
Abstract
Ricin toxin, a type 2 ribosome-inactivating protein and a category B bioterrorism agent, is produced from the seeds of castor oil plant (Ricinus communis). Chronic pathological changes in survivors of aerosolized ricin exposure have not been reported in primates. Here we compare and contrast the pathological changes manifested between rhesus macaques (RM) that succumbed to lethal dose of ricin (group I) and survivor RM exposed to low dose of ricin (group II). All animals in group I exhibited severe diffuse, necrotizing bronchiolitis and alveolitis with fibrinopurulent bronchointerstitial pneumonia, massive alveolar, perivascular and peribronchial/bronchiolar edema with hemorrhage, and necropurulent and hemorrhagic tracheobronchial lymphadenitis. All animals from group II had multifocal, fibrosing interstitial pneumonia with prominent alveolar histiocytosis and type II pneumocyte hyperplasia. Subacute changes like infiltration by lymphocytes and plasma cells and persistence of edematous fluid were occasionally present in lung and tracheobronchial lymph nodes. The changes appear to be a continuum wherein the inflammatory response shifts from an acute to subacute/chronic reparative process if the animals can survive the initial insult.
Introduction
Ricin toxin produced from the castor plant (Ricinus communis) is one of the most potent and lethal toxins known to man. Ricin is classified as a type 2 ribosome-inactivating protein (RIP). Type 2 RIPs have an A chain (RTA) which is a glycosidase covalently linked by a single disulfide bond to a B chain (RTB) which is a lectin. RTB is catalytically inactive, but serves to mediate entry of the A-B protein complex into the cytosol (Olsnes and Pihl 1973; Olsnes 2004; Endo and Tsurugi 1987; Endo et al. 1987). Endocytosis of the ricin toxin is followed by retrograde translocation of the toxin via the Golgi apparatus. The catalytically active RTA is an RNA N-glycosidase that depurinates large ribosomal subunits by removing specific adenyl residues from the site of interaction with elongation factors resulting in inhibition of protein synthesis and cell death (Olsnes and Pihl 1972; David, Wilkinson, and Griffiths 2009).
Ricin has been used in a wide array of biological applications by virtue of its anticancer properties (Mosinger 1951; Kreitman et al. 2011) and immunotoxicity (Olsnes et al. 1989; Frankel and Willingham 1998). However, ricin’s high potency, extremely small lethal dose, ease of aerosolization, and global availability of castor beans make it a potential biological weapon and a bioterrorism agent. Hence, it is classified as a category B bioterrorism agent and a Schedule 1 chemical.
Toxicity and resultant pathology subsequent to ricin exposure varies with route of exposure, dose, and species/strain of the laboratory animal used. Pathological changes resulting from lethal doses of ricin administered via parenteral, intratracheal instillation, and aerosolization have been reported in rodents, rabbits, and nonhuman primates (reviewed in Roy et al. 2012). Parenteral administration via intraperitoneal and intravenous routes resulted mostly in extrapulmonary necrotizing and hemorrhagic changes that affected lymphoid organs, heart, kidney, and intestinal tracts (reviewed in Salem and Katz 2006). Oral administration of ricin in mice was toxic only at very high doses (100 mg/kg), which resulted in mucosal erosions, necrosis, and hemorrhage in the intestinal tract and nasopharynx with widespread lymphoid depletion. Lethal doses of inhaled ricin in mouse (LCt50 [Lethal Concentration and time for 50% mortality] 4.54–11.21 mg/min/m−3) and rabbits (LCt50 4 mg/min/m−3) resulted in similar lesions across the species, which primarily affected lung and consisted of massive pulmonary edema and destructive inflammatory damage affecting bronchiolar and alveolar integrity (Brown and White 1997; Griffiths, Phillips, and Holley 2007). So far, there has been one study that specifically addresses the acute pathological changes in rhesus macaques (RM) following inhalation of supralethal doses (21–41.8 µg/kg) of ricin (Wilhelmsen and Pitt 1996). Lesions in this study resembled other species, were confined to the lung and tracheobronchial lymph nodes, and included fibrinohemorrhagic pneumonia, alveolar edema, and necrotizing alveolitis and bronchiolitis.
Most of the previous aerosolized ricin studies have used a lethal/supralethal dose of ricin and have documented the acute changes (within 30–48 hr postexposure) and then death. However, to our knowledge, lesions due to a sublethal dose of ricin that enables survival with different pathological changes have not been documented in a nonhuman primate model.
In this article, we have corroborated the pathological changes due to supralethal doses of aerosolized ricin based on a calculated LD50 (5.8 µg/kg, Roy et al. 2012), which is significantly lower than that published previously and have described the pathological changes in animals exposed to nonlethal dose of ricin.
Materials and Method
Animal Husbandry
RM (Macaca mulatta) monkeys of Indian origin were bred and born at the Tulane National Primate Research Center (TNPRC; Covington, LA), which is a U.S. Department of Agriculture (USDA) licensed and fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Animals were singly housed and fed primate chow (Harlan Teklad, Madison, WI) and water ad libitum. Animals were directly observed a minimum of twice daily for clinical changes. Animals determined to be in respiratory distress and those that survived until the end of each trial were euthanized by an overdose of sodium pentobarbital, consistent with the recommendation of the American Veterinary Medical Association’s (AVMA) Panel on Euthanasia, and submitted for necropsy. All methods were approved by the Tulane Institutional Animal Care and Use Committee (IACUC).
Experimental Design and Treatments
This study was performed as a part of a larger toxin therapeutic evaluation study that involved more than 40 RM. From these, tissues from a subset of 9 RM (Macaca mulatta) that had served as study controls and exposed to various doses of ricin were analyzed. The animals were classified into 2 groups: group I (n = 6) that received a supralethal dose of ricin (13.2–27.3 µg/kg), and group II (n = 3) that received a sublethal dose of ricin (1.9–5.2 µg/kg; Table 1). Animals in group I were euthanized and necropsied once they exhibited 2 or more of the response-based end points defined in the pain assessment guidelines for this study, which fell within 30 to 48 hr for all these animals. Group II macaques survived ricin exposure and were euthanized 11 days after exposure (Table 1).
Ricin treatment groups and dosage.
a Time to necropsy based on the manifestation of clinical signs and distress that warranted euthanasia.
Ricin Aerosolization, Dosing, and Calculation
Ricin aerosol exposures were performed within the infectious disease aerobiology laboratories at the TNPRC. The ricin toxin isolated from Ricinus communis var. zanzibariensis used in this study was supplied under contract from the Defence Science & Technology Laboratory (Dstl; Salisbury, UK). Aerosolization, dosing, and delivery of ricin were performed as described previously (Roy et al. 2012). Inductive plethsymography that measures volume of air breathed by each individual animal per minute was performed just before the exposure. Briefly, the animals were placed in a Class III Biological Safety Cabinet (BSC) with the head only in the exposure chamber. Ricin toxin was dissolved in 10-ml sterile phosphate buffer saline to the desired concentration for each animal based on plethsymography data for each animal obtained 2 days before the exposure. The ricin aerosols were generated directly in the head-only chamber using a Collision 3 jet nebulizer (BGI Inc., Waltham, MA) with fully automated management control system (Biaera Technologies, Hagerstown, MD). The nebulizer operated at 18 lb/in.2 equating to a flow of 7.5 L/min and produced 3.0E + 04 particles/cc with a mass median aerodynamic diameter (MMAD) of ∼1 µm. The aerosol exposure lasted 10 min for each animal. Air samples were continuously obtained during the exposure and the protein concentrations of these samples were determined using micro-bovine serum albumin (BSA) protein assay kit (Thermo scientific, Waltham, MA). The aerosol concentrations were determined and the inhaled dose of ricin for each animal was calculated by multiplying the empirically determined aerosol exposure concentration (µg/L air) in the chamber by volume of air estimated to have been breathed by the animal (via results of plethsymography just before exposure; Table 1). The LD50 for ricin calculated in our lab was 5.8 µg/kg body weight (Roy et al. 2012), and the aim in this study was to achieve approximately 3 times this concentration for all exposures that were considered lethal.
Tissue Collection, Histological Analysis, and Special Stains
After gross necropsy, tissues were collected in neutral buffered zinc formalin solution (Z-Fix Concentrate, Anatach LTD, Battle Creek, MI), and collected tissues were processed, cut at 5 µm, and stained with hematoxylin and eosin (H&E) for histopathological examination. Gamori’s one-step trichrome stain was done using the well-established, standard protocol with the exception that glacial acetic acid was replaced by hydrochloric acid.
Results
Pathology of Animals That Were Exposed to Lethal Doses of Aerosolized Ricin (Group I)
Gross and histological changes of 6 RM exposed to lethal doses of aerosolized inhaled ricin toxin were analyzed. The calculated doses range from 13.2 to 27.3 µg/kg body weight. The animals appeared normal immediately after the exposure, but within 12 to 30 hr postexposure, exhibited 1 or more of the following clinical signs: progressive anorexia, dyspnea, labored breathing, varying degrees of tachypnea, and hunched posture. Two of the 6 animals were found dead 36 and 42 hr later (confirmed by telemetry), while the remaining 4 were euthanized within 24 to 48 hr postexposure.
Gross and histological lesions in these animals were consistent with what was reported in RM previously (Wilhelmsen and Pitt 1996). Gross lesions were mainly confined to the lung and tracheobronchial lymph nodes. The thoracic cavity of all these animals contained variable amounts (10–80 cc) of serous to serosanguinous fluid. The fluid contained strands to sheets of fibrin that frequently coated the visceral pleural surfaces (Figure 1A). Lungs of all animals were wet, heavy, noncollapsed, firm, and exhibited prominent rib impressions. Variably sized, multifocal dark red to mottled areas were present on all lung lobes (Figure 1B). The cut surfaces of the lung frequently oozed frothy fluid. Frothy fluid was also present in the trachea and bronchi. Tracheal and mesenteric lymph nodes of 2 animals were twice the normal size.
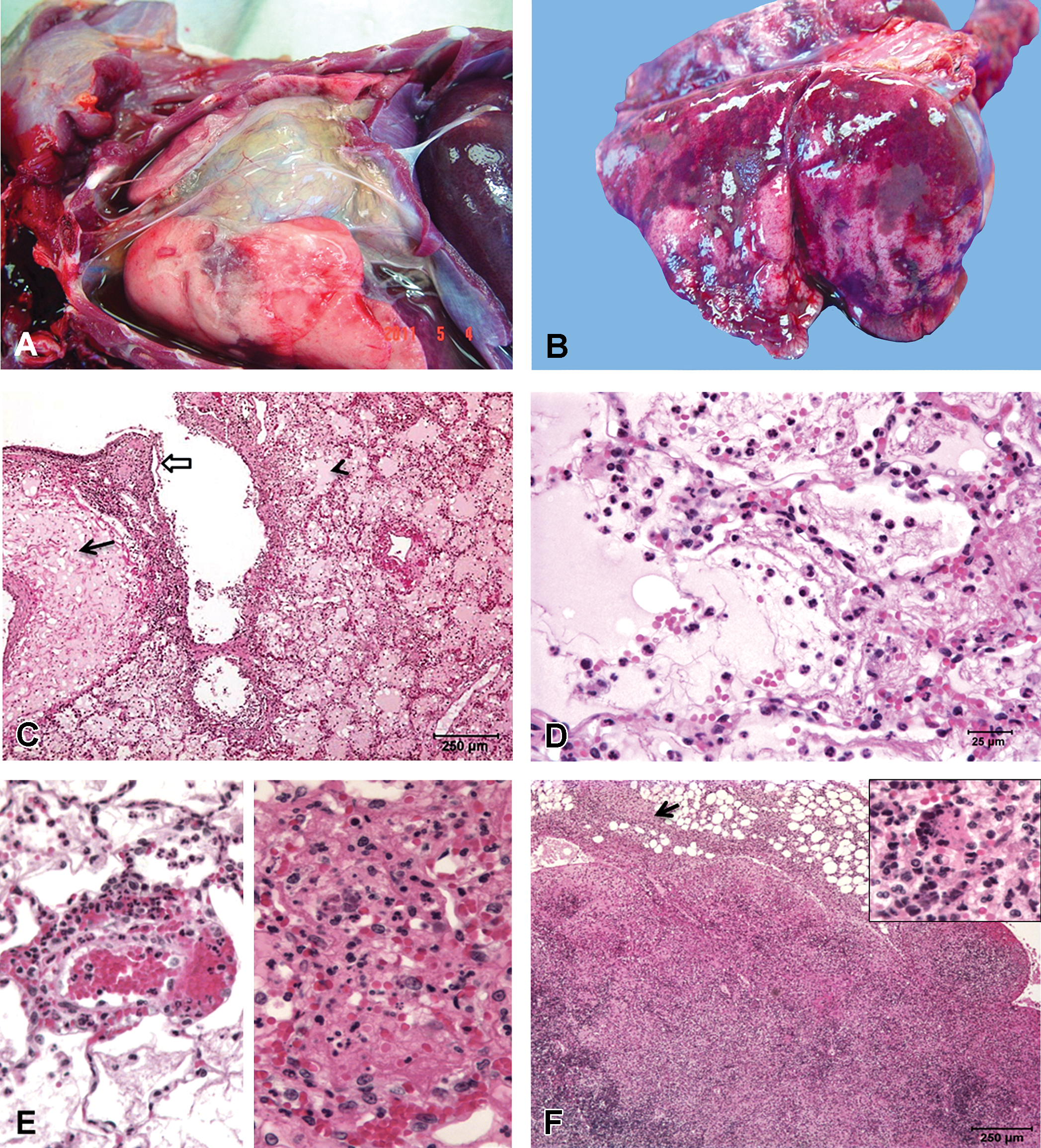
Acute lesions of lethal ricin toxicosis. Gross pathology: (A) Lung in situ: Marked effusion of protein-rich fluid into the thoracic cavity with abundant fibrin strands attached to the visceral pleura. (B): Lung from rhesus macaque (RM) HP95: wet, heavy, noncollapsed lung with multifocal dark red to mottled areas in all the lobes. Histopathological changes representative of all animals in group I: (C) Massive perivascular (solid arrow) and alveolar edema (solid arrow head) and partial denudation and necrosis of bronchiolar epithelium (block arrow). (D) Alveolar epithelial necrosis with hemorrhage, edema, fibrin, and degenerate neutrophils in the alveolar spaces. (E) Left panel: Blood vessels lined by leaky and reactive endothelial cells with perivascular leakage of erythrocytes and viable neutrophils; right panel: vascular necrosis with karyorrhexis, karyolysis, and loss of vessel wall integrity. (F): Bronchial lymph node: Lymphoid necrosis with loss of corticomedullary architecture and depletion of germinal centers. Necrotic cellular debris with degenerate neutrophils and fibrin (inset). The inflammation and necrosis extends beyond the capsule into the surrounding connective tissue (arrow).
Histologically, the most prominent feature was flooding of alveolar, perivascular, and peribronchial/bronchiolar spaces with large amounts of eosinophilic proteinaceous fluid (edema) containing abundant fibrin strands admixed with large numbers of degenerate and viable neutrophils and lesser numbers of lymphocytes, plasma cells, and alveolar macrophages (Figure 1C). These spaces frequently contained free erythrocytes (hemorrhage). The terminal bronchioles, alveolar epithelium, and alveolar spaces were the most affected. The alveolar septa were often expanded by fibrin, edema fluid, and aforementioned inflammatory infiltrate. The cellular debris and degenerate neutrophils were frequently present in the alveolar spaces admixed with the edema fluid. Multifocal to coalescing areas of necrosis with frequent, partial to complete loss of alveolar epithelial cells and alveolar architecture and loss of alveolar sepal integrity were present (Figure 1D). Edema frequently expanded the perivascular, peribronchial, and peribronchiolar spaces and contained neutrophils, lymphocytes, and less numbers of macrophages. The bronchiolar epithelium was occasionally and variably denuded and necrotic and bronchiolar and bronchial lumina contained cellular debris. Vascular necrosis (Figure 1E, right panel) and vessels lined by plump reactive endothelium surrounded by edema and granulocytes leaking into the perivascular spaces (Figure 1E, left panel) were present. However, large caliber vasculitis per se was not noted. Bronchus-associated lymphoid tissue was depleted in all the animals.
The tracheobronchial lymph nodes of all animals had moderate to severe loss of corticomedullary and follicular architecture (Figure 1F). Severe lymphoid depletion and replacement of the germinal centers with necrotic cellular debris was prominently present. These areas and sinuses were expanded by large numbers of degenerate and viable neutrophils, lymphocytes, plasma cells, macrophages, hemosiderophages, necrotic debris, and free erythrocytes admixed with edema and fibrin. The subcapsular sinuses were expanded by large numbers of neutrophils. These inflammatory and necrotic changes frequently extended beyond the capsule to the surrounding reticular connective tissue and adipose tissue. The upper respiratory tracts showed milder changes. Four of the 6 animals had mild suppurative tracheitis with submucosal edema. The tracheal lumen occasionally contained fibrin, cellular debris, and free erythrocytes. No discernible lesions were noted in the nasal cavity.
Other low-frequency lesions that might be related to ricin toxicosis included thymic depletion (3/6), lymphoid hyperplasia in tonsils, spleen, mesenteric lymph nodes with exhaustion matrix in the follicular centers (4/6), and unilateral adrenal medullary hemorrhage (1/6). Lesions considered unrelated to ricin toxicosis included unilateral adrenocortical mineralization (1/6), mild, unilateral interstitial nephritis (1/6), mild chronic hepatitis (1/6), and chronic colitis (1/6). A summary of main histological lesions noted in all animals involved in the study are presented in Table 2.
Summary of main histological lesions in RM treated with aerosolized ricin.
Note. 0 = absent, 1 = minimal, 2 = mild, 3 = moderate, 4 = severe.
Pathology of Animals Exposed to Sublethal Doses of Aerosolized Ricin (Group II)
Three animals given nonlethal dose of ricin toxin (Table 1) exhibited dyspnea, tachypnea, labored breathing, and anorexia initially like animals exposed to lethal dose of ricin. These signs decreased substantially within 48 to 60 hr, but mild sporadic signs persisted until termination of the study. Gross changes were limited to lung (2 animals) and lymph nodes (all animals). The congested lung of EM25 had prominent rib impressions and had multifocal to coalescing pale white to tan, firm areas in all lobes (Figure 2A). Posterior edge was gray and collapsed. The left lung of EE66 had extensive tan to dark red, rubbery, firm areas, and the right lung had a zone of collapse along the dorsal aspect of the lower lobe. The lung weights of all animals were higher than age-matched monkeys. All the lymph nodes were enlarged 2 to 5 times the normal size.
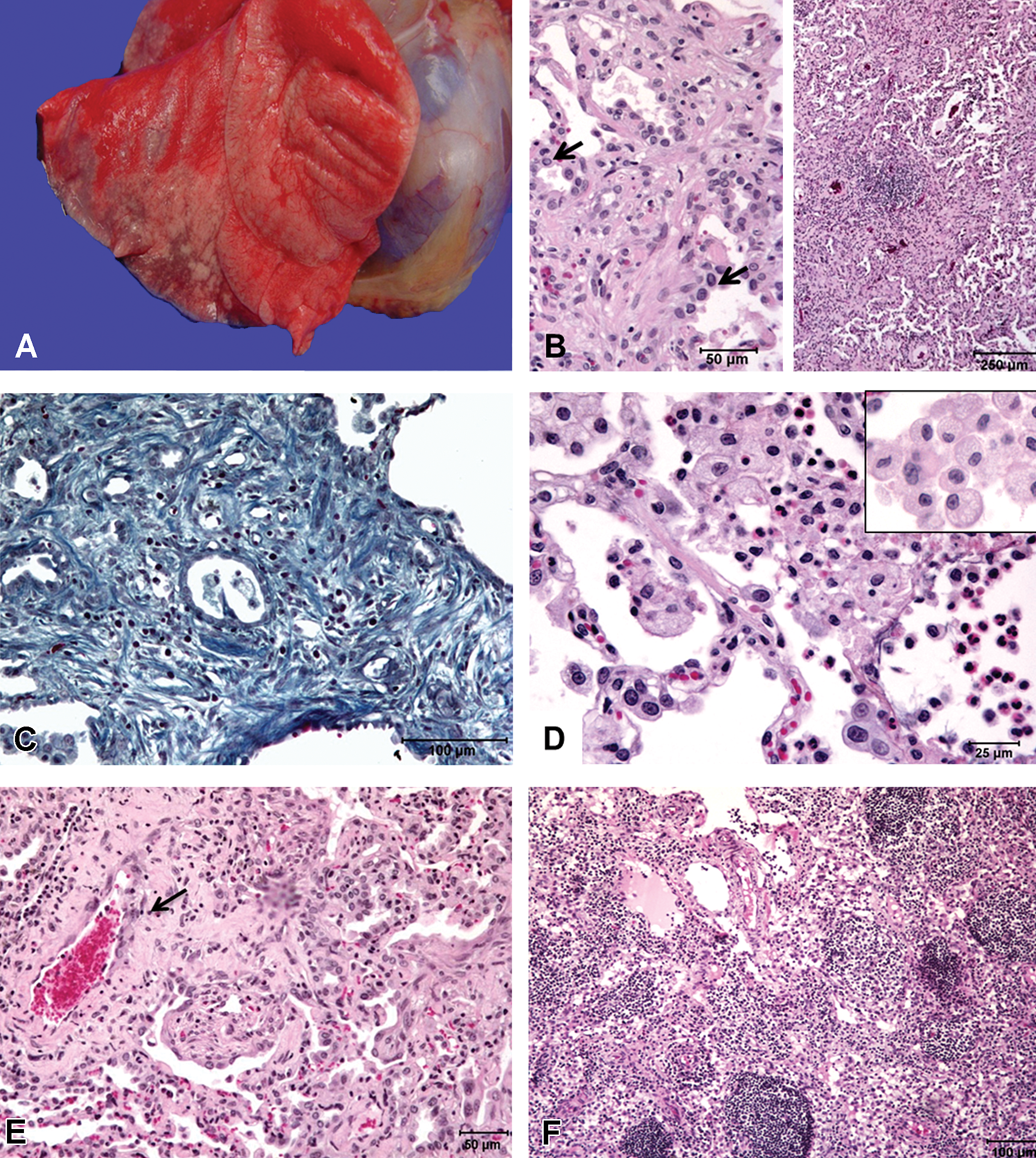
Lesions corresponding to sublethal doses of aerosolized ricin. Gross pathology: (A) Lung lobes are rubbery and noncollapsed with multifocal to coalescing gray to white firm areas. Histopathological changes: (B) Left panel: Alveolar interstitial fibrosis with type II pneumocyte hyperplasia (arrows); Right panel: Fibrosis obliterates the alveolar spaces and contain trapped cellular debris. (C): Gamori’s one-step trichrome stain revealed abundant collagenous stroma (blue stain) and sparse smooth muscle fibers. (D) Alveoli with large numbers of foamy macrophages and degenerate granulocytes. (E): Perivascualr fibrosis (arrow) with trapped degenerate inflammatory cells within the areas of fibrosis and type II hyperplasia. (F) The paracortical and medullary regions of a tracheobronchial lymph node with edema fluid containing foamy macrophages.
Histologically, similar changes were noted in all the 3 lungs in this group though the changes were more severe in EE66 (highest dose; Table 2). These changes were characterized by multifocal to coalescing, variably sized areas of fibroplasia and increased collagen deposition (fibrosis) often oriented around terminal bronchioles, replaced normal alveolar architecture, and contained trapped cellular and granulocyte debris (Figure 2B and E). Gamori’s one-step trichrome stain confirmed that these areas were predominantly composed of a collagenous stroma with sparse presence of smooth muscle cells (Figure 2C). Fibrosis often extended into the alveolar septa and expanded the interstitium (Figure 2B and E). Surrounding these areas of fibrosis and within the alveolar spaces were large numbers of foamy macrophages, lesser numbers of multinucleate giant cells, degenerate granulocytes, and occasional aggregates of lymphocytes, plasma cells, and siderophages (Figure 2D). Macrophages were also seen as small clusters or singly within the fibrotic nests or in close proximity to the areas of fibrosis. Fibrosis and inflammation were most prominent in alveoli around terminal bronchioles and in interstitium surrounding larger blood vessels. Prominent type II pneumocyte hyperplasia was noted multifocally (Figure 2B and E). Edematous perivascular areas noted in group 1 animals were mostly replaced by areas of fibrosis in this group (Figure 2E), although small lakes of proteinaceous fluid (edema) in alveoli, perivascular edema, and dilated lymphatics were occasionally present. The smooth muscle surrounding the larger airways appeared hyperplastic. Mild lymphoplasmacytic infiltration was noted in the tracheal mucosa and submucosa. Moderate lymphoplasmacytic rhinitis that affected the nasal septum was present in EM25 and EE66.
To see whether these changes were more pronounced in a particular lung lobe/lobes and to examine whether there is a dose-dependent increase in fibrotic response, the amount of fibrosis in each lobe was semiquantitatively graded by counting the number of fibrotic nests and their extent per 20× field for at least 5 fields in each lobe. No discernible, consistent difference was noted in the amount of fibrosis between different lobes of each individual monkey. Lobe to lobe comparison between EM25 and EF03 also did not reveal quantitative or qualitative differences. However, for all lobes, the changes were most severe in EE66 (highest dose) when compared to the other 2, which suggests that the highest sublethal dose in this study resulted in increased fibrosis and associated changes.
Tracheobronchial (3/3), mesenteric (3/3), inguinal (1/3), mandibular (1/3), and axillary (1/3) lymph nodes were examined. All the lymph nodes were reactive and had large lymphoid follicles, but with decreased lymphocytes in the germinal centers (exhaustion matrix). The sinuses and peripheral lymphatics were markedly dilated with numerous pigment-laden macrophages (which stained negative for iron), and fewer numbers of degenerate neutrophils and eosinophils. The paracortical and medullary regions of tracheobronchial nodes had occasional lakes of proteinaceous fluid (edema) containing foamy macrophages (Figure 2F). Mild to moderate lymphoid hyperplasia was noted in other lymphoid organs like spleen, thymus, and tonsils. Incidental adrenocortical mineralization (2/3) was noted.
Discussion
The gross and histopathological changes described in lethal and sublethal exposure to aerosolized ricin in RM were compared and contrasted in this study. The animals in group I exposed to lethal doses of aerosolized ricin had lesions nearly identical to those previously described in RM (Wilhelmsen and Pitt 1996). The consistent changes include severe, diffuse, necrotizing bronchiolitis, and alveolitis with fibrinopurulent bronchointerstitial pneumonia and massive alveolar, perivascular and peribronchial/bronchiolar edema, and necropurulent tracheobronchial lymphadenitis. However, adrenocortical necrosis described previously in 2 monkeys was not found in our study. The most significant difference in our study was the much smaller supralethal dose (∼2–5 times our LD50 of 5.8 µg/kg) given to the animals with a final dose range between 13.2 and 27.3 µg/kg; Roy et al. 2012). This LD50 is lower than the earlier reported dose of 15 µg/kg and the subsequent supralethal dosage (range between 21 and 41.8 µg/kg; Wilhelmsen and Pitt 1996; Anderson and Wannamacher 2005; Poli et al. 2007). The exposure to the lower doses produced similar lesions and clinical progression as seen in those exposed to the higher doses as seen in the previous report. The lower LD50 may be due to such variables as the source/potency of the toxin or possibly the overall efficiency of the inhalation delivery system. The methodologies used in the present study were modeled after those described in previous efforts with respect to aerosol characterization and delivery to the animal (Wilhelmsen and Pitt 1996) and as such this source of variation is discounted.
Massive pulmonary edema with necrotizing alveolitis that mainly affected the lower respiratory tract from terminal bronchioles to alveoli produced the clinical signs manifested by the animals in this study. The resultant, progressive impediment of oxygen exchange is thought to be the immediate cause of death. Inhaled particles that are 1 to 2 µm in diameter bypass the upper airways and bronchus and deposit in the bronchiolar–alveolar area producing similar, severe lesions in the lower respiratory tract of RM, Sprague-Dawley rats, and BALB/c mice (Brown and White 1997; Roy et al. 2003; Griffiths, Phillips, and Holley 2007; Roy et al. 2012; Benson et al. 2011; Anderson and Wannamacher 2005). Pulmonary changes observed in our macaques were similar to those reported for rats and mice with some exceptions. This includes 2 different studies involving BALB/c mice, CD-1 mice, and Sprague-Dawley rats where additional severe degenerative; necrotizing and apoptotic changes were noted in larynx and trachea and nasal turbinates (Benson et al. 2011; Poli et al. 1996) in contrast to the milder upper respiratory changes noted in our study and other reports in lab animal rodents. Also, vasculitis was reported in the lung of rats and mice (Benson et al. 2011) but was not prominent in RM. These differences seen in rodents might be due to the anatomical differences in the nasal cavities and the respiratory tree (reviewed in Griffiths, Phillips, and Holley 2007) together with a wider range of particle sizes that might have resulted in a broader deposition of the aerosol. Anatomical and methodological differences will produce some variation in the lesions observed in various animal models but, since there are few cases of human ricin toxicosis to compare, the range of lesions in animal models may prove useful and predictive.
Lesion formation in ricin toxicosis is attributed to the ability of ricin to inhibit protein synthesis via targeting eukaryotic ribosomes (Endo et al. 1987); however, the sequence of events that leads to the pulmonary and nodal lesions is not elucidated completely. We agree with the earlier reports (Wilhelmsen and Pitt 1996; Hertler and Frankel 1989) that the edema noted is primarily of permeability type based on the anatomic location, necrotic changes in the alveolar septa, and capillary endothelium and increased permeability of the endothelial lining of the larger vessels. Whether a cardiogenic component is present is one subject of our ongoing investigation. As speculated in the earlier report, necrotizing lymphadenitis that is localized in the tracheobronchial lymph node is most likely from the direct cytotoxic effect of ricin or its metabolites that are transported from the lung via the migrating leukocytes or lymph. Multiple in vitro studies and studies in mice have identified significant upregulation of various proinflammatory cytokines including tumor necrosis factor alpha (TNF-α), interleukin (IL)1β, IL-1 IL-8 c-FOS, c-JUN, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and pathways like cJun N-terminal kinase (JNK), extracellular signal-regulated kinases (ERK), and p38-mitogen-activated protein kinase (MAPkinase; Gonzalez, Farrant, and Mantis 2006; Licastro et al. 1993; Korcheva et al. 2005; Wong et al. 2007b) in response to ricin exposure. Two independent microarray studies in ricin-exposed lung tissues in BALB/c mice identified increased expression of genes that coded for proteins responsible for proinflammatory responses, formation of endothelial cell gaps, maintenance of membrane integrity and cellular adhesion, apoptosis, and early healing response (David, Wilkinson, and Griffiths 2009; DaSilva et al. 2003). So it appears that endothelial cell dynamics in this acute response is altered due to (1) direct cytotoxicity of ricin, (2) opening of the endothelial gaps with leakage of fluid, erythrocytes, and leukocytes and, (3) leukocyte-mediated endothelial injury via enzymes and proinflammatory mediators released during the acute inflammatory response. Our current studies are aimed at characterizing the molecular pathways involved in these processes.
To our knowledge, no study has examined lesions beyond the acute and early subacute stages of survivors after ricin exposure in a nonhuman primate model. The characterization of these lesions is of prime importance because of the following reasons. First, the authors are of the opinion that if a situation arises wherein aerosolized ricin is used as a bioweapon; the survivors of the attack will significantly outnumber the fatalities. The rationale behind this assumption is that the possibility of achieving a persistent lethal aerosol dose of ricin in unrestricted individuals in a relatively open environment would be less than achieving a sublethal aerosol dose. Second, there are various postexposure vaccine and antitoxin efficacy studies mostly in lab animal rodents (reviewed in Smallshaw and Vitetta 2012) that have shown different degrees of success in ensuring survival in animals exposed to ricin. So, in a scenario wherein an antitoxin is effective in preventing fatality, it still might produce some chronic lesions in the lung over time. Therefore, documentation of these changes and examining the underlying pathogenesis may be of significance in developing a treatment strategy and therapeutics for the same. We have used varying sublethal doses to identify lesions in survivors. The fibrosing interstitial pneumonia and type II pneumocyte hyperplasia with minimal edema and variable histiocytic and lymphoplasmacytic infiltration are indicative of an ongoing reparative process. The initial injury gives way to a subacute to chronic phase, where there is an influx of macrophages, clearing of alveolar spaces, and subsequent repair of the pulmonary architecture by a robust fibrotic response and type II pneumocyte hyperplasia. The reparative responses could be correlated with dose of ricin since among the 3 animals exposed to sublethal doses, the changes were more severe in the animal exposed to the highest sublethal dose (EE66). However, additional subjects are needed for statistical testing. From current observations, the fibrosis appears persistent but nonprogressive (data not shown). To our knowledge, effects of sublethal ricin exposure or survivors of inhaled ricin have not been characterized in primates. A vaccine efficacy study in New Zealand white rabbits using aerosolized ricin have reported occasional type II pneumocyte hyperplasia and interstitial fibrosis in the animals that survived the ricin challenge with occasional bronchiolitis obliterens, a feature not noted in our study (McLain et al. 2012). However, these animals were sacrificed 12 weeks postexposure and hence may reflect progressive reduction in the severity of damage over time when compared to our study. Also, tracheal instillation of sublethal doses of ricin resulted in mild to moderate interstitial fibrosis in another study in New Zealand white rabbits (Wong et al. 2007a). The increased numbers of foamy macrophages and their role in the fibrotic response in this study is uncertain. In vitro studies have shown that pulmonary inflammation triggered by ricin requires macrophages and IL-1 signaling (Lindauer et al. 2009). Also a growing body of evidence points toward macrophages as critical regulators of fibrosis through their ability to (1) produce a variety of profibrotic mediators, (2) recruit fibroblasts and inflammatory cells, (3) regulate activation and recruitment of myofibroblasts, and (4) antifibrotic response by distinct subpopulations (reviewed in Wynn and Barron 2010). Hence, the persistence of macrophages within and around the areas of fibrosis in our study might have a role in determining the speed and extent of fibrosis. Also, it was interesting to note that most of the lesions seen in lung were remarkably similar to those seen in bleomycin-induced lung injury, the most widely used model for pulmonary fibrosis in laboratory animals (reviewed in Moore and Hogaboam 2008). Recent studies have questioned the clinical relevance of these rodent models, especially in the idiopathic form of pulmonary fibrosis (Borzone et al. 2001; Gauldie and Kolb 2008) due to suspected differences in reparative responses of rodents and humans. A pulmonary fibrosis model in primates using ricin intoxication needs to be investigated.
The changes in the lymph nodes and other lymphoid organs were suggestive of responses to subacute to chronic inflammatory and reparative processes. The presence of pigment-laden macrophages in lymph nodes is a normal finding in RM. Though found in larger numbers in these macaques, its significance in this study is uncertain. The presence of edema and circulating granulocytes in tracheobronchial lymph nodes is indicative of the persistence of ricin-induced damage.
In summary, we describe the acute pathological changes in RM subsequent to aerosolized lethal doses of ricin and the chronic changes in survivors of sublethal doses of ricin. Further studies to elucidate the molecular mechanisms that control these processes and the validity of this treatment as a new model from human pulmonary fibrosis are in progress. The results from these studies may aid in the development of far more effective and reliable strategies for combating and/or eliminating 1 or more of the unfavorable outcomes associated with ricin toxicity and pulmonary fibrosis.
Footnotes
Acknowledgments
The authors acknowledge Stephanie Killeen and Rachael Redmann for their supporting roles in aerosolization studies and data collection. Drs. Nobuko Wakamatsu, Rudy Bauer, and Daniel B. Paulsen (Louisiana State University) are acknowledged for their input on the characterization of pathological changes. We also thank Maurice Duplantis for his assistance with necropsy and documentation of the pathological changes.
Author’s Note
Satheesh K. Sivasubramani's current affiliation is the University of Texas Medical Branch, Microbiology and Immunology, Galveston, Texas, USA.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the UK Ministry of Defence Contract Number DSTLX-1000036167 (Roy) and also supported in part by the National Institutes of Health, Office of the Director grant Number OD-011104-51. M. Bhaskaran is supported by National Institutes of Health Grant T32-OD011124.