current by ion flow, metal electrical devices by electron flow, and conductive polymer devices by movement of charges along a molecular backbone (Grill 2008; Kim et al. 2008).
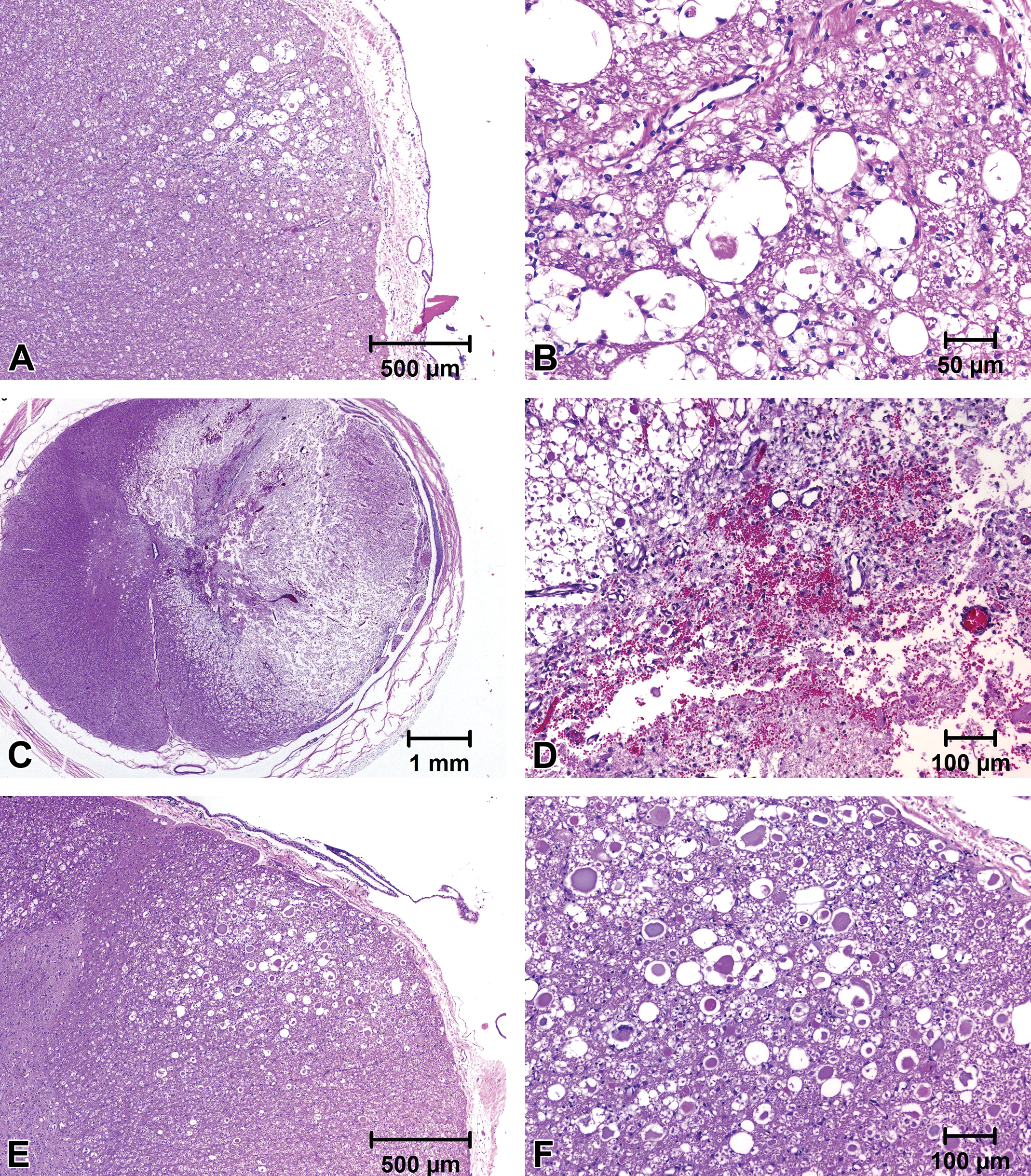
Spinal cord showing changes associated with high charge density after 5 days of electrical stimulation in a swine model. The cord shows degeneration and formation of spheroids/axonal degeneration (A and B; B is a higher magnification of A). Spinal cord showing acute thermal damage due to electrode overheating from an magnetic resonance imaging safety study (C–F; D and F are higher magnifications of C and E, respectively). In C and D, the cord adjacent to the electrode shows acute hemorrhage, inflammation, and edema. In E and F, the cord shows formation of spheroids/axonal degeneration (paraffin-embedded tissues, hematoxylin and eosin stain).
Nervous Tissue Responses to Devices
For safety study designs, it is imperative to separate changes that result from the placement or presence of the device from those that are due to administration of the therapy (Ortiz-Catalan et al. 2012; Sahyouni et al. 2017b). Delivered therapies can cause an exacerbation of tissue reactions (Butt 2011; Palazzi et al. 2016). Thus, it is critical to include appropriate negative and positive controls in studies assessing device safety.
Tissue Responses to Passive Devices: Central Nervous System (CNS)
For passive devices, tissue responses are dependent on location within the CNS or peripheral nervous system (PNS). Initial surgical implantation of a device into the CNS causes injury including damage to the blood–brain barrier with hemorrhage, edema, and acute inflammation (Stroncek and Reichert 2008; Sahyouni et al. 2017b; Polikov, Tresco, and Reichert 2005). Careful surgical technique, and sometimes imaging-guided navigation during surgery, is vital because trauma related to the surgery can make a study uninterpretable. Speed and method of insertion may affect the degree of damage (Bjornsson et al. 2006; Polikov, Tresco, and Reichert 2005; Edell et al. 1992).
Surgical trauma also incites microglial and astrocytic responses (Frontczak-Baniewicz, Chrapusta, and Sulejczak 2011), and microglial responses can be altered by postsurgical administration of certain antibiotics (Tikka et al. 2001). Glial cells react to injury and undergo morphologic changes, proliferate, alter surface expression of receptors, produce growth factors and cytokines, and migrate to the site of injury (Stroncek and Reichert 2008; He and Bellamkonda 2008; Polikov, Tresco, and Reichert 2005). Microglia and infiltrating macrophages phagocytize cellular debris (including axons and myelin) at implant sites (Stroncek and Reichert 2008). In the first 2 weeks postsurgery, the acute inflammation due to surgical trauma can be difficult to distinguish from device-associated tissue reactions (Polikov, Tresco, and Reichert 2005). This is a notable issue because the window to observe neuronal damage or death due to the device components (in biocompatibility studies) may occur only within this 2-week window (Switzer, Lowry-Franssen, and Benkovic 2011). Presence of necrotic neurons in ischemic infarcts caused by the device implantation has been observed for up to 4 weeks after implantation (J. P., personal experience).
Gliosis is a normal response to brain injury and is present in most CNS implants. Within two weeks of trauma, a compact astrocytic scar typically forms (Wanner et al. 2013). Glial scar formation can be exacerbated by chronic strain at the tissue–device interface (Polikov, Tresco, and Reichert 2005). Glial scars encapsulate the device and are composed of activated astrocytes with an inner layer of reactive microglia (Orlowski et al. 2017). Inflammatory cells may infiltrate the area, and infiltrating macrophages may be indistinguishable from reactive microglia. The glial scar may include fibrosis formed by fibroblasts inserted from the meninges or migrating from the perivascular space (Hovland et al. 2007; Stroncek and Reichert 2008). Fibrosis is common surrounding spinal cord intrathecal catheters and in the PNS (Figures 2A and B; Felice et al. 2011; Butt 2011). Glial scars can affect tissues up to approximately 500 μm from electrode tracks (Orlowski et al. 2017). If device implantation caused a large initial tissue defect, this may become a large cavity filled with CSF and rimmed by reactive astrocytes (Stroncek and Reichert 2008). After implantation, device migration can cause or exacerbate nervous tissue injury and may induce the formation of small cavities in the brain.
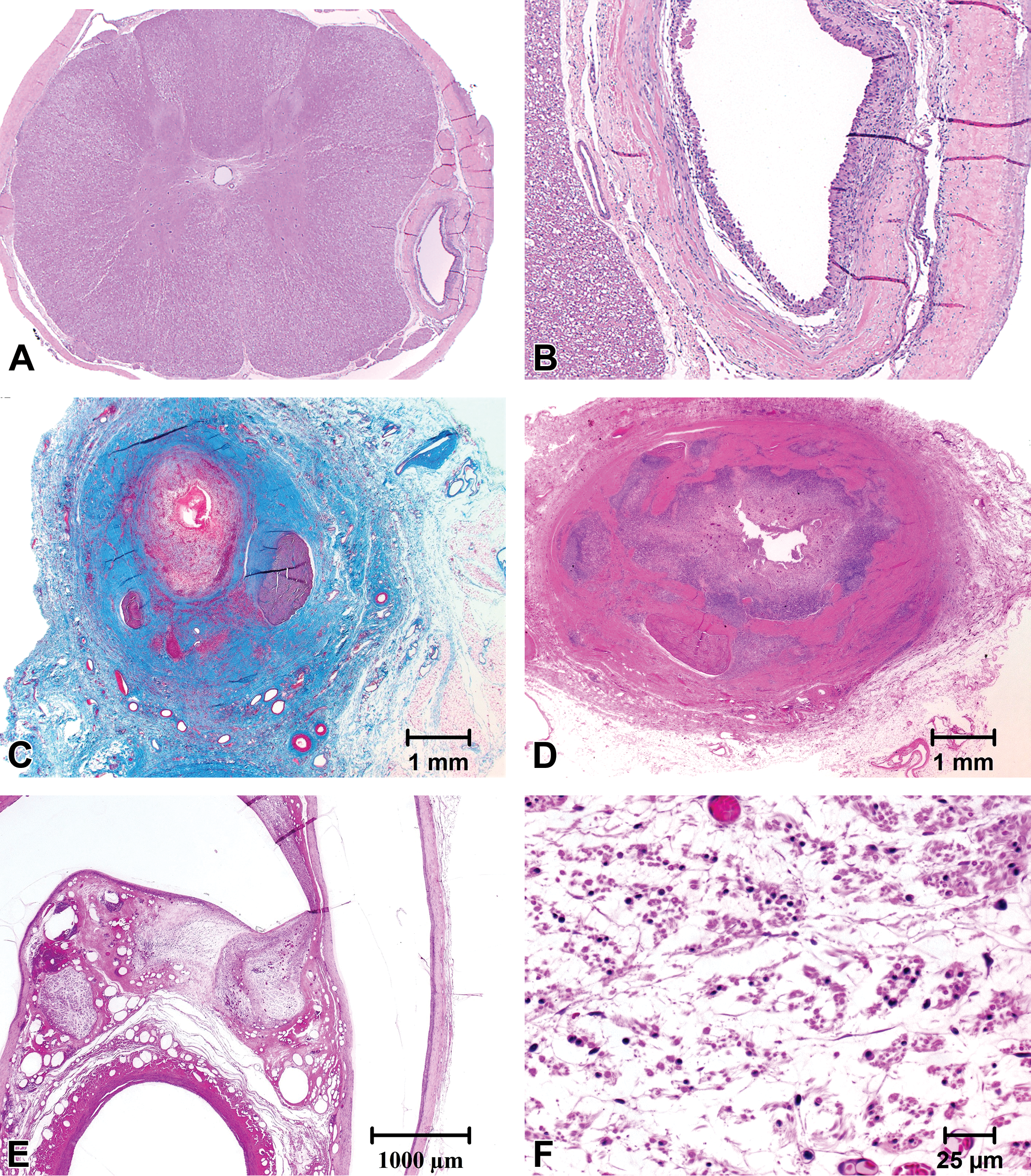
Spinal cord showing a chronic response to an intrathecal catheter in a drug safety study (saline control animal). Low magnification (A) shows compression of the cord adjacent to the catheter track. Higher magnification (B) shows the lumen of the catheter track surrounded by epithelioid macrophages, fibrosis, and few mononuclear cell infiltrates (paraffin-embedded, hematoxylin and eosin [H&E]). Vagosympathetic trunk with stimulator (straight lead) showing exuberant chronic inflammation and fibrosis surrounding the nerve and lead tip (C) and cathode (D) (paraffin-embedded, Masson’s trichrome (C) and H&E stains (D)). Vagosympathetic trunk implanted with a helical lead for chronic electrostimulation showing the lead in relationship to the nerve (E). Higher magnification (F) shows atrophy of the nerve (Spurr resin-embedded, H&E stain).
Additional CNS responses to an in-dwelling device may include foreign body reactions with mononuclear cell infiltrates and multinucleated giant cells (Hovland et al. 2007; Moss et al. 2004; Sahyouni et al. 2017b; Yaksh et al. 2003; Bijak et al. 2001). The device and/or tissue responses to the device may cause compression and degeneration of adjacent CNS tissues, particularly devices within the vertebral canal (Felice et al. 2011; Butt 2011). Tissue responses may increase separation between the electrode and neurons, changing the characteristics of the electrical field. Axonal damage in the brain and spinal cord will likely not undergo repair because these tissues lack the endoneurial scaffolding needed to guide axonal regeneration. Transection of axons in white matter tracts can sometimes produce detectable gliosis along these tracts that extends up to a few centimeters distant from the implant site.
Tissue Responses to Passive Devices: PNS
In the PNS, devices can cause fibrosis, axon degeneration, loss, and regeneration, myelin degeneration and loss, and foreign body reactions (Figures 2C–F; Agnew et al. 1989; Ortiz-Catalan et al. 2012; Koller et al. 1992; Sahyouni et al. 2017b). The presence of fibrosis misdirects and obstructs axonal regeneration. As long as the neuronal cell bodies survive, severed axons can regenerate if the endoneurial scaffold for that axon remains intact (as typically occurs with stretch injuries, but not with lacerations). PNS regeneration may begin as early as 3 hr after injury and can be maintained for at least 12 months. Regenerating PNS axons are thinner than normal axons (Koller et al. 1992). Neuromas may form if reinnervating axons cannot reestablish connections (Stroncek and Reichert 2008).
Additional Tissue Responses to Passive Devices
Implanted neurologic medical devices can cause additional complications, including changes in local CSF viscosity or flow; predisposition to infection and/or biofilm formation (Sahyouni et al. 2017b); abscess formation (Leblanc et al. 2013); intracerebral hemorrhage (Voges et al. 2007); skin erosion, lead fracture, misplacement, or migration (Hamani and Lozano 2006; Doshi 2011); and extension wire bow-stringing (Janson et al. 2010). In the spinal cord, intrathecal catheters may cause inflammatory masses/pyogranulomas (Butt 2011; Deer et al. 2008; Yaksh et al. 2003). Placement of the catheter in relatively small laboratory animal species like rats can be associated with inadvertent penetration of the spinal cord during surgery (Butt 2011).
Tissue Responses to Active Devices
Tissue responses to active neurologic medical devices depend on the therapy delivered and may vary (full discussion of the impact of myriad therapeutics on neurologic tissues is beyond the scope of this article). Electrical stimulation in the CNS may cause neuron death via the mechanisms described above (McCreery, Pikov, and Troyk 2010). Differentiating neuronal death due to device implantation from early effects of the therapy is a particular challenge, and this may not be possible in all studies. Indications that neuronal death may be due to the administered therapy may include consistent, asymmetrical zones of neuronal changes that can be reliably ascribed to a nonomnidirectional electrode, consistent death of a specific neuronal population, death of neurons that would not be expected to occur as a result of mechanical damage due to device placement, and/or death of neurons at the site of therapeutic administration that is notably increased over neuron death in areas distant from this site.
Stimulation of peripheral nerves can cause edema and axon degeneration (Agnew et al. 1989) as well as changes in myofiber components (Bijak et al. 2001). This underscores the importance of having negative controls as a comparison to active neurologic medical devices.
Designing a Neurologic Device Study
Designing a neurologic medical device study should include careful consideration of animal models, group design, device localization, euthanasia time points, tissue sampling and processing, histochemistry and immunohistochemistry, and communication of effects.
Animal Models
In vitro systems can provide some information about neurologic medical devices, but in vivo models are key for understanding tissue–device interactions (Polikov, Tresco, and Reichert 2005; Polikov et al. 2008). Animal studies should be performed in accordance with FDA Good Laboratory Practices or similar standards. Results from animal studies are not always directly translatable to humans (Kerlin et al. 2016). Many species have been used as models including dogs and rats (Butt 2011), rabbits (Barbolt et al. 2001), cats (Sahyouni et al. 2017a; Lemay, Grasse, and Grill 2009), sheep (Bijak et al. 2001), pigs (Hachmann et al. 2013), minipigs (Orlowski et al. 2017), and nonhuman primates (Felice et al. 2011; MacAllister et al. 2016; Capogrosso et al. 2016). The appropriate animal model(s) depends, in part, on the device to be tested. Variables including size, neuroanatomy, physiology, and other concerns may play a role in determining the appropriate model(s).
Size is especially important in determining the appropriate animal models for neurologic medical devices, since some devices may be too large to place in some species. ISO guidelines suggest neurologic implants in rats and rabbits should be limited to a size of 1 mm or less in diameter and 2 to 6 mm in length, while an 8-mm diameter disc is considered appropriate. When larger devices are being tested, other laboratory animal species should be used as models, and the preclinical safety assessment of clinically sized devices mandates that larger animal models be used. Pigs, minipigs, and sheep have typically been used to evaluate clinically sized devices, as they have similar neuroanatomy to humans, and they have large enough brains, spinal cords, and vertebral canals to accommodate DBS electrode arrays and intraspinal, epidural leads (Hachmann et al. 2013; Busscher et al. 2010; Orlowski et al. 2017). The swine skull anatomy, with its prominent nuchal crest and large sinuses, may be less desirable than the sheep for certain surgical procedures and magnetic resonance imaging (MRI). The flat frontal bone of the swine, however, provides advantages for external fixation of clinically sized devices compared to the convex sheep skull. Mature sheep and minipig strains (i.e., Yucatan) may be used for longer-term, chronic studies in which growth minimization is necessary to prevent changes in the position of the electrical field due to lead movement with spinal growth.
Cost, availability, physiologic similarity to humans, and ethical acceptance (particularly with regard to nonhuman primates and, in some countries, dogs) may also factor into the decision on which animal model(s) to use. Studies with time courses over a year should use animals with relatively long life spans. If accurate modeling requires active disease (i.e., a device to treat Parkinson’s disease), availability of animal disease models may be important (Schuh 2008); however, with medical devices, efficacy is usually tested during clinical trials.
Group Design
Compared to a general toxicity study, neurologic device studies may require larger groups, additional control groups, and additional euthanasia time points. It may be advisable to include additional/excess animals at the study onset to increase flexibility in group design, depending on study progress. Recovery groups may be important to assess potential resolution of effects (Hovland et al. 2007). Whether the study aims to address biocompatibility and/or safety will also inform the study design.
Although animals from both sexes would ideally be equally represented as recommended by ISO 10993-6, this may not occur in practice due to limited animal availability. The use of only one sex is likely to require justification. Intact male pigs are not typically used. Larger group sizes are important because the response to devices can be variable between animals (Polikov, Tresco, and Reichert 2005), and large groups enable better appreciation of a range of responses. In addition, devices can incite numerous tissue effects as described above, and these can obscure therapy-induced effects if the groups are too small. ISO 10993-6 provides guidelines for the traditional evaluation of biocompatibility of materials and is not always adequate to prescribe study design for the assessment of device safety with therapy delivery. Safety study design may diverge from ISO recommendations in regard to group design and device implantation strategy as detailed below.
For group design of an intrathecal catheter safety study, three control groups might be needed to discriminate tissue changes due to the passive catheter from those due to the active catheter. One control group is implanted with the catheter and administered saline (or a similar physiologically inert material) to identify tissue effects due to surgery and device presence. A second control group is implanted with the device and administered the vehicle to identify changes caused by the vehicle. A third control group is not implanted with the device but administered the test article systemically to identify systemic effects. Test groups include animals implanted with the device and administered the test article at multiple dosages (Felice et al. 2011).
For devices that deliver neurostimulation, implantation of a “control” in each animal will aid the pathologist in safety assessment as well as provide an acceptable and responsible approach for reducing the number of large animals used in the preclinical assessment (divergence from ISO 10993-6). Similar to drug delivery catheters, leads used for DBS and spinal cord stimulation (SCS) have effects on tissue due to implantation trauma and device presence within the tissue. Prudent study design for neurostimulation devices would include the implantation of a lead to deliver the “test” stimulation in one hemisphere and either nonactive control lead or a sham implantation in the contralateral hemisphere (divergence from ISO 10993-6). In the case of SCS leads, the control lead should be implanted a sufficient distance from the test lead (as possible) while still being in a suitably analogous location (i.e., thoracolumbar cord), and both locations should have electrode arrays as distant from the laminotomy/laminectomy or needle puncture site as possible.
In studies in which the objective is to evaluate the safety of a stimulation protocol rather than biocompatibility of materials, the appropriate control may also be an active lead. For instance, an appropriate control for evaluation of a high charge density stimulation protocol would be a stimulation protocol using the same electrode configuration as the test lead but at a charge density clinically accepted as safe. Another acceptable divergence from the ISO 10993 guidelines is the study designed to evaluate the safety of a clinical stimulation protocol or directions for use that prescribes bilateral stimulation or multisite stimulation. In that case, the purpose of the study would dictate that a single animal would receive implants in multiple locations.
Device Localization
For successful characterization of tissue reactions, the exact location of the device must be known. Taking samples that were within the electrical field is criticial in accurately assessing the effects of neurostimulation. In addition, tracking device migration throughout the study, or at least at implantation and termination, can add important information for interpretation of tissue injury.
Understanding device location can be aided by the study protocol, surgical notes, imaging records, and necropsy records including photographs taken during necropsy and/or at tissue trimming. Imaging may be especially helpful when extracting medical devices and techniques including the use of computed tomography (CT) scanning, MRI, radiography, and microradiography (Rousselle and Wicks 2008). Notably, device materials may prevent the use of some imaging techniques; for example, some metals are not compatible with MRI.
Radiography including fluoroscopy and/or CT is always a component of DBS and SCS implantations and follow-up examinations, and radiographic images should be used to determine the accurate location of the electrode arrays. The locations of specific electrodes should be assessed relative to depth from the cortical surface in DBS studies, and this can be accurately determined from surgical notes when frame-assisted implantations are performed. Follow-up radiography can be used to determine any lead retraction. CT or CT-equivalent rational fluoroscopy more accurately determines electrode location for spinal lead implantations than standard radiography or fluoroscopy, and interpreting standard radiography and fluoroscopy is inevitably confounded by parallax. CT allows for accurate measurements to be taken of electrode locations relative to specific vertebral landmarks.
Sampling sites for peripheral leads are usually straightforward, as the design and fixation of the leads results in encapsulation of the electrodes. However, some peripheral leads have a straight design and are implanted within the connective tissue sheath of the target nerve (i.e., carotid sheath for vagosympathetic trunk stimulation). For these types of leads, as well as certain intraspinal and intracerebral leads, in-life imaging is necessary for accurate sampling, as it is often practically impossible to prevent at least some lead movement during nerve, spinal cord, or brain access. In addition, angiographic landmarks can aid in determining the location of the electrode array or active electrode, and angiograms are often incorporated in peripheral stimulation studies to document the effects of the neural lead on adjacent arteries.
Euthanasia Time Points
Appropriate euthanasia time points depend on the device components and intended clinical applications. For biocompatibility assessments of local responses, ISO standard timelines depend on absorbability of device material. A less biodegradable material may reach quiescence in a tissue at earlier time points, whereas a more biodegradable material may require longer follow-up. General guidelines include an early time point to identify the initial response to the device (1–4 weeks), a middle time point to identify device degradation, and a late time point to identify chronic responses and/or degradation (of particular import with absorbable devices). Unless the chemical extraction profile is very satisfactory, devices intended for permanent implantation will need to be evaluated for chronic toxicity (FDA 2016).
For safety studies, prior to initiating therapy administration, animals should be given a reasonable amount of time to heal from device implantation and surgery. Safety studies for regulatory submissions of implanted neurologic devices typically include 30-day, 90-day, and 180-day time points. A 3- to 5-day time point after initial therapy administration may also be included to enable the detection of neuronal death. (In typical neurotoxicity studies, neuronal death is best assessed 2 to 5 days after the first exposure to a neurotoxicant [Switzer, Lowry-Franssen, and Benkovic 2011]; devices can incite similar rapid-onset neuronal necrosis, or neuronal necrosis may occur over a more prolonged and varied time course.)
Tissue Examination, Sampling, and Processing
Necropsy for neurologic medical devices that are novel and those that would be expected to cause significant tissue alterations should be performed with an experienced pathologist in attendance (Nikula 2016; Nikula and Funk 2016) or by a person with very specialized training. Suggestions for necropsy, fixatives, and handling of nervous system tissues in general toxicology studies have been published and are typically applicable to neurologic device studies (Fix and Garman 2000; Jortner 2011).
Optimal tissue preservation includes euthanasia via exsanguination and perfusion with a suitable fixative. Four percent methanol-free formaldehyde (aka paraformaldehyde) is generally considered optimal for preservation of perfused nervous tissues to be examined at the light microscopic level (Fix and Garman 2000), though 10% neutral buffered formalin may also be suitable. Perfusion fixation typically induces fewer artifactual changes than immersion fixation (Fix and Garman 2000), and exsanguination enhances the interpretation of some histochemical stains (notably the Fluoro-Jade-based stains). Perfused brain can be kept in situ for 12 to 24 hr before being removed from the skull. After removal, perfused tissues can be then immersed in fixative for a few additional days before further handling.
Gross examination of nervous tissues with an implanted medical device is often better performed after thorough fixation rather than at necropsy. Imaging data (described above) can be useful in locating the device, and photographs taken at necropsy and trimming can only improve the overall quality of the morphologic evaluation. The examination will vary, depending on whether the device will remain in situ or be removed prior to processing.
Device components must be considered when deciding whether to remove it or not. Most devices composed of polymers can be maintained in situ, paraffin-embedded, and sectioned on a standard microtome, as long as the components are compatible with processing (Rousselle and Wicks 2008). In contrast, maintaining devices composed of metal or other hard components in situ requires processing tissues in other media such as plastic resins. The choice of embedding material should take into consideration factors such as toxicity, shrinkage, compatibility with device materials, and infiltration time (Rousselle and Wicks 2008).
Removing a device at necropsy or after tissue fixation allows for benchtop evaluations of the device to facilitate evaluations of biostability. Examination should note the condition of the device and the surrounding tissue reaction. However, device removal risks introducing artifacts that can impact histopathologic examination. If the device is removed, cells adherent to the device can be analyzed separately, if needed (He and Bellamkonda 2008; Moss et al. 2004).
Leaving the device in situ enables better assessment of the tissue–device interface (e.g., a 3-mm stimulation field), particularly in situations where there is significant tissue in-growth into the device. In general, if there are adhesions or fibrous connective tissue around the device, dissection is not recommended (Rousselle and Wicks 2008). If the device remains in situ, the tissue–device interface should not be disturbed, and the region of interest should be collected with ample tissue borders.
Tissue trimming is an important study phase as it allows gross examination along the tissue–device interface, enabling specific regions to be trimmed and documented for inclusion in the pathology report. Extensive tissue sampling may be required to fully examine the response to the device. The Society of Toxicologic Pathology recommendations for sampling and processing the nervous system during general toxicity screenings (Bolon et al. 2013) may be useful, but this sampling strategy is not sufficient for complete assessment of neurologic medical devices. Given that the tissue reaction to a device may vary along different areas of the device as discussed above (e.g., more reaction along the tip of the device or more reaction in uninsulated areas), it is requisite to compare samples that were carefully sourced from the same location for each animal. Also, it is important to evaluate multiple portions of the device—solely examining the tissue surrounding a portion of an electrode shaft would miss increased tissue reactions occurring at the electrode tip, for example.
When designing a trimming scheme, it is important to consider the design of the device, location of components with different materials, the active therapy delivery sites versus passive components of the device, the location of the cathode(s) and anode(s), and the total area and shape of the electrical field for neurostimulation studies. In studies that include a catheter, it is ideal to have multiple samples taken from the region of the catheter tip. An attempt should be made to preserve the dura mater over CNS implantation sites for epidural and subdural devices.
Samples should be collected to include unaffected tissue surrounding the device, and nervous system tissues distant from the site should also be examined. For example, for a study investigating a peripheral nerve stimulator, sections of brain may also need to be examined. Depending on the device, examination of draining lymph nodes may be appropriate. The components of the device system that are not in contact with neural tissue must also be considered, particularly when conducting safety assessment of a clinically sized system for ISO 10993-6 biocompatibility purposes. The pathway of the catheter and lead body outside the nervous system should be sampled as well as components of the implantable pulse generator or the compound infusion pump.
Histochemistry and Immunohistochemisty
Hematoxylin and eosin (H&E)-stained histology slides are useful for interpreting most of the neural tissue changes associated with devices such as necrotic neurons, axon degeneration, glial cell reactions, vascular changes, edema, hemorrhage, inflammatory cell infiltrations, and others. Additional histochemical stains and immunohistochemical (IHC) techniques can provide important information for nervous tissue histopathologic examinations (Table 1), including the ability to assess distribution of cellular reaction such as gliosis. The International Harmonization of Nomenclature and Diagnostic Criteria for Lesions in Rats and Mice document provides details about many possible auxiliary stains/labels, and some more commonly used techniques are detailed below (Kaufmann et al. 2012).
Basic Strategies for Sampling and Processing Nervous System Tissues from Neurologic Device Studies.
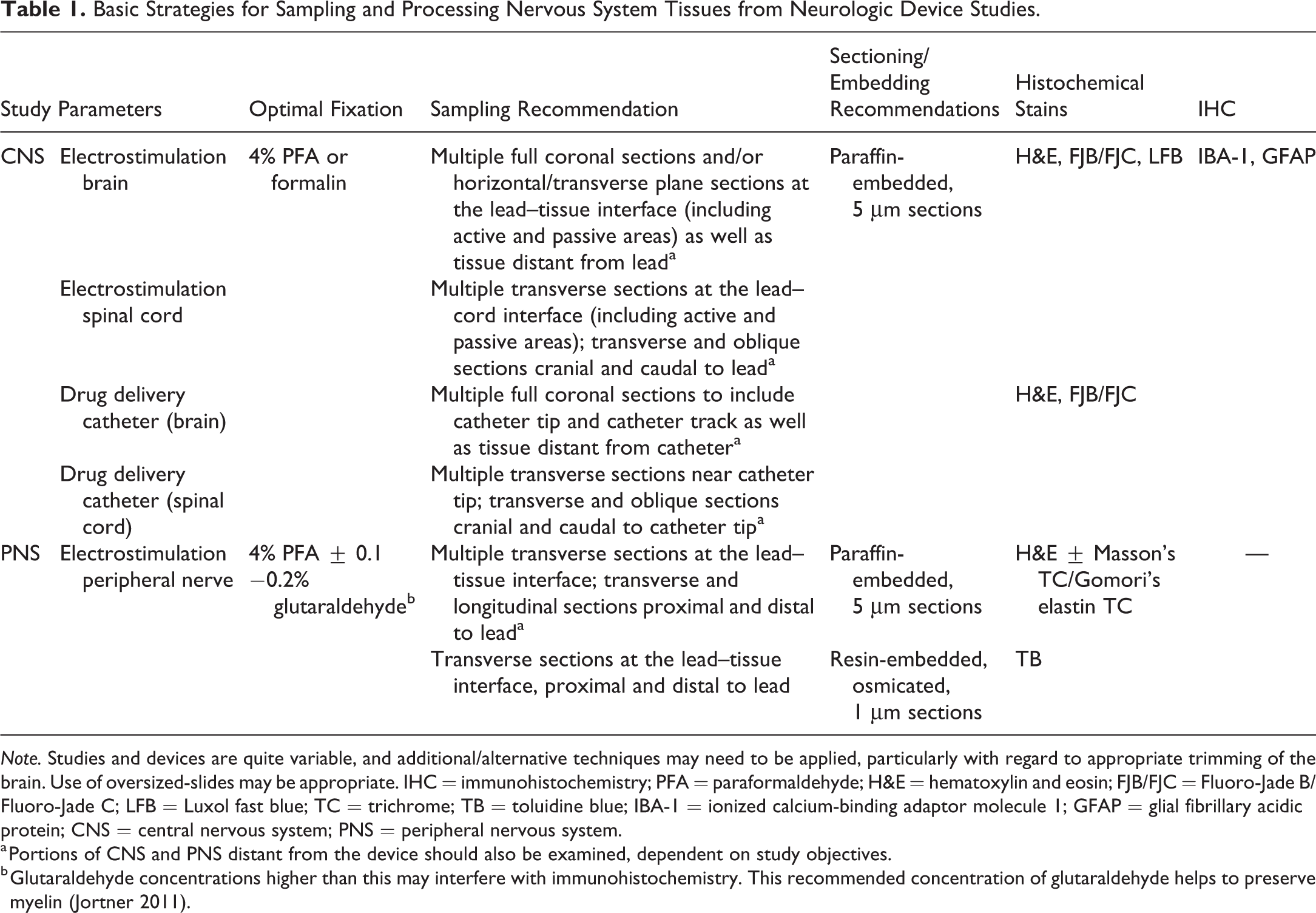
Note. Studies and devices are quite variable, and additional/alternative techniques may need to be applied, particularly with regard to appropriate trimming of the brain. Use of oversized-slides may be appropriate. IHC = immunohistochemistry; PFA = paraformaldehyde; H&E = hematoxylin and eosin; FJB/FJC = Fluoro-Jade B/Fluoro-Jade C; LFB = Luxol fast blue; TC = trichrome; TB = toluidine blue; IBA-1 = ionized calcium-binding adaptor molecule 1; GFAP = glial fibrillary acidic protein; CNS = central nervous system; PNS = peripheral nervous system.
a Portions of CNS and PNS distant from the device should also be examined, dependent on study objectives.
b Glutaraldehyde concentrations higher than this may interfere with immunohistochemistry. This recommended concentration of glutaraldehyde helps to preserve myelin (Jortner 2011).
Necrotic neurons can be identified with Fluoro-Jade B (Schmued and Hopkins 2000) or Fluoro-Jade C (Schmued et al. 2005) on paraffin-embedded sections or amino cupric silver on frozen sections (Kaufmann et al. 2012; Switzer 2000). These stains are better performed on tissues from exsanguinated animals because red blood cells stain positively and interfere with interpretation. Time point is vital to visualizing necrotic neurons, as the window for observation of neuronal death may be brief (Switzer, Lowry-Franssen, and Benkovic 2011). If neuron loss is suspected, but the study design did not allow for an evaluation during the window of neuronal loss/death, neuron density can be assessed using quantitative methods including stereology (Kaufmann et al. 2012; Brown 2017). Identification of neurons may be enhanced using Nissl stains (e.g., cresyl violet) or IHC methods. The use of anti-NeuN antibodies labels most but not all neurons (McCreery, Pikov, and Troyk 2010), while specific subpopulations of neurons can also be labeled, for example, using antityrosine hydroxylase antibodies to identify dopaminergic neurons or anticholine acetyltransferase antibodies to identify cholinergic neurons.
In paraffin sections, assessment of myelin may be enhanced using luxol fast blue staining or antimyelin basic protein IHC (Kaufmann et al. 2012). Osmium fixation and resin/plastic embedding with toluidine blue staining produces superior samples for myelin evaluation in the central (on small areas such as corpus callosum) and peripheral portions of the nervous system.
Axons can be highlighted using Bielschowsky’s silver or Bodian’s silver stains or IHC for neurofilament proteins. (Kaufmann et al. 2012; Felice et al. 2011). Identification of reactive microglia and macrophages can be facilitated with IHC for ionized calcium binding adaptor molecule 1, and reactive astrocytes can be visualized with IHC for glial fibrillary acidic protein. These can be performed on paraffin-embedded or frozen sections (Felice et al. 2011; Kaufmann et al. 2012; Yaksh et al. 2003; Ito et al. 2001). Light microscopic assessment of peripheral nerves would ideally be performed on osmicated, resin-embedded nerves sectioned at 1 μm thickness and stained with toluidine blue. This method has proven optimal for preserving and highlighting peripheral nerve morphology (Jortner 2011). For cost savings, osmicated, paraffin-embedded, H&E-stained sections can also be used to highlight myelin (Di Scipio et al. 2008), though these do not yield the same degree of detail as resin sections.
Communication of Effects
In addition to standard background content, the pathology report on a neurologic device should address, when applicable, device retrieval procedures, histologic preparation of the device, device degradation/biostability/structural changes, and tissue remodeling. Microscopic evaluations of neurologic medical devices should include assessments of tissue responses with quantitative or semiquantitative estimates of the area affected by tissue reactions. The following general reactions should be assessed when present: degeneration, inflammation/cellular infiltration, necrosis, device material changes (if visible), and tissue ingrowth. Specific to nervous system devices, the following should be semiquantitatively scored: implant disruption of neuronal processes, reactive astrocytes and/or connective tissue surrounding the implant, capsule formation, neovascularization, inflammatory cell infiltration, microglial reactions, myelin loss/changes, multinucleated giant cells, and mineralization. Grading schemes for intrathecal catheter changes have been published (Butt 2011). As with other reports, neurologic device reports should indicate, when appropriate, which changes were attributed to the presence of the device and which were attributed to therapy delivery.
The pathology report should also state, if possible, which effects are considered adverse with respect to intended clinical use (Kerlin et al. 2016). It is not possible to introduce a device into the brain or spinal cord without causing some degree of local damage (Frontczak-Baniewicz, Chrapusta, and Sulejczak 2011). Determining effect adversity is often complex, and the unique requirements of neurologic medical devices further confound this (Butt 2011). General recommendations for determining adversity have been published (Kerlin et al. 2016; Palazzi et al. 2016). An adverse effect can be defined: “In the context of a nonclinical toxicity study, an adverse effect is a test item-related change in the morphology, physiology, growth, development, reproduction or life span of the animal model that likely results in an impairment of functional capacity to maintain homeostasis and/or an impairment of the capacity to respond to an additional challenge” (Palazzi et al. 2016). For neurologic medical devices, it is important to dissect adverse effects arising from the nonactive device, the active device, and the implantation surgery. In some situations, small reactions that would not ordinarily be considered adverse can impact device function. For example, a small area of fibrosis that might not be considered adverse in a general toxicity study could separate an electrode from neurons, reducing device functionality (i.e., resulting in an increase in amplitude of stimulation necessary for capture of a clinical response).
Conclusions
With expanding technological capabilities, new neurologic medical devices are being developed to enable treatments using previously inaccessible therapeutic targets. Pathologists can provide guidance and feedback at many stages of device development from study design through interpretation and communication of tissue changes. Closely collaborating in multidisciplinary teams, pathologists are vital for successful development and approval of neurologic medical devices.
Footnotes
Acknowledgments
Victor Perez and Bridgette Glass deserve thanks for help with manuscript preparation.
Author Contributions
Authors contributed to conception or design (SC, JL, JP, WS); data acquisition, analysis, or interpretation (SC, JL, JP, WS); drafting the manuscript (SC); and critically revising the manuscript (JL, JP, WS). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential, real, or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article. SC, JL, MB, and WS work in contract research laboratories, and a subset of their work includes evaluation of medical devices from different sponsors. JP works at Medtronic, and a subset of his work includes evaluation of Medtronic medical devices.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.