
Abstract
The inability to unequivocally predict translatable drug-induced kidney injury in nonclinical studies during pharmacological development is evidenced by drug attrition in human clinical trials. Eight urinary proteins have been qualified as renal safety biomarkers for limited context of use in nonclinical drug development studies in rats. Formal qualification of human renal safety biomarkers is pending the submission of data from prospective clinical trials and analyses of biomarker performance to the Food and Drug Administration and European Medicines Agency by the Foundation for the National Institutes of Health and Predictive Safety Testing Consortium’s Nephrotoxicity Working Group. In vitro kidney platforms may be leveraged to investigate the potential risk of compound-induced acute kidney injury and/or dysfunction. The early assessment of drug-related kidney safety profiles using biomarker-level changes in animal models and in vitro platforms could significantly reduce renal safety-related drug attrition; yet, there are no well-validated in vitro systems to enable comprehensive investigations of compound-induced nephrotoxicity. Thus, histopathology remains the gold standard for diagnosing nephron-specific damage. Traditional and emerging biomarker panels should be combined with histopathology and/or cytopathology to enable early identification of compound-induced kidney injury.
Introduction
Under healthy conditions, the kidneys receive approximately 25% of total cardiac blood flow. The kidneys control fluid (volume, electrolyte, and mineral levels); acid–base balance; synthesize hormones that help produce red blood cells; promote bone health; and regulate blood pressure. The functional unit of the kidney is the nephron, and there are approximately 1 million nephrons per kidney. The key components of the nephron include the glomeruli for passive filtration of the blood and tubules for both active reabsorption and secretion of solutes. Both endogenous and exogenous toxicants may induce nephron-specific damage and subsequent alterations in detoxification and excretion patterns. The traditional biomarkers, serum creatinine (sCr) and blood urea nitrogen (BUN) lack sensitivity and specificity for monitoring alterations in glomerular filtration rate (GFR) that is the best indicator of kidney function. Rises in sCr and/or BUN and decreases in urine output may occur only when greater than 50% of the nephrons are damaged.
The etiology of inducible nephrotoxicity is often poorly understood; however, such may include but are not limited to the following: interstitial nephritis, glomerulonephritis, rhabdomyolysis, thrombotic microangiopathy, altered intraglomerular hemodynamics, crystal nephropathy, tubular cell toxicity, and glomerulosclerosis (Mårtensson, Martling, and Bell 2012). Given the multitude of factors, it is not surprising that candidate drug-induced acute kidney injury (AKI) is associated with significant attrition (Benjamin et al. 2015). Kidney toxicity accounts for 2% of drug attrition during nonclinical studies and 19% in phase 3 clinical studies (Redfern et al. 2010). Several studies have revealed transcriptional mechanisms associated with drug-induced kidney injury (DIKI) in amphotericin B (AmpB)-treated male Jcl:ICR mice (Kondo et al. 2012), vancomycin-treated rats (Dieterich et al. 2009), gentamicin-treated dogs (McDuffie et al. 2013), and gentamicin-treated monkeys (Davis et al. 2004). DIKI may occur following distinct intracellular compound accumulation (Awdishu and Mehta 2017), resulting in acute nephropathies: AKI, chronic kidney disease, and end-stage renal disease (ESRD). To establish AKI in humans, the Risk, Injury, Failure, Loss (RIFLE) and ESRD (Bellomo et al. 2004) and the Acute Kidney Injury Network (AKIN; Mehta et al. 2007) criteria were posited. The diagnostic biomarkers used to define AKI using the RIFLE criteria are relevant increases in sCr and/or loss of urine output. In accordance to the AKIN criteria, AKI is defined by an abrupt (within 48 hr) reduction in kidney function, when an absolute increase in sCr at ≥0.3 mg/dl or ≥26.4 μmol/L, a percentage increase in sCr of more than or equal to 50% (1.5-fold from baseline), or a reduction in urine output based on recorded oliguria of less than 0.5 ml/kg/hr for more than 6 hr. Compounds may also cause dose-limiting AKI in animal models; therefore, dose stopping criteria based on reference values and monitorable increases in sCr concentrations in circulation are often leveraged during nonclinical studies (McDuffie et al. 2010).
Nonclinical Assessment of DIKI
Nonclinical studies conducted as part of regulatory packages for candidate drugs are defined within guidances. As examples, the International Conference on Harmonisation (ICH) M3(R2) (Anonymous 2009) and ICH S7A (Anonymous 2000). The ICH M3(R2) guidance recommends that candidate drug sponsors routinely include sCr and BUN in clinical pathology panels (Table 1) for 28-day (1 month) nonclinical toxicology studies to support clinical trials. Within the ICH S7A guidance, the assessment of kidney function is considered supplementary.
To gain insights regarding the strategies used to investigate kidney safety, the lead safety pharmacologists of the top 15 pharmaceutical companies (defined by 2012 research and development portfolio size) responded to two short anonymous surveys of which a 67% and a 60% response rate was obtained, respectively (Benjamin et al. 2015). Nine of the ten respondent companies conduct renal excretory measurements based on measurements of quantitative urinalysis parameters (also referred to as renal excretory parameters, Table 1) in good laboratory practice (GLP) and/or non-GLP nonclinical toxicology studies. Only five of the ten conduct specific renal safety pharmacology studies include renal excretory measurements (Table 1). On a case-by-case basis, these companies use the regulatory qualified biomarkers that include urinary (Ur) kidney injury molecule 1 (Ur KIM-1), urinary trefoil factor-3 (Ur TFF3), urinary clusterin (Ur CLU), urinary renal papillary antigen 1 (Ur RPA-1), urinary total protein (Ur TP), Ur albumin (Ur ALB), urinary beta 2 microglobulin (Ur β2M), and/or urinary cystatin C (Ur CYS C) and/or nonqualified DIKI biomarkers such as urinary neutrophil gelatinase-associated lipocalin (Ur NGAL), urinary osteopontin (Ur OPN) and serum Cystatin C (sCysC) in nonclinical studies conducted as part of regulatory packages (Table 1). The survey also revealed that renal vascular resistance, mean arterial pressure, renal blood flow via imaging, para-aminohippurate (PAH) clearance, direct GFR measurements via inulin clearance and implantable telemetry, and/or an integrated platform consisting of automated blood sampling and telemetry (Chen et al. 2013) are leveraged to investigate mechanisms responsible for compound-induced AKI. Yet, multiorgan histopathology remains the “gold standard” by which compound-induced AKI is established. Additionally, microscopic analysis, including but not limited to molecular pathology techniques (e.g., immunohistochemistry [IHC] staining, in situ hybridization, Raman imaging), imaging mass spectroscopy (Van Acker et al. 2016), and/or other platforms may be used, especially when a compound induces kidney injury and related measurable changes in soluble biomarker concentrations in the absence of alterations in sCr and/or BUN. Furthermore, DIKI biomarkers that appear to be more sensitive than sCr or BUN but are minimally detectable or undetectable in biological fluids from control animals warrant characterization of the biomarker origins within the kidney by way of molecular pathology.
GLP and/or Non-GLP Nonclinical Toxicology Study Endpoints for Identifying Drug-induced Kidney Injury.

Note: DIKI = drug-induced kidney injury.
a For quantitative urinalysis parameter analysis (also referred to as renal excretory measurements), urine is collected from animals that are individually housed in metabolism cages with ad libitum access to water but without access to food. Up to 16 hr urine collection period for rats and large animals (e.g., dog, monkeys). Up to 6 hr urine collection period for mice.
The Evolution of Mouse Kidney Safety Biomarkers—AmpB Nephrotoxicity in Mice
Most investigations of AmpB-related DIKI biomarker changes in rodents have been carried out in mice (Tonomura et al. 2009; Kondo et al. 2012). AmpB induces AKI in male Jcl:ICR mice following intravenous dosing (Tonomura et al. 2009). The mechanisms responsible for AmpB renal injury include acute binding to tubular epithelial cells and subsequent swelling, lysis/increased tubular permeability, and tubular dysfunction. Subclinical AKI occurs in mice that receive a single dose of AmpB, with increases in urine volume (Ur Vol) and urinary lactate dehydrogenase (Ur LDH) activity and decreases in creatinine clearance (CrCl) and GFR when compared with concurrent controls. The single dose of AmpB at 2 mg/kg induced mild tubular necrosis in the Henle’s loop of the medullary outer layer. Following single intravenous dosing of AmpB at 4 mg/kg, deep breathing and pale pinnae were observed yet disappeared after about 5 min. Significant decreases in terminal body weight due to a reduction in food consumption (although insignificant); significant increases in plasma levels of creatinine, BUN, TP, ALP; significant increases in Ur Vol; and significant decreases in CrCl and GFR were identified from the AmpB (4 mg/kg)-treated mice when compared to controls. The single dose of AmpB at 4 mg/kg induced mild-to-moderate congestion in the medulla, hyaline casts in the cortex, and mild dilatation and necrosis of proximal cortical tubules. No clinical signs or changes in traditional blood chemistries were observed following once-daily intravenous dosing of AmpB at 1 or 2 mg/kg for 1 week. Repeated dosing of AmpB at 2 mg/kg induced mild-to-moderate tubular regeneration in more than half of the animals, and one animal showed mild tubular necrosis in the medulla. Significant increases in Ur Vol and significant decreases in CrCl and Ur TP and urinary N-acetyl-β-
The Evolution of Kidney Safety Biomarkers—AmpB Nephrotoxicity in Rats
Unfortunately, most studies of kidney safety biomarkers have included only male rats; therefore, acute biomarker panel changes were examined in female Sprague-Dawley (SD) rats that received repeated intravenous injections of AmpB at 3 mg/kg/day or vehicle for 10 consecutive days (McDuffie et al. 2016). As expected, AmpB-induced kidney histopathology findings were characterized by slight to moderate cortical and medullary tubular alterations, interstitial inflammation, intratubular granular and inflammatory cell casts, acute pelvic inflammation, and tubular mineralization. Until 2008, the eight fully qualified urinary protein DIKI biomarkers had not been systematically investigated in AmpB-treated rats (Thukral et al. 2005). Hence, McDuffie et al. (2016) demonstrated the value of early elevations in Ur CLU, Ur NGAL, Ur ALB, Ur CYS C, Ur TP, Ur KIM-1, and Ur OPN for detecting subclinical AmpB nephrotoxicity in female SD rats, thereby providing the basis for inclusion of female rats on a case-by-case basis in nonclinical toxicology studies designed to investigate DIKI. Besides expected AmpB binding to tubular epithelial cells and subsequent swelling and lysis/increased tubular permeability and tubular dysfunction, the nature of DIKI in female SD rats remains unclear. Constitutive RPA-1 protein is expressed primarily in medullary collecting ducts, while inducible RPA-1 protein expression may be detected in response to gentamicin-induced proximal tubular regeneration in male SD rats (Zhang et al. 2009). Increased Ur RPA-1 has also been described as an indicator for N phenylanthranilic acid-induced medullary collecting duct degeneration/necrosis. More robust elevations in Ur RPA-1 were detected from in male Han-Wistar rats with more severe lesions when compared to male SD rats (Harpur et al. 2011). Ur RPA-1 protein levels appeared to gradually increase in control male SD rats during the 14-day period (Vinken et al. 2012). Conversely, another study showed stable baseline concentrations of Ur RPA-1 in naive male and female SD rats (Gautier et al. 2010) or variations at different time points over a 44-day period (Rouse et al. 2011). In spite of microscopic evidence of AmpB nephrotoxicity, Ur RPA-1 did not show sensitivity for inflammatory kidney injury in female SD rats treated with AmpB. Future longitudinal studies should include the assessment of RPA-1 messenger RNA (mRNA) expression level changes and/or protein immunolocalization in nephron segments in kidneys from female and male SD rats to better understand the utility of Ur RPA-1 when monitoring for potential sex-dependent mechanisms responsible for AmpB-induced kidney inflammatory injury in different rat strains.
DIKI Biomarkers: The First Formal Qualification of Safety Biomarkers for Regulatory Decision-Making
The U.S. Food and Drug Administration (U.S. FDA), European Medicines Agency (EMA), and Pharmaceuticals Medical Devices Agency, Japan (PMDA), have acknowledged the “first” formal qualification of novel Ur protein biomarkers that are highly sensitive and specific for monitoring DIKI progression in male and female rats (Dieterle et al. 2010). The first formally qualified rat DIKI biomarkers included Ur KIM-1, Ur CLU, Ur ALB, Ur TP, Ur β2M, Ur CYS C, and Ur TFF3. The U.S. FDA and EMA later acknowledged urinary renal papillary antigen 1 (Ur RPA-1) as the “second” singly qualified DIKI biomarker in male and female rats as well as increased level of evidence for and clarified context of use for Ur CLU in male rats (Harpur et al. 2011). The first and second rat DIKI biomarker qualification submissions were from the Predictive Safety Testing Consortium (PSTC)/Nephrotoxicity Working Group (NWG) within the Critical Path Institute (C-Path) and the Health and Environmental Sciences Institute (HESI)/Committee on Biomarkers of Nephrotoxicity within the International Life Sciences Institute (ILSI), respectively.
Steps toward Qualifying Kidney Safety Biomarkers for Limited Context of Use in Clinical Drug Development Studies
It is of interest to further characterize novel biomarker profile changes in situations where compound-induced AKI is present but not detectable by sCr in various animal models (including nonrodents) and humans. Recent successes in the identification, development, and qualification of translational next-generation kidney injury biomarkers have been summarized (Ennulat and Adler 2015). The kidney safety biomarker performance data are paramount for clinical qualification; however, nonclinical data are used to underpin the clinical data by anchoring biomarker profile changes to kidney histopathology findings (Sauer, Walker, and Porter 2015). The Safer and Faster Evidence-Based Translation (SAFE-T) Consortium and C-Path’s PSTC/NWG generated data from healthy human volunteers as well as exploratory cisplatin and contrast agent studies in humans. On the substantiated basis of the qualification advice, respective Letters of Support were issued by the FDA (Anonymous 2016) and EMA (Anonymous 2016) to encourage the further development and exploratory use of urinary α -glutathione S-transferase (Ur α GST), Ur CLU, Ur CysC, Ur KIM-1, Ur NGAL, Ur OPN, Ur ALB, Ur TP, and/or sCysC as biomarkers of kidney tubule injury in early clinical trials. More recently, the Biomarkers Consortium’s Kidney Safety Project aimed to qualify novel biomarkers of drug-induced AKI in humans. The biomarkers were first evaluated via “learning” phase studies. The learning phase studies included a Phase I study in healthy volunteers (Brott et al. 2014) and retrospective analyses of biomarker profile changes in mesothelioma patients (Anonymous 2013). Separate sample aliquots were sent to Pacific Biomarkers (Seattle, WA), Myriad RBM, Inc. (Austin, TX), and a central laboratory at AstraZeneca for analysis. At the central laboratory, urine was analyzed using single analyte assays for osmolality, urinary creatinine (Ur Cr), Ur NAG, urinary potassium, urinary sodium, and Ur TP; and plasma was analyzed for creatinine, potassium, sodium, osmolality, and TP. At Pacific Biomarkers, urine was evaluated using single analyte assays for Ur β2M, creatinine, cystatin C, KIM-1, NAG, and NGAL. At Myriad RBM, Inc., urine was analyzed using the Human KidneyMAP (Multi-Analyte Profile)® v. 1.0 multiplex panel that includes human-specific reagents for detection of urinary alpha-1 (Ur α1M), urinary calbindin D-28k (Ur CALB), urinary IL-18 (Ur IL-18), urinary α-glutathione S-transferase (Ur α-GST), urinary γ-glutathione-S-transferase μ (Ur GSTYb1), urinary retinol-binding protein 4 (Ur RBP4), urinary Tamm–Horsfall Protein also known as uromodulin (Ur THP or Ur UMOD), urinary vascular epithelial cell growth factor (Ur VEGF), urinary connective tissue growth factor (Ur CTGF), Ur β2M, Ur CLU, Ur Cr, Ur CYS C, Ur NGAL, Ur OPN, Ur KIM-1, Ur ALB, Ur TFF3, and sCysC. From the Phase 1 study, confidence intervals (pilot reference intervals), intersubject and intrasubject variability, effects of food intake, effect of sex, and vendor assay comparisons were generated (Brott et al. 2014). The retrospective analyses of biomarker changes in mesothelioma patients enabled prioritization for the novel biomarkers that seem most promising for downstream “confirming” phase studies. The prioritized biomarkers are currently being evaluated in prospective observational studies in adult cystic fibrosis patients receiving tobramycin (Anonymous 2012) and adult head and neck cancer patients receiving cisplatin therapy (Anonymous 2013). The prioritized biomarkers being evaluated to validate whether they outperform sCr and/or BUN for predicting AKI include Ur CLU, Ur CysC, Ur KIM-1, Ur NGAL, Ur OPN, Ur ALB, Ur NAG, and/or Ur TP (Sauer, Walker, and Porter 2015). In August 2018, the U.S. FDA acknowledged that the Kidney Safety Project team (composed of The Foundation for the National Institutes of Health and C-Path's PSTC/NWG) had achieved the first ever qualification of clinical safety biomarkers previously qualified for use in toxicology studies in rats, for use now in Phase 1 clinical trials (Anonymous 2018).
Utilization of Novel Kidney Safety Biomarkers in Toxicology Studies in Beagle Dogs
The primary data warranted for clinical qualification would come from human clinical studies with cisplatin and tobramycin. However, nonclinical data are necessary to underpin the clinical data and anchor biomarker-level changes to morphologic evidence of DIKI incidences and/or severities (Sauer, Walker, and Porter 2015). Unlike the rat, no biomarkers have been qualified for predicting DIKI in other nonclinical animal models. Several mechanisms responsible for cisplatin nephrotoxicity have been described (Wei et al. 2007), leading to its common use as a reference compound to systematically evaluate comparative novel DIKI biomarker level changes in various animal models treated with proprietary compounds with unknown kidney safety liabilities. Cisplatin-related novel DIKI biomarker profile changes in dogs were once underappreciated in the literature; therefore, a 28-day study was designed to evaluate traditional kidney safety biomarker changes in male beagle dogs post intravenous administration of cisplatin (0.75 mg/kg) or vehicle (0.9% saline) for 5 consecutive days (McDuffie et al. 2010). Animals were euthanized when sCr levels measured at 1.9 mg/dl, indicating significant loss of renal function (decreased GFR). Histologically, cisplatin-related renal changes were characterized as proximal tubule dilatation, vacuolization, degeneration, regeneration, and interstitial inflammation. Prestudy sCr levels ranged from 0.6 to 0.8 mg/dl. Post-treatment, on days 1 to 4, sCr concentrations ranged from 0.5 to 1.1 mg/dl; and terminal sCr concentrations ranged from 0.6 to 6.6 mg/dl. Using the canine cytokine/chemokine LINCOplex kit (Millipore/LINCO Research, St. Charles, MO), increases in urinary interleukin (IL)-2, IL-8, monocyte chemoattractant protein-1, granulocyte-macrophage colony-stimulating factor (GMCSF), and keratinocyte-derived chemokine were detected on days 3 to 4 and increases in urinary IL-7 on day 4. This study showed for the first time that inflammatory cytokines/chemokines in urine positively identified acute proximal renal tubular injury in dogs at time points earlier than sCr.
Additional next-generation kidney injury biomarker changes have been evaluated from the male beagle dog model of experimental cisplatin nephrotoxicity (McDuffie et al. 2016) using the previously reported dosing regimen (McDuffie et al. 2010). Most notably, increases in Ur OPN and Ur CLU on day 5 correlated with enhanced OPN and CLU immunopositive staining in damaged cortical epithelium in the proximal tubules, while enhanced KIM-1 staining in damaged corticomedullary tubular epithelium appeared in the absence of rises in Ur KIM-1 (as measured using the second-generation single-plex ELISA kit from ICL, Inc., Portland, OR). This study showed changes in Ur OPN and Ur CLU associated with acute cisplatin nephrotoxicity in dogs and possibly in humans. To further support the clinical qualification of DIKI biomarkers, the PSTC/NWG has commenced investigational studies with tobramycin in SD rats (data not shown), beagle dogs (data not shown), and Cynomolgus monkeys (data not shown).
An integrative overview of known mechanisms underlying gentamicin nephrotoxicity has been described previously (Quiros et al. 2010). To gain further insights, a genomic profiling study was conducted in male beagle dogs that were intramuscularly administered gentamicin (40 mg/kg/day) or vehicle (saline) for 10 consecutive days (McDuffie et al. 2013). Gentamicin-induced acute kidney histopathology changes were localized to the proximal convoluted tubules and characterized as slight-to-marked, diffuse corticomedullary tubular epithelial degeneration/necrosis. Interestingly, gentamicin induced increases in Ur KIM-1 mRNA on day 6, preceding detectable elevations of sCr and/or BUN on days 7 to 8. The increased Ur KIM-1 mRNA correlated with multifocal KIM-1 immunostaining in the corticomedullary tubular epithelial cells. Microarray analysis revealed changes in additional mRNA expression products detected in urine and/or kidney that should be further investigated for use as potential biomarkers for acute gentamicin-related nephrotoxicity in dogs, suggesting the utility of noninvasive urinary genomic parameters for monitoring acute DIKI in dogs. At the time of this study, there were no suitable commercially available canine-specific Ur KIM-1 ELISA kits.
Zhou et al. (2014) reported on the evaluation of the utility of canine-specific single-plex ELISAs for measurements of Ur CLU (Biovender, Czech Republic) and Ur NGAL (Bioporto, Denmark) along with traditional clinical pathology parameters (sCr, BUN, Ur TP, and Ur NAG) and histopathology in beagle dogs treated by intramuscular injection of gentamicin for 9 days. Ur NAG exhibited early changes in concentrations and/or activity levels on day 1 which persisted up to day 9 when compared to the control group. Ur CLU and Ur NGAL levels were significantly elevated in dogs on days 1 and 3, providing insights into the use of Ur CLU with Ur NGAL for detection of DIKI in dogs. Wagoner et al. (2017) evaluated temporal changes in urine-based novel/traditional DIKI biomarkers and metabolomics in gentamicin-treated male and female beagle dogs. The first-generation Canine KidneyMAP® multiplex panel (Myriad RBM, Inc.) was used to measure Ur ALB, Ur CLU, Ur NGAL, and Ur KIM-1 proteins. Additionally, the first-generation Milliplex MAP Canine Kidney Toxicity Panel 1 (Millipore, St. Louis, MO) was used to simultaneously measure Ur ALB, Ur CLU, Ur NGAL, Ur OPN, Ur Cys C, and Ur KIM-1 proteins. A custom KIM-1 sandwich immunoassay was established at GlaxoSmithKline (GSK) using commercial canine-specific KIM-1 reagents (rabbit monoclonal capture antibody, rabbit polyclonal detection antibody, recombinant canine HAVcr-1/tissue injury molecule 1 (TIM-1) protein as calibrator from Sino Biological, Wayne, PA) and the MesoScale Discovery® platform (Meso Scale Discovery, LLC, Rockville, MD). In this study, gentamicin-induced kidney lesions in male dogs were characterized by moderate to severe proximal tubular epithelial cell degeneration/necrosis, epithelial cell regeneration, and dilitation. Urinary metabolite changes included time and treatment-dependent increases in lactate, taurine, glucose, alanine, and citrate as well as 9 other known metabolites. As early as 3 days post-dose, gentamicin induced increases in Ur ALB, Ur CLU, Ur NGAL, and Ur TP. KIM-1 is a type I transmembranous protein whose expression is markedly shed from injured proximal tubule epithelial cells in gentamicin-treated rats (Lou et al. 2016). Various assays were used to assess gentamicin-induced changes in Ur KIM-1 protein. Leveraging these assays across three independent research laboratories revealed the same mild elevations in urinary KIM-1 protein. Interestingly, when compared to the custom KIM-1 sandwich immunoassay (established at GSK), increases in Ur KIM-1 concentration were less reliable based on utilization of the first-generation canine KidneyMAP multiplex panel (Myriad RBM, Inc.) and the first-generation Milliplex MAP Canine Kidney Toxicity Panel 1 (Millipore). Immunohistochemical identification of KIM-1 protein was detected along the lumen of the proximal tubules in the outer medulla of the kidneys from gentamicin-treated dogs, in line with the localization of the treatment-related tubular degeneration. In situ hybridization also showed weak or mild elevations in KIM-1 mRNA that was localized in the tubules of the outer medulla. These findings suggest a more blunted KIM-1 biomarker response to gentamicin-induced proximal tubular injury in beagle dogs differs from changes seen with gentamicin nephrotoxicity in rats.
Utilization of Novel Kidney Safety Biomarkers in Toxicology Studies in Cynomolgus Monkeys
Davis et al. (2004) first reported on confirmation of rodent gene expression changes associated with nephrotoxicity in a 7-day nonhuman primate study, providing preliminary evidence for using gene transcript levels to predict the onset of drug-induced proximal convoluted tubular damage in female cynomolgus monkeys after exposure to the aminoglycoside gentamicin, the experimental oligosaccharide antibiotic everninomicin, or a combination of gentamicin and everninomicin for 7 days. Monkeys receiving these drugs in combination developed kidney injury as early as day 1. By day 7, monkeys dosed with everninomicin alone developed kidney lesions; however, animals that received both compounds had more severe lesions. On day 1, urinary γ-glutamyl transpeptidase (Ur GGT) activity was significantly greater than controls in the everninomicin-treated animals as well as those in the combination group. On day 3, Ur GGT activity was significantly greater than controls in gentamicin-treated animals. Ur NAG activity increased in a similar manner at all time points in treated versus control groups. The everninomicin group had significantly greater sCr on days 1, 3, and 7. Of note, Clu, Spp1, and Havcr-1 mRNA expression level changes and other genes (wild-type p53-activated fragment 1 [waf-1], matrix metalloproteinase-9 [MMP-9], vimentin [Vim]) indicated tubular toxicity on days 1 or 7 postdosing. At the time of this study, commercially available ELISA kits that cross-react with Ur CLU, Ur OPN, and/or Ur KIM-1 from nonhuman primates did not exist.
Multiplex biomarker panel assays were developed (in part) to support monitoring for compound-related target organ safety liabilities. Guha et al. (2011) first reported on the usefulness of the Human KidneyMAP v. 1.0 multiplex panel (Myriad RBM, Inc.) for detection of urine-based protein biomarkers in combination with histopathology, traditional urinary biomarker measurements, and IHC staining to reliably detect (1S,2S)-3-(methylamino)-2-(naphthalen-2-yl)-1-phenylpropan-1-ol (PRC100-SS)-induced dose proportional distal tubule and collecting duct injury in male and female cynomolgus monkeys. At doses higher than those tested in the monkey (noted below), PRC200-SS did not cause nephrotoxicity in Wistar rats when dosed at 100 and 70 mg/kg/day for 7 days and 1 month, respectively. The exact mechanism by which PRC200-SS induces kidney injury in the cynomolgus monkey remains unknown. PRC200-SS (a triple reuptake inhibitor), administered via nasogastric intubation, resulted in a maximum tolerated dose (0.4–100 mg/kg with 6 or 7 days of drug-free periods between each dose), 7-day repeat dose pilot study in naive animals (10, 25, or 50 mg/kg/day), and a 1-month repeat dose study (vehicle, vehicle, 5 mg/kg/day [low dose], or 20 mg/kg/day [high dose with a drug-free recovery period of 4 weeks]). Segment-specific kidney lesions were demonstrated using triple IHC staining for CALB, aquaporin 2 (APQ2), and aquaporin 1. Three soluble markers (Ur α-GST, Ur KIM-1, and Ur TFF3) in the multiplex panel did not cross-react well with the cynomolgus monkey urine samples as expected (also reported by the manufacturer). The multiplex panel revealed a dose proportional increase in the excretion of Ur CALB and Ur CLU in the PRC200-SS-treated cynomolgus monkeys with levels returning to baseline during the drug-free recovery period. This study provided evidence of Ur CALB and Ur CLU as potential DIKI biomarkers with utility in the clinic.
More recently, Chen et al. (2017) summarized perspectives on using the Human KidneyMAP v. 1.0 multiplex panel (Myriad RBM, Inc.) to detect cisplatin-induced acute proximal cortical tubular toxicity in male and female cynomolgus monkeys that received a single intravenous administration of cisplatin at 2.5 mg/kg. The cisplatin-induced kidney injury was characterized by acute and progressive proximal cortical tubular degeneration/necrosis, regeneration, tubular dilitation and proteinaceous casts in the absence of statistically significant changes in traditional plasma biochemistry and urinalysis parameters. When normalized to urinary creatinine, cisplatin induced significant increases in Ur KIM-1 in females on day 4, Ur CALB in males and females on day 4, decreases in Ur THP in males on days 1, 4 and 9, and increases in Ur CLU in females and males on days 15 and 20, respectively, when compared to concurrent controls. This study revealed the usefulness of the Human KidneyMAP v. 1.0 multiplex panel (Myriad RBM, Inc.) when measuring for changes in Ur KIM-1, Ur CALB, and Ur CLU to detect cisplatin-induced acute/progressive cortical tubular injury in male and female cynomolgus monkeys. To further underpin the clinical qualification of DIKI biomarkers, more definitive studies with cisplatin in cynomolgus monkeys are ongoing within the PSTC NWG.
Due in part to the lack of sensitivity (poor cross-reactivity) by the Human KidneyMAP v. 1.0 multiplex panel (Myriad RBM, Inc.) to detect protein biomarkers in urine from nonhuman primates, conventional single-plex ELISAs (summarized in the next paragraph) have been developed to aid the detection of DIKI in this species. Gautier et al. (2016) first reported on the usefulness of histopathology, conventional clinical pathology parameters in parallel with single-plex ELISAs, the Human KidneyMAP v. 1.0 multiplex panel, and an immunoprecipitation-LC/MS multiplex assay for determination of DIKI biomarker changes from male cynomolgus monkeys given daily intramuscular injections of gentamicin at dose levels of 10, 25, or 50 mg/kg/day for 10 days. Gentamicin induced a dose-dependent increase in kidney tubular cell degeneration/necrosis. The novel urinary biomarkers, Ur ALB, Ur α1M, Ur CLU, and Ur OPN as well as the traditional clinical pathology parameters, Ur TP and Ur NAG, were more sensitive than sCr and BUN. This study gave more confidence in the noninvasive DIKI biomarkers of proximal tubule injury in cynomolgus monkeys which may be potentially useful in humans.
The PSTC NWG has conducted studies to further evaluate single-plex ELISAs (Ur KIM-1 [Meso Scale Discovery, LLC]; Ur Clu [BioVendor-Laboratorni medicina a.s., Brno, Czech Republic or R&D Systems, Minneapolis, MN; IBL-America, Minneapolis, MN]; Ur NGAL [BioPorto, Denmark], and Ur CYS C [BioVendor-Laboratorni medicina a.s.]. They also evaluated an immunoprecipitation-LC/MS multiplex assay for simultaneous detection of Ur RBP4, Ur CLU, Ur OPN, and Ur α1M (Signatope GmbH, Reutlingen, Germany) using urine and kidney tissue samples from male and female cynomolgus monkeys with gentamicin-induced acute experimental nephrotoxicity. In control kidneys, IHC staining showed KIM-1 localization that appeared as a granular cytoplasmic signal in the proximal convoluted tubules. In kidneys from gentamicin-treated monkeys showed the KIM-1 translocated to an apical (luminal) membranous and/or cytoplasmic distribution in tubules with degenerative changes of variable severities.
The immunolocalization of α1M, KIM-1, OPN, and NGAL proteins in male cynomolgus monkeys with AmpB-induced acute corticomedullary tubule injury have been reported (McDuffie et al. 2016). AmpB-related immunoreactivities included α1M in damaged proximal tubule epithelium, KIM-1 in damaged medullary tubule epithelium, OPN mostly in the infiltrating cells of cortical tubule interstitium, and NGAL in the granular and cellular casts in dilated cortical tubules. In a follow-up study, gene and/or proteins as candidate DIKI biomarkers in urine and/or kidneys; non-specific kidney safety biomarkers in serum and/or urine; and histologic indices of kidney injury were evaluated in male cynomolgus monkeys that received AmpB (data not shown, author’s unpublished data). Adaptable dose range finding and definitive study designs for evaluating traditional versus novel DIKI biomarker profiles in animal models treated with nephrotoxic compounds are summarized in Table 2. In general, the dose range finding study is intended to explore potentially dose-dependent DIKI biomarker changes when anchored to kidney (and other tissue) histopathology findings and define a minimally nephrotoxic dose level that would be evaluated via a definitive study.
Adaptable In Vivo Drug-induced Kidney Injury Study Designs.
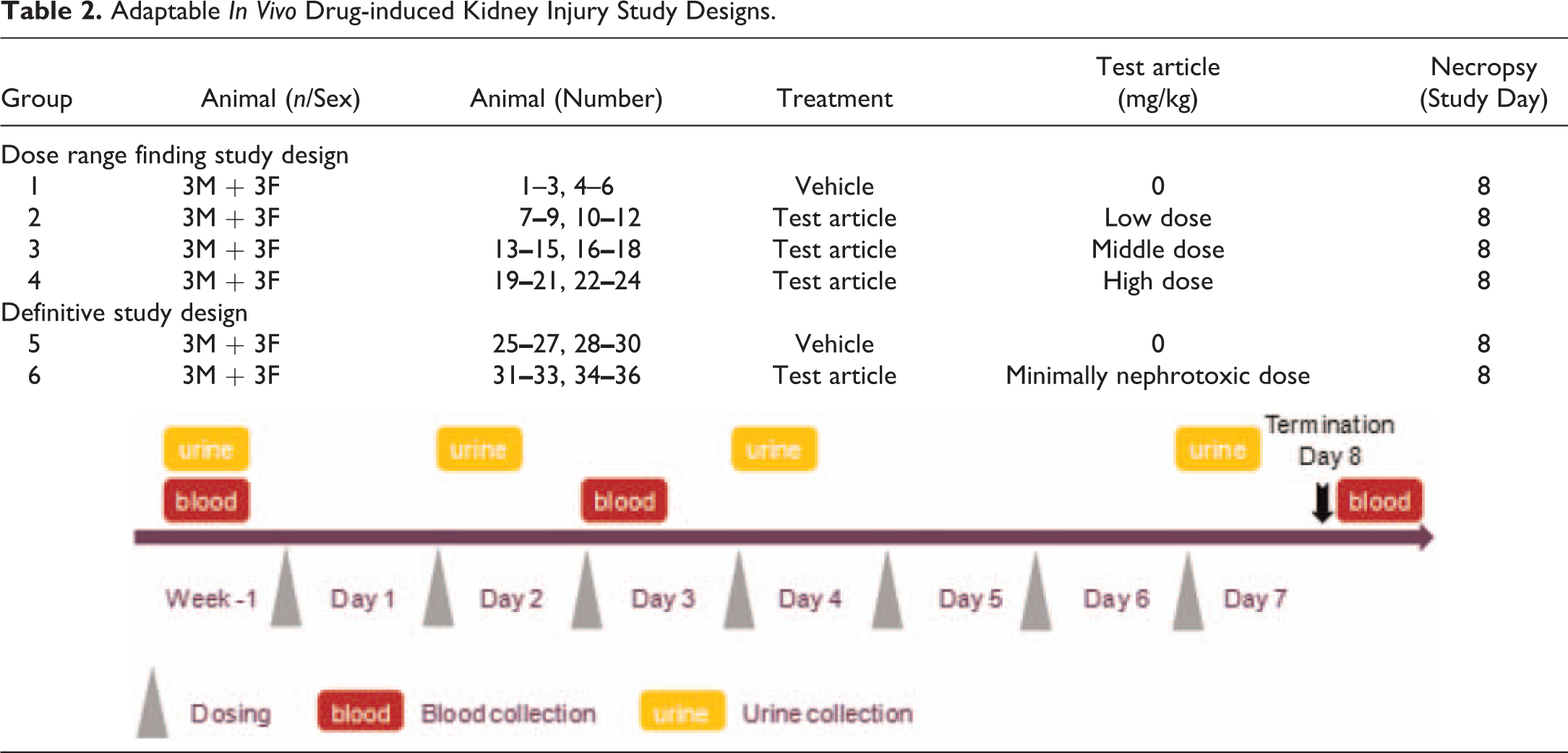
miRNAs as Next-generation Kidney Safety Biomarkers
Mechanistic context of use is emerging for miRNAs that regulate over 50% of all protein-coding genes at the post-transcriptional level. The HESI/Committee on Biomarkers of Nephrotoxicity conducted a benchmark study that revealed miRNA-34c-3p as an early predictive biomarker for doxorubicin-induced glomerular injury progression in male SD rats (Church et al. 2014). Let-7d, miR-203, and miR-320 appeared as promising novel urinary biomarkers for gentamicin-induced kidney tubular epithelial injury in male SD rats (Nassirpour et al. 2014). Pavkovic et al. (2016) reported on Ur KIM-1 and miRs (-21, -200c, and -423) in malignant mesothelioma patients who received intraoperative cisplatin therapy. Evidence for human proximal tubular epithelial cells (HPTECs) as the source of miRNAs in urine was obtained by in situ hybridization as well as in the medium of cultured 2-dimensional (2D) HPTECs treated with cisplatin and 4-aminophenol. A panel of miRNAs as potential cortical tubular toxicity biomarkers in cynomolgus monkeys has been described; however, additional validation studies are ongoing (Veeranagouda et al. 2015). Additionally, the PSTC NWG and HESI/ Committee on Biomarkers of Nephrotoxicity have partnered to systematically evaluate the utility of urine and/or plasma-based miRNAs as novel DIKI biomarkers in dogs and in cynomolgus monkeys following exposure to standard of care drugs commonly associated with nephrotoxicity in humans (e.g., cisplatin and AmpB).
The Utilization of In Vitro Platforms to Predict Compound-induced Nephrotoxicity
The assessment of compound-induced AKI and related biomarker changes are often studied in animal models; however, animals are sometimes referred to as outdated tools with limited predictability (Dehne, Hasenberg, and Marx 2017). In vitro kidney platforms require fewer live donors (animals and/or humans) and minimal amounts of test compound to possibly recapitulate studies performed in vivo. Yet, validated and accepted in vitro platforms for predicting compound-induced nephrotoxicity are emerging. Physiological conditions lacking in 2D cultures are more evident in newer 3-dimensional (3D) rapidly bioprinted platforms (King et al. 2017) and innovative microfluidic chip platforms (Jang et al. 2013). The 3D bioprinted human proximal tubule tissues (referred to as “3D PT ExViveTM Human Kidney Tissue” from Organovo, San Diego, CA) appeared to recapitulate characteristics of human kidney in vivo and enabled toxicity studies post daily (14-day) treatment with cisplatin in culture media supplemented with a final concentration of 2.5% v/v fetal bovine serum (FBS) in both the apical and basolateral compartments showed decreased cell viability and GGT activity and increased LDH release. The 3D PT ExViveTM Human Kidney Tissue also enabled fibrosis studies following daily (7-day) treatment with transforming growth factor beta 1 (TGF-β1), showing concentration-dependent decreases in GGT activity and increases in collagen 1 (COL1A1), connective tissue growth factor (CTGF), fibroblast-activating protein, and platelet-derived growth factor receptor β. Conversely, novel DIKI biomarker characterizations in 3D models remain limited to measurable LDH released from primary human proximal tubular epithelial cells (Biopredic Inc., Rennes, France) cultured under flow conditions in the microfluidic chip device including when compared to biomarker profile changes under static condition (24-well Transwells, Corning; Tewksbury, MA), indicating cytotoxicity (e.g., cisplatin). The utilization of both in vivo models and in vitro platforms including immortalized models (Wieser et al. 2008) would give confidence when predicting compound-induced nephrotoxicity. Yet, there are limitations to consider relative to commonly used mouse strains (e.g., C57BL6) when studying dose-limiting nephrotoxicities (Wei et al. 2007). Case in point, in C57BL6 mice treated with cisplatin moribundity and mortality occurs within 2 to 4 days following single high-dose exposure; however, Sharp and Siskind (2017) recently reported on the development of a better mouse model to study cisplatin-induced kidney injury using an albino, inbred mouse strain that is named after its susceptibility to Friend leukemia virus B (FVB). Wei et al. (2007) described the unique activation and involvement of p53 in cisplatin-induced nephrotoxicity in C57BL6 mouse as well as in 2D cultured primary proximal tubular cells from this strain.
In vitro Evaluation of DIKI Biomarkers Using 2D Rat NRK-52E Cells
The NRK-52E cell line (de Larco and Todaro 1987) is an epithelioid clone of the parental NRK cell cultures that were derived from rat proximal tubule epithelium (Duc-Ngugen, Rosenblum, and Zeigel 1966). 2D NRK-52E cells have limited use for identifying changes in selected biomarkers that are shed from damaged kidney proximal tubular epithelium in rats. Dose- and time-dependent increases in Havcr-1, Timp-1, Lcn-2, Spp1, Clu, and Vim mRNA expression levels were detected in male F344/N rat kidneys but not in 2D NRK52E cell cultures following exposure to ochratoxin (Rached et al. 2008). We investigated the potential in vitro context of use for select DIKI biomarker changes in cisplatin-treated 2D NRK-52E cells using Western blot analysis (author’s unpublished data). Cisplatin (30 μM) induced apoptosis of 2D NRK-52E cells showing downregulation of Bcl-2 and activation of cleaved caspase-3. The protein levels of KIM-1 and α-GST were significantly increased in the conditioned media of 2D NRK-52E cells treated with cisplatin. The levels of Ur KIM 1 and Ur α-GST were significantly increased in cisplatin-treated SD rats (data not shown, author’s previously unpublished data), providing in vivo validation of the in vitro results. These results demonstrated that KIM-1 and α-GST can be used as biomarkers for cisplatin-induced nephrotoxicity in 2D NRK-52E rat cells.
In Vitro Evaluation of Autophagic Responses to DIKI Using 2D Rat Proximal Tubule Cell Models
Ishihara et al. (2013) first demonstrated two pathways associated with autophagy and mitophagy in NRK-52E cells after oxidative stress. Specifically, p53-sestrin-2 and HIF-1-BNIP3 resulted in protection of renal tubules during acute hypoxia in vitro and ischemia-reperfusion (I/R) acute kidney injury in rats. These phenomena highlight a caveat for rat kidneys, which are highly susceptible to vacuolation that may precede degeneration/necrosis in proximal tubules, distal tubules, and/or ductal epithelium (Frazier et al. 2012). Interestingly, unconjugated polyethylene glycol (PEG) of high molecular weights accumulate in rat tubular epithelium and induce cytoplasmic vacuolation (Rudmann et al. 2013). In rat following intravenous administration of 100 mg/kg unconjugated PEG at 40 kDa for 3 months, tissue distribution and the vacuolation were observed as an adaptive change that was further characterized by the localization of cytoplasmic PEG immunoreactivity. Male and female SD rats that received the dimeric PEG linked tumor necrosis factor-binding protein (TNF-bp) of <70 kDa at 20 or 40 mg/kg for 3 months, tubular vacuolation showed only partially reversible after a 2-month recovery period (Bendele et al. 1998). The cases of unconjugated PEG and PEG-linked protein-induced tubular vacuolation appeared in the absence of changes in traditional or novel DIKI biomarkers or soluble indicators of kidney function in serum and/or urine and were considered non-adverse. In male and female rats, kidney tubular vacuolation occurred following intravenous administration of the 40 kDa PEG moiety of macugen® (pegaptanib), an anti-angiogenic aptamer for the treatment of neovascular age-related macular degeneration (Anonymous 2012). Slight-to-mild chronic progressive nephropathy was observed in all repeated dose group animals and concurrent controls however, in male rats administered 10 mg/kg/day, a slight decrease in serum total protein and albumin was evident, suggesting chronic progressive nephropathy. Mechanisms responsible for tubular vacuolation induced by a proprietary PEG-linked peptide in SD rats and in 2D NRK-52E cells have been investigated (data not shown, authors’ unpublished data). To potentially establish fit-for-purpose for 2D NRK-52E cells, we considered that Periyasamy-Thandavan et al. (2008) described autophagy as a cytoprotective response to cisplatin-induced stress that occurred hours earlier than cell death of rat kidney proximal tubular cell line (Dr. Hopfer, Case Western Reserve University, Cleveland, OH) transfected with the anti-apoptotic gene, B-cell lymphoma 2 (Bcl-2). In the Bcl-2 transfected rat proximal tubular cells, cisplatin caused autophagy in a concentration- and time-dependent manner prior to apoptosis. In our study, concentration- and time-dependent cisplatin nephrotoxicity (stress and cell death) was assessed in 2D NRK-52E cells at 0, 1, 3, 6 and 12 hr post-treatment (author’s unpublished data). Cell stress (perinuclear cytoplasmic vacuolation) was visualized by routine microscopy. The subcellular localization and redistribution of light chain 3 (LC3) from cytosol to mature autophagosomes was visualized by confocal microscopy. Cisplatin (30 μM) induced minimal vacuolation by 1-hr post-treatment. Light chain 3 subunits (LC3-I and LC3-II) and LAMP2 (lysosomal-associated membrane protein-2) were used as indicators of autophagy induction and autophagic flux. LC3-II is the autophagic form of LC3. Cisplatin-induced LC3-I, LC3-II, and LAMP2 accumulation was revealed using Western blot analysis. We developed a green fluorescent protein (GFP)-LC3 puncta assay to visualize autophagosomes in 2D NRK-52E cells. Increases in punctate LC3-I expression staining were observed starting between 3 and 6 hr, while increases in localized LC3-II and LAMP2 expression levels were evident as early as 1 hr after cisplatin-induced cell stress and remained increased thereafter. The cisplatin-induced increases in LC3-II puncta were suppressed by the inhibitor of autophagy and apoptosis 3-methyladenine (3-MA) at 5 µM. This study showed cisplatin-induced autophagy in a dose- and time-dependent manner prior to apoptosis represents an important cytoprotective mechanism for 2D NRK-52E cell survival.
In Vitro Evaluation of DIKI Biomarkers Using 2D Human Primary Kidney Cells
2D human primary kidney cell cultures may emulate intact tissue to provide more translatable findings (Glynne 2000); however, they often show fewer characteristic properties following repeated passaging. Selected human renal cell lines have been developed by either continuously or conditionally introducing viral oncogenes, for example, the human kidney-2 (HK-2) cell line was derived from human normal kidney proximal tubule epithelial cells and established by transduction via exposure to a recombinant retrovirus containing the human papilloma virus (HPV) 16 E6/E7 genes. 2D HK-2 cells can be cultured in serum-free media for more than one year, while retaining select functional characteristics of proximal tubular epithelium and freshly isolated proximal tubule cells, including but not limited to brush border enzyme activity, pH-dependent ammoniagenesis and Na+-dependent/phlorizin-sensitive sugar transport (Ryan et al. 1994). These characteristics suggest some utility when assessing compound-induced nephrotoxicity in vitro. Conversely, 2D HK-2 cells lack a stable transcriptome due to the inactivation of p53, creating a non-quiescence cancerous cell phenotype that cannot form domes, showing low transepithelial electrical resistance, and does not exhibit full contact-dependent growth inhibition yet grow from monolayers postconfluence. Sohn et al. (2013) extensively evaluated cisplatin-induced DIKI biomarker expression (gene/protein) levels in 2D HK-2 cells. There was detection of significantly increased Havcr-1, Calb, and Timp-1 mRNA in HK-2 cells as well as increased expression levels of KIM-1, CALB, TIMP-1 proteins but not β2-M, CysC, NGAL, or clusterin proteins in the conditioned media. Additionally, in vivo studies in rats revealed significant increases in Ur KIM-1, Ur CALB, and Ur TIMP-1 proteins in cisplatin-treated rats.
In Vitro Evaluation of DIKI Biomarkers Using Canine Kidney Cell Lines
In September 1958, Madin-Darby canine kidney (MDCK) cells were isolated from epithelial cells from the kidney tubules of an adult female Cocker Spaniel by S. H. Madin and N. B. Darby (ATCC). These cells are the most widely used canine kidney cell line and are suitable for 3D culturing (Ananymous 1958; O’Brien, Zegers, and Mostov 2002). MDCK cells represent spontaneously immortalized cells, where immortalization was coincidental, and the immortalization-inducing agent is not known. 2D MDCK cells show upregulated expression levels of an osteopontin-related 20-kDa polypeptide protein postexposure to choroquine at 100 µM (Ullrich et al. 1991), clusterin mRNA due to apoptosis-inducing stimulus (Flach, Cattaruzza, and Koch-Brandt 1995), and KIM-1 mRNA/protein following ∊-toxin-induced cytotoxicity (Ivie et al. 2011). More recently, epithelial–mesenchymal transition in MDCK cells was characterized using high-throughput transcriptome and miRNAome profiling methods; and select differentially-expressed genes were validated using real-time quantitative polymerase chain reaction: epithelial cadherin 16 (CDH16) and slit homolog 2 (SLIT2) as well as cfa-miR-194, hsa-miR-675-3p, and cfa miR 802 (Shukla et al. 2015). Additional functional studies are warranted to the roles for these three miRNAs relative to DIKI in vitro.
Summary
Both sCr and BUN levels are traditional kidney safety biomarkers that are insensitive and non-specific indicators of DIKI. In response to this concern, novel biomarkers have been identified in rats, of which eight (see Steps toward Qualifying Kidney Safety Biomarkers for Limited Context of Use in Clinical Drug Development Studies section) have been qualified by the FDA, EMA, and PMDA for use in nonclinical drug development studies as well as clinical trials to guide regulatory decisions (case by case). Next-generation DIKI biomarkers are under further investigation in in vitro platforms, animal models, and humans (Table 3) to support clinical DIKI biomarker qualifications by regulatory authorities. Systematic evaluations of kidney-enriched miRNA panels as candidate DIKI biomarkers in plasma and/or urine as DIKI biomarkers are ongoing. 3D kidney organoids express DIKI biomarkers (e.g., KIM-1, CLU, OPN) in response to cisplatin and doxorubicin (Wilmer et al. 2016). Yet, the accuracy of predicting and identifying an early nephrotoxic (cisplatin, tenofovir, cyclosporin A, and tobramycin) events (expression-level changes in miRNAs: mir-29a, mir-34a, and mir-192; enzymes: NAG, LDH, and GGT; and/or proteins: e.g., KIM-1) remain under investigation using emerging in vitro microfluidics platforms such as that developed by Mimetas in the Netherlands, utilizing the human proximal tubule epithelial cell line (ciPTEC-OAT1) cultured on the three-lane OrganoPlate® positioned on an interval rocker platform (Suter-Dick et al. 2018). Notably, parallel tissue chip studies and traditional 2D culture conditions using two different renal proximal tubule epithelial cell types (primary human kidney cells designated as “HIM-31 cells” from the University of Washington and human renal proximal tubule epithelial cells also referred to as RPTECs [CC-2553, Lot #0000581945] from Lonza), revealed the importance of developing microphysiological platforms using renewable cell sources to monitor for expression level changes in KIM-1 protein (Sakolish et al. 2018). Overall, iterative improvements to the tactical strategies intended to enable early detection of DIKI would potentially reduce the incidence of dose-limiting nephrotoxicity in patients.
Mouse, Rat, Dog, Monkey, and/or Human Drug-induced Kidney Injury Biomarkers: Current Status and Context of Use during In Vivo and/or In Vitro Studies.

Footnotes
Author’s Note
This article is based upon selected work supported by C-Path’s/PSTC.
Acknowledgments
The author appreciate both the PSTC/NWG members and Janssen colleagues for their support of relevant research activities described within this manuscript, as well as the input from FDA and EMA scientists who serve as advisors. The author also thank Sinae Lee, PhD, for establishing the GFP-LC3 puncta assay to evaluate mechanisms responsible for inducible proximal tubular vacuolation in NRK-52E cells.
Author Contributions
JM contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. The author gave final approval, and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential, real, or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.