
Abstract
Safety (“biocompatibility”) assessment of medical devices has evolved along a different path than that of drugs, being historically governed more by the considerations and needs of engineers rather than chemists and biologists. As a result, the involvement of veterinary pathologists has been much more limited—almost entirely to evaluating tissue responses in tissues in direct contact with implanted devices. As devices have become more complex in composition, structure, placement, and use, concerns as to adverse systemic responses in patients have called for more comprehensive and thoughtful evaluations of effects throughout the body. Further complexities arise from the increasing marriage of devices and drug/biologic therapeutics to achieve either better dose control and, specifically, in delivery to target organs/tissues or better tolerance of the body to medical devices (i.e., minimization of the foreign body response). The challenge to pathologists is to integrate in new technologies (such as in vivo imaging and immunology) and ways of viewing interactions with patient bodies. To fail to do so will allow the methods and standards for medical device safety evaluation to be based on chemical analysis and then the limited details inherent in literature-based risk assessments.
Introduction
Historically, medical devices have been viewed as inert constructs of polymers, metals, structural tissue components from donor animals and ceramics, expressing only a limited range of interactions with the body, almost exclusively limited to those host tissues in direct contact with the medical devices. The authors believe that this has resulted in the involvement of pathology in evaluating the medical devices being extremely limited in both scope and extent. Since the early 1980s, the utilization, composition, structure, and function of medical devices have advanced dramatically, particularly with the advent of nanodevices, surface treatments on devices, and combination products serving to provide focused and controlled delivery of therapeutics. The authors believe that pathology has not likewise come to utilize the full range of its available technologies and critical thought to fill the increased needs for evaluating medical device safety. Evolving regulatory guidances are focused on chemical analysis of extractable substances from new products, followed by literature-based risk assessments, and not on thoughtful and knowledgeable evaluation of changes in tissues by those having the experience and expertise to do so. Current guidances minimize potential histopathology evaluations.
Role of Toxicologic Pathologists
Unlike medical pathologists and PhD scientists, the veterinary and toxicologic pathology community has been very slow to see the need for and embrace histopathological evaluation of medical devices as a valid part of safety evaluation. Coupled with the resistance of medical device companies and innovators to perform broader and more extensive safety evaluations, this has led to toxicologic pathologists not being well represented nor leaders in determining nomenclature and testing standards or regulations for evaluation of biomaterials and finished medical devices (i.e., those entering the marketplace and ready for patient use). Regardless, toxicologic pathologists are ideally suited to provide safety assessment for biocompatibility, efficacy of medical devices in animal models, and to act as an important member of medical devices development teams. Currently, much of the required evaluation of the potential deleterious effects of biomaterials and medical devices are done by modeling, analytical chemistry, and paper risk assessment, but increasingly, veterinary pathologists are being asked to participate in more complex evaluations. Toxicologic pathologists new to medical device evaluation may encounter difficulties in understanding the types and categories of biomaterials and medical devices, study design, terminology applicable to biomaterial and medical device testing, and appropriate histologic and histopathology evaluation procedures. A more active role of toxicologic pathologists and the Society of Toxicologic Pathology in adopting the use of new technologies and developing nomenclature and international evaluation standards for materials and medical devices is long overdue.
The current trend by Food and Drug Administration (FDA) and other regulatory agencies is to depend on chemical analysis of extracts from medical devices and literature-based risk assessments. In the authors’ opinion, this trend is limiting the involvement of pathologists to evaluation of direct tissue contact effects and not involving them in new technologies. Given the widening range of technology involved in the development of new medical devices and their clinical applications, it will be essential for pathologists to become further involved.
Current Regulatory Guidances and Points to Consider
The medical device industry is globally a third the size of the pharmaceutical industry in revenues, and the assessment of patient biologic safety (“biocompatibility”) has followed a different regulatory acceptance path than that for small- and large-molecule pharmaceuticals due to the differences in the nature and use of the biomaterials and medical devices and of potential safety, risks to patients, as well as product life cycles and economics. Historically, it was only in the early 1960s that the potential for constituent materials or medical devices to have adverse effects on patients became of concern (Hutt 1989). Modern medical device regulatory safety assessment has evolved into general adherence to the current International Standards Organization (ISO) 10993 guidelines for Biological Evaluation of Medical Devices (http://www.iso.org; Gad and Gad-McDonald 2016). While toxicologic pathologists involved in the evaluation of medical devices should be familiar with all relevant ISO 10993 and related guidances (e.g., ISO parts 1 to 7, 9 to 20, 22, and 33), ISO parts of particular interest are summarized in Table 1 due to their relevance to histopathology and safety evaluation.
International Standards Organization 10993 Standards Emphasizing in Vivo Studies and Histopathology.

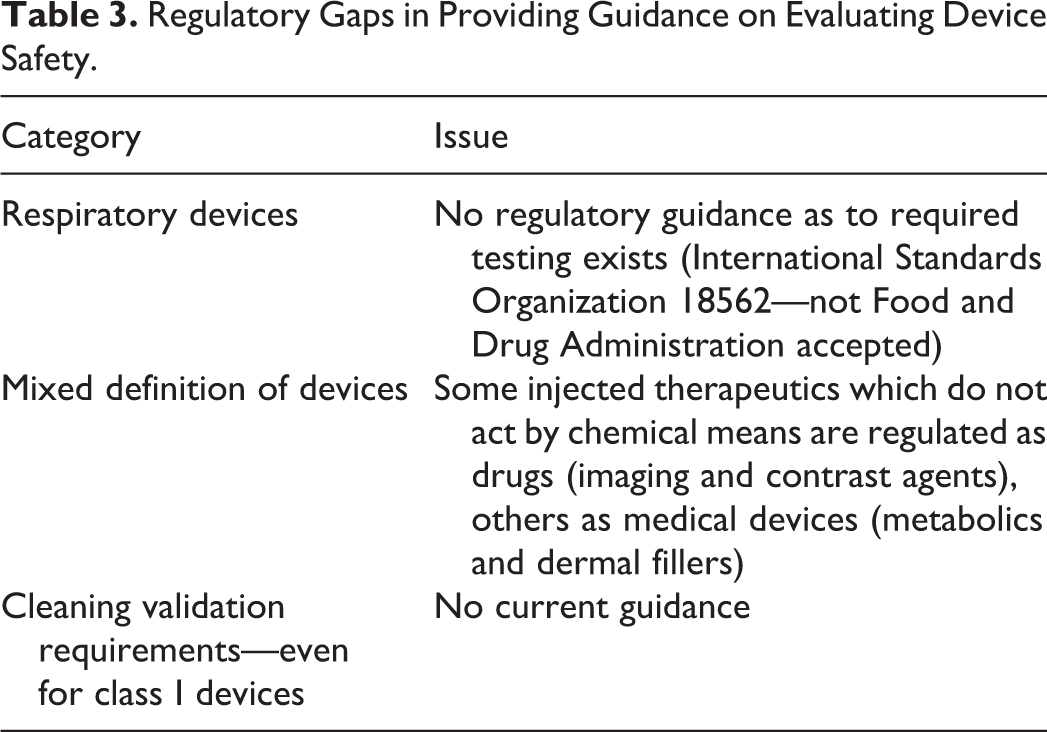
There are a range of other organizations which have regulatory/quasi-regulatory impact (Table 2), but there are also a number of gaps in current guidances, which complicate biocompatibility program design and study selection/design (Table 3).
Regulatory Bodies Involved in Assessing Device Safety.

Note. CDRH = Center for Devices and Radiological Health; CFDA = Chinese Food and Drug Administration; MHW = Ministry of Health and Welfare.
Regulatory Gaps in Providing Guidance on Evaluating Device Safety.
Other regulatory points: Unlike with drugs and biologics, all required biocompatibility evaluations must be performed before any clinical evaluations are performed. No testing is done afterward. As a logical requirement for thoroughly evaluating safety before human exposure, testing must be performed on the device as it is intended to go in or on humans—that is, the “finished” product. The FDA usage guide for ISO 10993 (as summarized in Table 1), which outlines concurrence and differences to that contained within ISO 10993-1:2009, has been recently updated (U.S. Food and Drug Administration 2016b). The FDA and European Medicines Agency (EMA) have a preference that medical device biocompatibility testing be conducted under relevant Good Laboratory Practices (GLPs) as described in 21 CFR Part 58 (USFDA, 2016a) or equivalent. Compliance with GLPs is required for studies with nonclinical safety endpoints required to support an Investigational Device Exemption (IDE) for clinical evaluation of the device. FDA routes to marketing approval for a device include 510(k) Premarket Notifications (the most common route), Premarket Approvals (PMAs, similar to a New Drug Approval—that is, an NDA), and Humanitarian Use Device (HUD). Other standards and scientific organizations with publications relevant to biological evaluation include The American Society for Testing and Materials (http://www.astm.org), U.S. Pharmacopeia Convention (http://www.usp.org), and the ISO.
The Organisation for Economic Co-operation and Development (2015) Principles of GLP also allows for jurisdictional inclusion of medical devices. The International Medical Device Regulators Forum (http://www.imdrf.org) attempts to standardize global regulations (http://www.imdrf.org/documents/documents.asp), but ISO and other guidance documents remain the primary reference for safety and efficacy assessment. FDA marketing submissions include Premarket Notification 510(k), IDE, PMA (similar to an NDA), and HUD.
Terminology Applicable to Biomaterial and Medical Device Testing
A key part of the FDA definition of medical device is that it does not depend on chemical action and metabolism to achieve its desired therapeutic effect. However, some therapeutic modalities, which achieve their intended therapeutic effects without chemical interaction (imaging and contrast agents), are considered drugs if injected into the body, while others (such as metabolics and dermal fillers) are medical devices.
The FDA defines a medical device as an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory which is: recognized in the official National Formulary, or the United States Pharmacopoeia, or any supplement to them, intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or intended to affect the structure or any function of the body of man or other animals, and which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of any of its primary intended purposes (U.S. Food and Drug Administration 2014).
Other global regulatory agencies use similar definitions, but for unique medical devices, each agency may need to be consulted to obtain a determination of what constitutes a device. Additional definitions can be found in a glossary (Attachment G) of the FDA 10993-1 usage guide (U.S. Food and Drug Administration 2016b) or guidance documents released by other countries.
The FDA defines biocompatibility as the ability of a medical device material to perform with an appropriate host response in a specific situation. Biocompatibility testing of medical devices requires an early assessment of medical device performance (equivalent to efficacy for a drug) and safety, unlike testing of drugs and biologics where safety but not efficacy is required.
Types and Categories of Biomaterials and Medical Devices
Medical devices include a single biomaterial or a combination of biomaterials (e.g., polymer on metal supports for replacement joints), diagnostic medical devices, and may include microelectronics, computer components, and software. Major biomaterials include metals, ceramics, textiles, polymers, nanomaterials, and animal-derived materials or tissues.
Most medical devices are solid entities, but there are also injectable liquids (e.g., dermal fillers, hyaluronic acid lubricants for knee joints) and suspensions of particles or liquids (e.g., metabolics). Likewise, medical devices may be nonresorbable (permanent) or resorbable, and the rate of resorption by the body can usually be “tuned” to a desired period.
In terms of regulatory concern, medical devices are classified according to potential risk as Class I (minimal risk), Class II, Class III (high risk), and active implantable medical device (most risk); but other countries may have additional subcategories based on whether they are “active” (include energy sources or are potentially influenced by external energy sources).
Program and Study Design
There is no standard development pathway, as regulatory guidances depend on the material(s), type and site of patient contact and cumulative duration of patient contact for the intended medical device, clinical indication, and expected extent of global marketing. The current (2016) FDA version of ISO 10993-1 provides general guidance (in most cases—respiratory devices being a notable exception) as to which biocompatibility studies are or may be required and (in ISO 10993-21) provides very general guidance as to study designs. Testing must be conducted on final finished medical devices after sterilization by the mode to be used for clinical products (including ethylene oxide, heat, steam, radiation, or other chemical sterilants).
Medical device materials may need to be tested for cytotoxicity, extractables and leachables, sensitization, irritation, hemocompatibility, genotoxicity, pyrogenicity (bacterial endotoxins), particulates and contaminants, local tissue tolerance/biocompatibility (implants), acute through chronic toxicity, safety pharmacology, reproductive and developmental toxicity, immunotoxicity, carcinogenicity and manufacturing and packaging sterilization, and contaminants. Gad and Gad-McDonald (2016) should be referenced for study design requirements. Clinical observations, body weights, organ weights, and clinical pathology determinations are relevant to evaluation of medical devices (see Gad and Gad-McDonald 2016). Dose responses are seldom relevant to medical devices, as the maximum implantable dose is often the final finished medical product or multiples thereof.
Testing for functionality (efficacy) and for the finished medical device may require testing in an animal model of “disease” (e.g., pacing model of congestive heart failure in dogs, rat long bone fractures, and wound healing models in pigs).
Carcinogenicity studies are usually required only for a lifetime-implanted medical device, which include novel component materials or treatment modalities. But tumorgenicity may be caused by mechanisms other than chemical interactions with genetic materials. Solid-state carcinogenesis (nongenotoxic tumor induction due to shape and texture of the material—the “Oppenheimer effect” in rodents, for example) may complicate interpretation of the results. The recent identification of breast implant-associated anaplastic large-cell lymphoma (a peri-implant lymphoid tumor) in women (U.S. Food and Drug Administration 2017) appears texture related and additional chronic or carcinogenicity nonclinical testing may be a future requirement.
Histologic and Histopathology Considerations
There are a number of considerations for pathologists when it comes to the method and evaluation of medical devices.
The toxicologic pathologist should be involved early in the testing program and study design to help choose appropriate species, endpoints, sacrifice intervals, specimen and tissue sampling, histologic preparation, histopathology scoring methods, and application of advanced evaluation techniques (Nikula and Funk 2016). Handling of samples and evaluations not usually used for safety assessment of drugs and biologics may be required. Tissues such as rabbit vagina, hamster cheek pouch, and chinchilla ear may be the objective of investigations, and lymph node draining sites serving the region of implants are a valuable tissue supplement to the evaluation of the inflammatory response and biodegradation of biomaterials and medical devices.
Evaluation of intramuscular or surgical implants can be complicated by procedural trauma, sutures or clips (secondary medical device implants), and wound contamination. Further, evaluation of local and systemic toxicity in some species (large hound dogs, calves, and sheep) not commonly evaluated may be complicated by the lack of available histopathology information on background or spontaneous findings in these species and strains. Special procedures will be required for energy-based medical devices or nanoparticles intended to receive specific light frequencies and transmit heat.
Objective means of evaluation and documentation of the results (such as digital photographic documentation of in-life observations and at necropsy) should be employed when applicable. In-life ultrasonography, fluoroscopy, radiography or microradiography, optical computer tomography, magnetic resonance imaging, and microcomputed tomography (µ-CT) may improve accurate sample collection. Evaluation of the tissue–device interface in its correct orientation is critical. The medical device should be retained with the tissue unless this precludes histologic preparation.
Biodegradable materials may not survive histologic processing, and an irregular and inappropriate empty space with or without an inflammatory reaction may be the only evidence of the implanted material. Accordingly, embedment in plastic resins and tissue section grinding may be required to properly evaluate metallic or complex implants and to retain the tissue–device interface. Electron microscopy (transmission and scanning), histochemistry, immunohistochemistry (IHC), and cryomicrotometry may be required, but appropriate fixatives and samples need to be planned in advance.
Hematoxylin and eosin staining is generally adequate for most evaluations. Special histochemical stains to differentially stain-specific tissue components (blood vessels, bone, cartilage, and nervous system) may be required but will rarely stain biomaterials. An algorithm to select suitable embedment, cutting tools, and stains has been described (Alves, Metz, and Render 2012).
Making a determination of complete biodegradation may require the use of interim biopsies and necropsies, and extending the study duration to as long as 2 years, depending on how long complete device degradation/resorptions requires.
While some materials and medical devices induce little to no tissue response, the toxicologic pathologist needs a current understanding of the inflammatory (cells, signaling and secretory molecules), foreign body, and fibrotic responses (Anderson, Rodriguez, and Chang 2008; Goad and Goad 2013; Klopfleisch and Jung 2017).
Guidance as to expected nomenclature and scoring templates for histopathology of medical devices are found in ISO 10993-6:2016 (International Organization for Standardization 2016). However, the toxicologic pathologist should develop a medical device and tissue-matched semiquantitative scoring system including pilot studies and refinement over multiple studies of increasing duration rather than adhere only to templates contained in ISO 10993-6:2016. Standardization nomenclature systems such as the International Harmonization of Nomenclature and Diagnostic Criteria may be applicable, but these standards have not considered findings (e.g., intramuscular fatty infiltration) associated with biomaterials and medical device implants (Greaves et al. 2013). Additional tissue changes not captured by a standardized scoring system should be footnoted or included as a comment, and may require inclusion in the interpretive summary to properly communicate risk assessment for the material or medical device, only if a novel material is employed.
ISO 10993-6 (2016) Annex E now indicated that conclusions for scoring should use “reaction scores” rather than “irritancy scores.” Nonbiodegradable implants (vascular stent and bone) commonly employ published (Rentsch et al. 2014; Schwartz et al. 2004) or implant-specific quantitative or morphometric grading systems. The need for numerous and consistent sample locations and the need to maintain the tissue implant interface are practical limitations that may not allow morphometric and stereology evaluation.
Surface treatments on devices can evoke significant and unusual responses, which require careful evaluations of local and, potentially, systemic effects. An example would be textured (as opposed to smooth-surface) breast implants causing anaplastic large-cell lymphoma (Swanson 2017).
Some medical devices either have energy sources or are designed to selectively absorb or respond to energy. Tissue effects of transient energy must also be considered and precisely characterized. This may require changes in or additions to existing nomenclature.
Evaluations Using New Technologies
Low-resolution radiography and high-resolution in vivo imaging, such as computed tomography (CT), µ-CT, optical coherence tomography, magnetic resonance imaging, and confocal microscopy have been used to characterize location, movement, and in situ measurements of nonbiodegradable or slowly degrading materials, but for practical reasons, these techniques are seldom used outside of research settings and are generally not applicable to rapidly degrading materials.
IHC to identify specific cell or tissue antigens may be useful but will require sourcing of appropriate antibodies, which are often limited for test species used for biomaterials and medical devices, and will require extensive method development with optimization (Rentsch et al. 2014; Diller, Audet, and Kellar 2015). Use should be on a case-by-case basis.
Conclusion
Evaluation requirements for biomaterials and medical devices continue to grow and evolve. There are similarities and differences between the evaluation of drugs, biologics, and medical devices, but inclusion of a toxicologic pathologist on the product development team provides a valuable understanding of tissue–host responses. Thoughtful application of their basic skill portfolio allows a toxicologic pathologist to provide a tailored safety and efficacy evaluation and communicate risk assessment of novel biomaterials and finished medical devices. Toxicologic pathologists interested in this field need to obtain fluency with regulatory aspects, novel biomaterials, and medical devices, and understand the complexity of medical devices, new materials, and interventions to modulated undesirable host responses to implant biomaterials.
Footnotes
Authors’ Contribution
All authors (SG, JS) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.